

Abstract
We have previously reported that chaperone-rich cell lysates (CRCL) derived from the BCR-ABL+ 12B1 leukemia activate dendritic cells (DCs) and stimulate leukemia-specific immune responses. Because CRCL contain a variety of heat shock/chaperone proteins, we theorized that CRCL obtained from BCR-ABL+ leukemias are likely to chaperone BCR-ABL–derived fusion peptides and that DCs pulsed with 12B1 CRCL could cross-present BCR-ABL fusion peptides to T cells. We found that splenocytes from mice vaccinated with BCR-ABL+ leukemia-derived CRCL secreted interferon-γ (IFN-γ) when restimulated with a BCR-ABL peptide, GFKQSSKAL, indicating that BCR-ABL peptides are chaperoned by leukemia-derived CRCL. We next eluted peptides from 12B1 leukemia-derived CRCL and used high-pressure liquid chromatography (HPLC) fractions to restimulate splenocytes harvested from mice vaccinated with DC/GFKQSSKAL or DC/12B1 CRCL. We found that the same peptide fractions derived from 12B1 CRCL and from “refractionated” GFKQSSKAL stimulated IFN-γ production, suggesting the presence of BCR-ABL peptides in the peptide repertoire of 12B1 CRCL. We also demonstrated that immunization with DCs loaded with leukemia-derived CRCL induced BCR-ABL–specific cytotoxic T lymphocytes (CTLs) in vivo. Moreover, mice immunized with DCs pulsed with 12B1-derived CRCL had superior survival (60%) when compared with those immunized with DCs pulsed with BCR-ABL peptide (20%), indicating that CRCL vaccines provide additional immune stimulus over and above individual peptide vaccination.
Introduction
Tumor vaccines composed of multiple chaperone proteins generated by a free-solution iso-electric focusing (FS-IEF) technique have been shown to elicit potent antitumor immunity.1–4 We have termed these preparations as chaperone-rich cell lysates (CRCL) and found that tumor-derived CRCL activate dendritic cells (DCs) and stimulate tumor-specific, T-cell–mediated immune responses.2
CRCL contain at least 4 major immunogenic chaperone proteins: heat shock protein 70 (HSP70), HSP90, glucose-regulated protein 94 (GRP94), and calreticulin.1–3 We have further described CRCL as large, multimember entities wherein chaperones coseparate in high-molecular-weight fractions despite dissociative conditions used in size-exclusion chromatography.3
Srivastava and Maki5 postulated that chaperone protein “tumor antigens” could be carriers of immunogenic moieties such as peptides. In essence, the chaperones take on the immumnologic identity of the tissue in which they resided. This concept has become largely entwined with the antigenic properties of tumor-derived chaperones.6 We reasoned that tumor-derived CRCL could chaperone antigenic peptides. However, due to the isofocusing conditions used in the vaccine generation (ie, 6 M urea), one could argue that the peptide repertoire of CRCL would be lost. Also, recent works have questioned both the ability of chaperone proteins to maintain interactions with peptides in vivo7,8 and the necessity of tumor-specific peptides for chaperone protein vaccine effects.9,10 To study if, in fact, CRCL do chaperone tumor-specific antigens, we generated CRCL from 2 BCR-ABL+ murine leukemia tumors, 12B1 and 32Dp210.11,12 We theorized that CRCL obtained from these tumors were likely to chaperone BCR-ABL–derived fusion peptides. We further hypothesized that DCs could cross-present these peptides to T cells and induce specific immune responses in vivo. In this report, we show that BCR-ABL peptides are indeed carried by leukemia-derived CRCL and are presented by DCs to generate specific T cells; such cross-presentation is not major histocompatibility complex (MHC) class I restricted. Mice immunized with DCs loaded with leukemia-derived CRCL display superior survival compared with those vaccinated with DCs loaded with BCR-ABL peptide alone. This marks our first attempts to characterize the antigen spectrum from tumor-derived CRCL vaccine, and it re-enforces the notion that a broadened antigenic peptide repertoire found in chaperone protein–based vaccines might contribute to their overall effectiveness.
Materials and methods
Peptides
Synthetic BCR-ABL chimeric peptide, GFKQSSKAL, and control peptide, HYLSTQSALSK, were purchased from Sigma-Genosys (Sigma-Genosys, The Woodlands, TX). The 9-mer GFKQSSKAL (the bolded lysine represents the novel amino acid at the fusion junction) will also be referred to as “BCR-ABL peptide” within this work. The 11-mer HYLSTQSALSK peptide is processed by DCs and the resulting 9-mer HYLSTQSAL is predicted to bind with high affinity to H-2Kd.13
Bcr-abl–positive leukemia cell lines
12B1 is a murine leukemia cell line derived by retroviral transformation of BALB/c bone marrow cells with the human bcr-abl (b3a2) fusion gene, and these cells express the p210 BCR-ABL protein. This is an aggressive leukemia, with the 100% lethal dose (LD100) being 102 cells after tail-vein injection and 103 cells after subcutaneous injection.1,2,14 The 12B1 cell line was kindly provided by Dr Wei Chen (Cleveland Clinic, OH). 32Dp210 (American Type Culture Collection [ATCC], Rockville, MD) is another murine leukemia cell line derived by transformation of interleukin 3 (IL-3)–dependent 32D myeloid cell line with the BCR-ABL gene. The cell lines were tested monthly and found to be free of Mycoplasma contamination.
Mice
Six- to 10-week-old female BALB/c (H2d) mice or C3H/HeJ (H-2k) mice (Harlan Sprague Dawley, Indianapolis, IN) were used for the experiments. The animals were housed in a dedicated pathogen-free facility and cared for according to the University of Arizona Institutional Animal Care and Use Committee guidelines, Tucson.
Tumor generation
All tissue/cell culture reagents were purchased from Invitrogen (Gaithersburg, MD). 12B1 and 32Dp210 cells were cultured at 37°C and in 5% CO2 in RPMI medium containing 10% heat-inactivated fetal calf serum and supplemented with 2 mM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin sulfate, 0.025 μg/mL amphotericin B, 0.5 × minimal essential medium nonessential amino acids, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. Cells were prepared for injection by washing and resuspending in Hanks balanced salt solution. The cells were counted and adjusted to a concentration of 25 × 106 cells/mL. Female BALB/c mice or C3H/HeJ mice were injected with 0.2 mL (5 × 106) 12B1 or 32Dp210 cells, respectively, subcutaneously in both flanks and were monitored for tumor development. Tumors more than 1 cm in diameter were harvested from euthanized mice. In vivo passaging of tumors involved harvesting and mincing the tumor to produce a cell suspension. The cell suspension was filtered through a cell strainer (BD Biosciences Discovery Labware, Bedford, MA) to remove debris and was then centrifuged. The cell pellet was resuspended, washed, counted, and injected into mice.
Generation of chaperone-rich cell lysate (CRCL) vaccines
12B1 and 32Dp210 tumor tissue grown in vivo was harvested from mice. CRCL were generated as described previously.1–3,15 Briefly, tumor was homogenized in lysis buffer and a 100 000g supernatant was obtained and quantified (bicinchoninic acid [BCA] protein assay; Pierce Endogen, Rockford, IL). The high-speed supernatant was subjected to FS-IEF in a Bio-Rad Rotofor cell (Hercules, CA) for 5 hours at 15 W constant power. Twenty fractions were harvested, and each fraction was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot probing with specific antibodies for the chaperones HSP60, HSP70, HSP72 (inducible form of HSP70), HSP90, GRP94, and calreticulin. Fractions from FS-IEF that contained substantial amounts of the above chaperone proteins, as determined by SDS-PAGE and Western blotting, were pooled and dialyzed stepwise out of urea and detergents. Pooled fractions were then concentrated using Centricon devices (Millipore, Bedford, MA). Detergents were removed by passage over an Extractigel matrix (Pierce Endogen). The fractions were reconstituted in phosphate-buffered saline (PBS), quantified and stored at 70°C until use.
Generation of bone marrow–derived DCs
BALB/c mouse bone marrow DCs were generated using a slightly modified protocol from that described previously.16 Bone marrow was harvested from femurs and tibiae and filtered through a Falcon 100-μm nylon cell strainer (Becton Dickinson Labware, Franklin Lakes, NJ). Red blood cells were lysed in a hypotonic buffer and the marrow was cultured in complete RPMI medium (therapeutic grade; Invitrogen) containing 10% fetal calf serum, l-glutamine, human serum albumin, 50 μg/mL streptomycin sulfate, and 10 μg/mL gentamicin sulfate. Murine granulocyte macrophage–colony-stimulating factor (GM-CSF; 10 ng/mL; Peprotech, Rocky Hill, NJ) and IL-4 (10 ng/mL) were added to the culture. After 6 days, the nonadherent and loosely adherent cells were harvested, washed, and used for in vivo and in vitro experiments. Fewer than 10% of these cells were contaminated by macrophages (CD14+ cells).
Immunization strategy
Day-7 DCs were pulsed with 100 μg/mL HYLSTQSALSK or GFKQSSKAL in the presence of 5 μg/mL β2-microglobulin (Accurate Chemical and Scientific, Westbury, NY) for 3 hours, or pulsed with 50 μg/mL leukemia-derived CRCL overnight. DCs were then harvested, washed with PBS 3 times, and resuspended in PBS. For interferon γ (IFN-γ) production and cytotoxicity assay, a total of 5 × 105 DCs were injected per mouse subcutaneously into the left groin on day −14 and day −7.
ELISPOT assays
Enzyme-linked immunospot (ELISPOT) assays were performed to assess IFN-γ production of splenocytes from vaccinated mice following in vitro stimulation with CRCL or peptides. Splenocytes (106) were cultured with 50 μg/mL CRCL or 5 μg/mL peptides for 48 hours on Millipore MultiScreen-HA 96-well plates (MAHA S45; Millipore, Bedford, MA). The plates had been previously coated overnight with anti–IFN-γ capture antibody (10 μg/mL, clone R4-6A2, rat monoclonal antibody (mAb) antimouse IFN-γ; BD PharMingen, San Diego, CA). Cells were then washed out with copious amounts of PBST (PBS plus 0.05% Tween 20). Biotinylated anti–IFN-γ antibody (2 μg/mL, clone XMG1.2, rat mAb antimouse IFN-γ; BD PharMingen) was added for 2 hours. Free antibody was washed out, and the plates were incubated with horseradish peroxidase (HRP)–linked avidin (ABC Elite reagent, 1 drop each of Reagent A and Reagent B per 10 mL PBS; Vector Laboratories, Burlingame, CA) for 1 hour, following extensive washing with PBST, and then washing with PBS. Spots were visualized by the addition of the HRP substrate 3-amino-9-ethylcarbazole (AEC; Sigma Chemical, St. Louis, MO) prepared in acetate buffer (pH 5.0) with 0.015% hydrogen peroxide. Spots were examined using a dissecting microscope. Wells of interest were photographed with a microscope-mounted Cool SNAP CCD camera (RS Photometrics, Tucson, AZ), and images captured with RS Image, Version 1.07 (Roper Scientific, Tucson, AZ). The image of each well was electronically optimized to visualize the maximum number of spots. The number of spots above background was plotted.
Cytotoxicity assay
BALB/c mice were immunized as indicated in “Immunization Strategy.” Seven days after the second immunization, splenocytes from the immunized mice were harvested. The in vivo–primed splenocytes were cultured in complete RPMI for 6 days in the presence of 10 μg/mL 12B1 CRCL and 20 U/mL IL-2. Viable cells were then collected by Ficol density centrifugation and used as effector cells. Dendritic cells loaded with peptide or CRCL were used as target cells. Day 5 bone marrow–derived DCs were incubated with 5 μg/mL HYLSTQSALSK or GFKQSSKAL for 3 hours; alternatively, DCs were cocultured with 50 μg/mL 12B1 tumor-derived CRCL overnight. Cells were collected and used as target cells. Stimulated effector cells were incubated with 2 × 104 target cells at indicated ratios for 6 hours and cytolytic activity was measured by nonradioactive cytotoxicity assay (Promega, Madison, WI) following the instructions provided. The percentage of cytotoxicity was determined according to the formula provided in the kit instructions.
Reverse-phase high pressure liquid chromatography (rpHPLC) fractionation of 12B1 CRCL eluted peptides and identification of fractions containing BCR-ABL peptide. Peptides were stripped from CRCL by procedures similar to those used to elute peptides from MHC molecules.17,18 CRCL (3 mg-10 mg) were acidified by addition of trifluoroacetic acid (TFA; all HPLC-grade solvents were from Mallinkrodt Baker, Phillipsburg, NJ; HPLC grade) to 0.2% and left on ice for 1 hour. Peptides dissociated from CRCL were harvested by centrifugation through a Centricon-3 device (3-kDa cutoff membrane; Millipore). The centricon device was precleared prior to addition of peptides by passage of 0.2% TFA in PBS through the membrane. One mL HPLC-grade water was added to the retentate, and was centrifuged through the membrane. The initial flow-through material was pooled with the water-wash flow-through, and the combined materials were vacuum evaporated (Speed Vac; Thermo-Savant, Farmingdale, NY). The peptides were resuspended in 0.1% TFA in HPLC-grade water (TFA/H2O, solvent A) and chromatographed over a YMC ODS-A S5 C18 reverse-phase column, 3.0 × 250 mm (5 μm beads, 120-angstrom [12 nm] pore size; Waters, Milford, MA). The chromatography was performed using a WatersAlliance 2690 separation module equipped with a PDA 996 photodiode array detector (Waters). The column was run at a flow rate of 0.4 mL/minute with a solvent gradient of 0% to 30% solvent B (0.1% TFA in HPLC-grade acetonitrile) over 30 minutes, then to 100% B by 40 minutes. Fractions (0.4 mL) were collected each minute, and eluent was monitored from 190 nm to 400 nm, with results shown for 214-nm wavelength. Between samples, the column was extensively washed with solvent B before returning to equilibration in solvent A.
Fractions were dried (Speed-Vac), washed once with tissue-culture–grade water, and dried again. Fractions were frozen at −20°C until used as restimulators in ELISPOT assays. For those experiments, fractions were resuspended in complete RPMI media, and each fraction was used in duplicate to restimulate splenocytes from mice that had been vaccinated with DCs pulsed with (1) 12B1-derived CRCL, (2) GFKQSSKAL peptide, or (3) HYLSTQSALSK peptide. IFN-γ ELISPOT assays were conducted as described in “ELISPOT assays.” Control HPLC separations and restimu-lators included the GFKQSSKAL peptide alone, the HYLSTQSALSK alone, PBS made to 0.5% TFA and treated as were the CRCL peptides, murine serum albumin (10 mg, fraction V; Sigma) treated the same as CRCL, and peptide retention standards (Pierce Endogen).
Immunoblotting
The absence of p210 BCR-ABL protein in the 12B1 CRCL or 32Dp210 CRCL was confirmed by Western blotting and compared with the 12B1 or 32Dp210 tumor lysates. CRCL from BCR-ABL− murine B-cell lymphoma, A20, were used as negative control. Tumor lysates refer to the starting material of FS-IEF that were prepared as described in “Generation of CRCL vaccines.” An equivalent amount of protein was loaded onto the gel as determined by BCA assay and examination of the Ponceau Red–stained blots. Following SDS-PAGE, the gels were electroblotted to nitrocellulose using an Idea Scientific electroblotter (Minneapolis, MN). Gels were transferred in 25 mM Tris, 200 mM glycine, 20% methanol overnight at 60 V, stained with Ponceau Red, and destained in TBST (50 mM Tris-Cl, 150 mM NaCl, 0.1% Tween 20, pH 7.4). Blots were blocked in 3% nonfat dried milk in TBST for 20 to 60 minutes, followed by three 5-minute rinses in TBST. The protein of interest was identified using the mAb to bcr (AB-2) clone no. 7C6 (Oncogene Research Products, Cambridge, MA). Primary antibody solutions were prepared in blocking solution, and blots were incubated in primary antibody for 1 hour at room temperature or 12 hours at 4°C, followed by three 5-minute rinses in TBST. Peroxidase-conjugated goat antimouse secondary antibody was applied for 1 hour at room temperature or 12 hours at 4°C followed by chemiluminescent detection (Super Signal; Pierce Endogen).
In vivo tumor growth experiments
Mice were injected with 103 viable 12B1 cells in the right groin on day 0. DCs were pulsed with 100 μg/mL HYLSTQSALSK or GFKQSSKAL in the presence of 5 μg/mL β2-microglobulin (Accurate Chemical and Scientific) for 3 hours, or pulsed with 50 μg/mL 12B1 tumor-derived CRCL overnight. DCs were then harvested, washed with PBS 3 times, and resuspended in PBS followed by subcutaneous injection into mice. On day 2, a total of 5 × 105 DCs were injected per mouse subcutaneously into the left groin. As control, mice were injected with an equal number of nonpulsed DCs. To exclude potential CRCL toxicity on DCs, day 6 DCs were plated in 96-well flat-bottom plates (50 000 cells/well) in the presence of increasing concentrations of 12B1 tumor-derived HSP70 or CRCL (0 μg/mL to 100 μg/mL) for 24 hours. MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], stock solution 5 mg/mL; Sigma) at 10 μL per well as added for an additional 4 hours. The supernatant was aspirated and the formazan crystals were solubilized in dimethylsulfoxide, followed by determination of optical densities at 560 nm and 690 nm using a microtiter plate reader. Tumor size was measured every other day with calipers once the tumors became palpable. Tumor volume was calculated using the following formula: length × width2 × π/6. Differences in mean tumor volume between groups were compared using an unpaired t test. In other experiments the Kaplan-Meier product-limit method was used to plot survival curves and the log-rank statistic was used to test differences between groups.19,20 Mice with tumors were euthanized at the end point listed.
Results
Immunization of mice with BCR-ABL+ leukemia-derived CRCL induces BCR-ABL–specific IFN-γ production
To determine whether or not BCR-ABL peptides were major antigenic components of BCR-ABL+ leukemia-derived CRCL, IFN-γ production of splenocytes from immunized mice was measured by ELISPOT assays. The rationale behind this is that DCs will process the peptides chaperoned by CRCL and in turn present them to T cells. If BCR-ABL peptide is, in fact, carried by CRCL, the in vivo tumor CRCL-primed T cells will release IFN-γ when restimulated in vitro with BCR-ABL peptide. Mice were therefore immunized with DCs pulsed with leukemia-derived CRCL, a BCR-ABL peptide, GFKQSSKAL, or control peptide, HYLSTQSALSK, on days −14 and −7. On day 0, splenocytes were harvested and restimulated in vitro with the CRCL or individual peptides. We found that splenocytes from mice primed with 12B1- or 32Dp210-derived CRCL secreted substantial amounts of IFN-γ when restimulated with BCR-ABL peptide, GFKQSSKAL (Figure 1). This finding indicates that BCR-ABL peptides from 2 different murine leukemias are immunologically prominent components of CRCL. It also indicates that the peptide can be presented by DCs irrespective of the MHC haplotype of the source of CRCL (eg, H-2d for 12B1 CRCL, and H-2k for 32Dp210 CRCL). Similarly, when splenocytes from mice immunized with GFQKSSKAL-loaded DCs were restimulated with either GFKQSSKAL peptide or with 12B1 or 32Dp210 CRCL, there was substantial IFN-γ release. CRCL-primed or GFKQSSKAL-primed splenocytes failed to secrete IFN-γ when restimulated with an irrelevant peptide, HYLSTQSALSK (Figure 1), or with CRCL derived from a BCR-ABL− tumor, A20 (data not shown). Moreover, when splenocytes from mice primed with A20 CRCL were restimulated with GFKQSSKAL or with 12B1 CRCL, there was no significant amount of IFN-γ production (data not shown). These findings confirm that GFKQSSKAL is a component of 12B1 CRCL and 32Dp210 CRCL.
Figure 1.
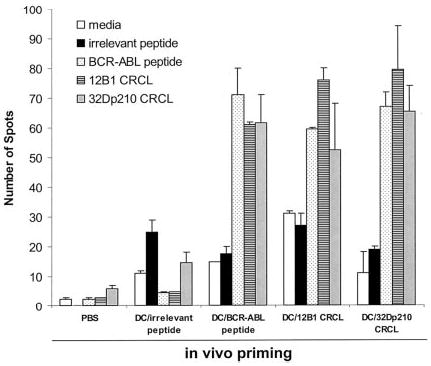
Immunization of mice with DCs loaded with bcr-abl+ leukemia-derived CRCL induces BCR-ABL–specific IFN-γ secretion. Mice were immunized with DCs (5 × 105) loaded with 12B1- or 32Dp210-derived CRCL, or with a BCR-ABL peptide, GFKQSSKAL, or with an irrelevant peptide, HYLSTQSALSK, on days −14 and −7. Control mice were immunized with equal volumes of PBS or equal numbers of empty DCs. On day 0, splenocytes were collected and restimulated with indicated peptide or CRCL for 48 hours. IFN-γ production was determined by ELISPOT assay. Error bars represent SEM.
Fractionation of antigenic peptides in 12B1 CRCL
One of the tenets of chaperone protein–based vaccines is that such proteins should have access to essentially the entire cellular peptide repertoire due to their chaperone activities.6 CRCL vaccines, consisting of multiple chaperones from various subcellular compartments, should theoretically sequester a diverse variety of peptide antigens from the tumor or tissue of origin. To examine the peptide content of 12B1-derived CRCL vaccines, we acid-stripped peptides from the vaccine and fractionated the peptides via rpHPLC (Figure 2A). We then used the fractionated peptides as restimulators in IFN-γ ELISPOT assays of primed splenocytes from mice that had been vaccinated with DCs pulsed with 12B1 CRCL, or with the GFKQSSKAL peptide, or with the irrelevant HYLSTQSALSK. The objective was to determine which fractions from the peptide separation would stimulate IFN-γ release from the appropriately primed splenocytes. As shown in Figure 2B, rpHPLC fractions 10 to 12 from peptides derived from 12B1 CRCL could induce IFN-γ secretion from splenocytes of mice that had been vaccinated with either DC/12B1 CRCL or DC/GFKQSSKAL. As a positive control, we subjected GFKQSSKAL to rpHPLC separation under identical conditions as for the 12B1 CRCL-derived peptides. Again, fractions 10 to 12 provided the best restimulation for the splenocytes. These data indicate that fractions 10 to 12 from 12B1 CRCL peptide chromatography and from GFKQSSKAL chromatography may contain some common antigenic components, that is, BCR-ABL peptides that are likely present in the peptide repertoire of 12B1 CRCL. As a negative control, PBS was subjected to the same acid treatment and rpHPLC as were the peptides. No fractions from the faux “peptide” separation from PBS were capable of inducing IFN-γ output from splenocytes from any of the vaccinated animals (Figure 2B and data not shown). Other controls included rpHPLC of HYLSTQSALSK, peptide separations from peptide retention standards, and materials stripped from murine serum albumin. Only the HYLSTQSALSK was able to stimulate IFN-γ responses from splenocytes of DC/HYLSTQSALSK vaccinated mice; all other controls were negative (data not shown). It should furthermore be noted that fractions 20 to 24 from 12B1 CRCL peptides also stimulated significant amounts of IFN-γ secretion from splenocytes primed with DC/12B1 CRCL. Thus, fractions 20 to 24 from 12B1 CRCL may contain some other antigenic peptides that contribute to the overall immunogenicity of 12B1 CRCL.
Figure 2.
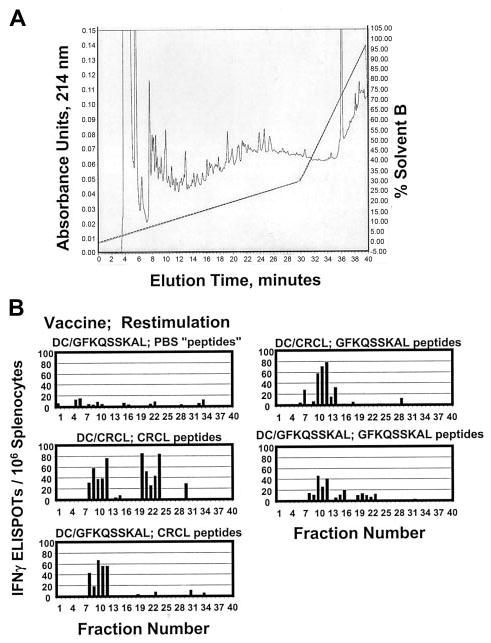
Fractionation of antigenic peptides in 12B1 CRCL. (A) Peptides were acid-stripped from 12B1 CRCL and applied to a reverse-phase C18 HPLC column (rpHPLC). Peptides were separated with a 0% to 30% acentonitrile gradient over 30 minutes, followed by a 30% to 100% acetonitrile gradient over the next 10 minutes (40 minutes total, dotted line shows solvent B gradient). Elution was monitored at 214 nm (shown from 0–0.15 arbitrary units). In addition, GFKQSSKAL peptide was likewise subjected to HPLC (data not shown). Phosphate-buffered saline (PBS) was treated identically to CRCL as a faux peptide separation and as a negative control (data not shown). (B) Forty HPLC fractions from each peptide separation were collected, dried, washed, reconstituted in media, and used to restimulate the splenocytes from mice vaccinated with DC/GFKQSSKAL or DC/12B1 CRCL. IFN-γ production was determined by ELISPOT assay. PBS “peptides” refers to materials derived from PBS and fractionated by rpHPLC.
Immunization of mice with 12B1-derived CRCL–loaded DCs induces BCR-ABL–specific CTL activity
Previous studies in our laboratory have shown that splenocytes from mice immunized with DCs pulsed with BCR-ABL peptide or 12B1 CRCL specifically lysed 12B1 cells but not BCR-ABL− A20 cells.4,21 In this study, we wished to verify and extend our hypothesis that DCs effectively cross-present 12B1 CRCL–chaperoned BCR-ABL fusion peptides in a manner capable of inducing specific cytotoxic T cells (CTLs). We therefore examined whether immunization with 12B1 CRCL–loaded DCs induce BCR-ABL–specific CTLs. Mice were immunized with DC/12B1 CRCL, DC/GFKQSSKAL, or DC/HYLSTQSALSK. In vivo–primed splenocytes were restimulated in vitro with 12B1 CRCL for 6 days. Viable cells were then collected by density centrifugation and used as effector cells. These effectors were a mixed population of T cells containing both CD4+ T cells and CD8+ T cells. GFKQSSKAL- or 12B1 CRCL–loaded DCs served as target cells. For controls, DCs were loaded with irrelevant peptide. Primed splenocytes from mice immunized with either DCs loaded with GFKQSSKAL or with 12B1 CRCL displayed significant cytolytic activity against DCs pulsed with either GFKQSSKAL or with CRCL (Figure 3). These results again indicate the presence of GFKQSSKAL in CRCL and further suggest that GFKQSSKAL peptides are cross-presented to CD8+ T cells by DCs. Splenocytes from mice vaccinated with either saline or unpulsed DCs displayed no cytotoxicity against any of the target cells. In addition, no significant killing was observed against HYLSTQSALSK-pulsed DCs (Figure 3), demonstrating the specificity of CRCL or GFKQSSKAL immunization.
Figure 3.
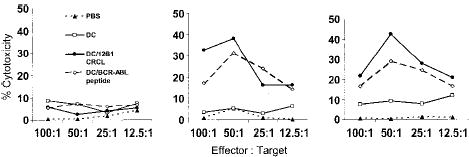
Immunization of mice with 12B1-derived CRCL-loaded DCs induces BCR-ABL–specific CTL activity. Mice were immunized as described in “Materials and methods.” On day 0, splenocytes were harvested and cultured in the presence of 10 μg/mL 12B1 CRCL and 20 U/mL IL-2 for 6 days. Viable cells were then harvested by Ficol density centrifugation and used as effector cells. Day-5 bone marrow–derived DCs were incubated with 5 μg/mL HYLSTQSALSK or GFKQSSKAL peptide for 3 hours; alternatively, DCs were cocultured with 50 μg/mL 12B1 tumor-derived CRCL overnight. Cells were then collected and used as target cells. Effector cells were tested for cytolytic activity against indicated target cells by nonradioactive cytotoxicity assay kit (representative data from 2 experiments are shown). ▴ indicates PBS; □, DC; •, DC/12B1 CRCL; and ⋄, DC/BCR-ABL peptide.
Absence of BCR-ABL p210 protein in leukemia-derived CRCL
Western blotting was performed to exclude the possibility that BCR-ABL–specific IFN-γ secretion or CTL activity of splenocytes from vaccinated mice was due to the coseparation of BCR-ABL p210 protein in leukemia-derived CRCL preparations. BALB/c-derived antigen-presenting cells (APCs) can present BCR-ABL peptides on their MHC class I molecules (H-2Dd).21 Therefore, if BCR-ABL protein was present in the CRCL preparations, DCs could potentially process the protein and present BCR-ABL peptides on their MHC class I molecules to CD8+ T cells. An antibody against c-ABL that recognizes the c-terminal portion of ABL protein (but not BCR-ABL fusion peptides) was used. BCR-ABL protein was undetectable by Western blotting in 12B1 CRCL or 32Dp210 CRCL (Figure 4), but was present in the lysates from both tumors (ie, the starting material processed by FS-IEF). This confirms that the BCR-ABL specificity of leukemia-derived CRCL immunization was not an artifact of BCR-ABL protein coseparating into fractions pooled for CRCL vaccine preparation but likely due to BCR-ABL peptides chaperoned by CRCL.
Figure 4.
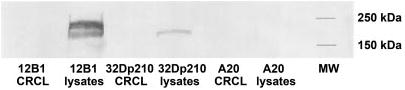
Absence of BCR-ABL p210 protein in 12B1 or 32Dp210 CRCL. 12B1 or 32Dp210 tumor lysates and CRCL from each of the lysates were prepared and separated on SDS-PAGE, followed by transfer to nitrocellulose for Western blotting. Presence of p210 BCR-ABL protein was probed using an antibody for c-ABL. A20 tumor lysates and CRCL served as negative controls.
Immunization of mice with 12B1-derived CRCL–loaded DCs provides superior therapeutic effects
BCR-ABL peptide–based vaccines have been explored in a variety of clinical studies.22,23 Given that other antigenic components may also be present in 12B1 CRCL (Figure 2), in vivo experiments were then conducted to compare the efficacy of CRCL-loaded DCs with that of GFKQSSKAL-loaded DCs. Mice were inoculated subcutaneously with a lethal dose of 12B1 cells on day 0. Bone marrow–derived DCs were pulsed with 12B1-derived CRCL or with GFKQSSKAL, or with HYLSTQSALSK. The DCs were washed and resuspended in PBS and each mouse was immunized subcutaneously with 5 × 105 DCs on day 2. Immunization with CRCL-loaded DCs significantly inhibited tumor development when compared with all other groups. This vaccine resulted in eradication of tumor growth in 60% of mice. DCs pulsed with GFKQSSKAL were less effective albeit also significantly better than saline or DCs pulsed with HYLSTQSALSK, with about 20% of mice remaining tumor free (Figure 4). These findings suggest that 12B1 tumor-derived CRCL may offer an advantage as a source of tumor antigens for pulsing DCs when compared with a single peptide such as GFKQSSKAL.
Discussion
Vaccination with heat shock proteins isolated from tumor has been shown by a variety of studies to elicit specific T-cell immunity against the tumor of origin.1–3,24,25 It is been proposed that the immunogenicity of HSPs relies on the peptides that are chaperoned by HSPs. Chaperone proteins have been purified from cells containing known peptide antigens, including viral epitopes,26–29 minor histocompatibility antigens,30 or artificial ones transfected or transduced into the cells such as β-galactosidase30 or chicken ovalbumin.31,32 The presence of these peptide antigens in chaperone protein preparations has been demonstrated via direct and indirect assays. However, the presence of an antigenic (T-cell) epitope derived from an oncogene as critical as BCR-ABL within HSP/chaperone preparations has not been reported.
Our laboratory has demonstrated that CRCL, which contain multiple chaperone proteins, elicit potent antitumor immunity.1–3 Specificity experiments have shown that CRCL vaccine from one tumor does not protect animals against challenge from a different tumor type.2 Those results argue that antigenic peptides are indeed present in CRCL vaccines. In the current study, we used BCR-ABL+ murine leukemia models to study the antigenicity of CRCL. Since 12B1 and 32Dp210 tumor cells express BCR-ABL protein, we reasoned that BCR-ABL fusion peptides are likely to be components of the antigenic repertoire of leukemia-derived CRCL. To test this hypothesis, we examined whether immunization of mice with DCs loaded with CRCL induced BCR-ABL–specific immune responses in vivo.
We found that splenocytes from mice immunized with DCs loaded with CRCL secreted large amounts of IFN-γ when restimulated with a BCR-ABL peptide, GFKQSSKAL, indicating that BCR-ABL peptides, either this specific 9-mer peptide, GFKQSSKAL, and/or its longer precursors, are chaperoned by 12B1 tumor-derived CRCL. Similarly, splenocytes from mice vaccinated with CRCL derived from 32Dp210 were able to respond to either GFKQSSKAL or CRCL from 12B1 (and vice versa), indicating that cross-priming over MHC restrictions is possible. rpHPLC fractions 10 to 12 appear to contain the GFKQSSKAL peptide since these fractions reproducibly stimulated IFN-γ production from GFKQSSKAL or 12B1 CRCL–immunized mice. Moreover, fractions 20 to 24 from 12B1 CRCL may contain some additional antigenic peptides since they stimulated IFN-γ production of 12B1 CRCL–primed splenocytes but not GFKQSSKAL-primed splenocytes (Figure 2). The potential candidate antigens chaperoned by CRCL may include Wilms tumor antigen (WT-1),33 minor histocompatibility antigens,34,35 and protease 3 antigen (PR1),36 which have been documented to be potential chronic myeloid leukemia (CML) tumor-associated antigens. 12B1 cells express WT-1 protein by Western blotting (Y.Z., unpublished data, October 2003). We have found that among a number of interesting peptides there are several that may have roles in cancer, including thymosin beta 4, glutathione transferase, and triosphosphate isomerase (M.W.G., unpublished data, March 2004). More experiments are ongoing in our laboratory to identify the antigenic components of CRCL chaperoned peptide repertoire.
We have demonstrated herein that there are numerous peptides confined within CRCL (Figure 2), and that some of those peptides are antigenic. Despite recent reports implying that chaperone adjuvant effects are of primary importance in generating antitumor immunity following chaperone protein immunization,9,37,38 the specificity of CRCL vaccine draws attention to CRCL’s peptide repertoire. Because CRCL contain multiple chaperone complexes, we do not have clear evidence as to the nature of the peptide-chaperone protein interactions within CRCL, nor do we propose that such interactions must occur readily in vivo and prior to cell lysis.39,40 Nonetheless, the presence and immunogenicity of tumor-derived peptides shown here undoubtedly contribute to CRCL vaccine efficacy.
DCs are professional APCs, known to be critical activators of T-cell responses. Although the mechanisms are not completely clear, an increasing body of data suggests that DCs take up chaperone-peptide complexes through specific receptors and re-present the peptides on MHC class I molecules.39,41–44 Following uptake, the chaperone protein–escorted peptides are processed and re-presented on MHC class I molecules to generate antigen-specific CTLs. BCR-ABL peptides, which are chaperoned by 12B1 CRCL as indicated by data shown in Figures 1–3, have been previously reported to be able to bind MHC class I and MHC class II molecules.45–48 Other studies in our laboratory have shown that DCs take up CRCL and present peptides associated with CRCL on their surface (K. L. Kislin, M.W.G., D. F. Lake, E.K., manuscript in preparation). We therefore hypothesized that BCR-ABL peptides are cross-presented on MHC class I molecules for CD8+ T-cell priming. We found that splenocytes from mice immunized with CRCL-loaded DCs specifically lysed GFKQSSKAL-loaded target cells, but not HYLSTQSALSK-pulsed DCs. Similarly, splenocytes from mice immunized with GFKQSSKAL-loaded DCs lysed CRCL-pulsed targets (Figure 2). These findings clearly demonstrate that immunization with 12B1-derived CRCL induces BCR-ABL–specific CTL responses (Figure 5). They further indicate that DCs process CRCL-chaperoned antigens and cross-present the escorted antigenic peptides, including GFKQSSKAL, to T cells in vivo.
Figure 5.
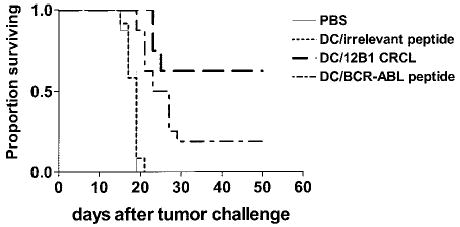
Immunization of mice with 12B1-derived CRCL-loaded DCs provides superior therapeutic effects. BALB/c mice were injected with 3 × 103 12B1 cells subcutaneously in the right groin on day 0. On day 2, mice were immunized with 5 × 105 DCs that had been loaded with 12B1-derived CRCL or indicated peptides as mentioned in “Materials and methods.” Survival of mice was monitored and displayed in the Kaplan-Meier plot (n = 8–16 mice per group; pooled data from 2 experiments are shown). PBS versus DC/CRCL, P < .05; PBS versus DC/BCR-ABL peptide, P <.05; DC/irrelevant peptide versus DC/CRCL, P <.05; DC/irrelevant peptide versus DC/BCR-ABL peptide, P <.05; DC/CRCL versus DC/BCR-ABL, P <.05.
The efficacy of BCR-ABL peptide–based immunotherapy has been widely studied.22,49 We have previously shown that immunization of mice with DCs loaded with the synthetic BCR-ABL chimeric nonapeptide, GFKQSSKAL, generates BCR-ABL–specific CTLs in vivo.21 The translocation resulting in the Bcr-abl fusion gene is the primary mutation that leads to malignant transformation; however, secondary mutations often occur.50 Therefore, leukemic cells can easily escape immune responses generated by single peptide vaccination. However, since CRCL likely carry multiple antigenic peptides as indicated in this study (Figures 1 and 2), immunization with CRCL may induce more widespread immunity against the tumor when compared with single peptide immunization. In addition, CRCL have been shown to activate DCs2 and to enhance the immunogenicity of apoptotic tumor cells.51 With these adjuvant effects, CRCL are likely to confer superior immunogenicity compared with peptide vaccinations. We tested this by comparing the potency of immunization with DCs loaded with GFKQSSKAL or with 12B1-derived CRCL. We showed that vaccination with DCs loaded with CRCL led to significantly higher survival rates, confirming the superiority of CRCL vaccination. The lack of DC activation by peptides, followed by disappointing immune responses by single peptide vaccines, has been a concern recognized by cancer immunotherapists.52 CRCL may circumvent the problems associated with single peptide vaccines by providing both large antigen repertoires as well as adjuvant effects that stimulate APCs.
Taken together, we have shown in this study that BCR-ABL peptides are chaperoned by 2 MHC haplotype-variant BCR-ABL+ leukemia-derived CRCL and are cross-presented by DCs to generate BCR-ABL–specific CTLs in vivo. One may argue that the ability to make a response to the human sequence in a mouse may not predict an ability of the same method to make a response to a self antigen in a human. It is true that the BCR-ABL fusion protein is human protein and therefore presumably may have higher antigenicity in mice. We have demonstrated that pulsing of DCs with 100 μg/mL of BCR-ABL peptide alone was less effective in overall antitumor response than were DCs pulsed with 50 μg/mL leukemia-derived CRCL. Since CRCL contain multiple protein components,1–3 and the BCR-ABL peptide makes up a small fraction of the overall peptide (ie, we did not detect it in mass spectrometric studies; M.W.G., unpublished data, March 2004), the amount of BCR-ABL peptide supplied to DCs by CRCL must be quite small. Nonetheless, those CRCL-based vaccines are extremely effective against this very aggressive 12B1 tumor, and in generating specific responses against cells presenting the BCR-ABLpeptide. In agreement with our studies on mice, we have also found that CTLs generated by using CRCL derived from human tumor cells are able to specifically kill the tumor cells of origin (G. Li, Y.Z., M. A. Brewer, E.K., manuscript in preparation).
CRCL have consistently been demonstrated to be able to activate specific T-cell responses.1–4 The potent immune-mediated antitumor effects of CRCL as well as the superiority of CRCL vaccination over single HSP70 or antigenic peptide vaccination implore many interesting questions that need to be answered. Aside from the cell biologic mystery of CRCL, ie, how and why chaperone proteins interact with peptides in vivo, we were also faced with a biochemical mystery of how peptides can possibly remain in association with the chaperone conglomerate that is CRCL, given the stringent isoelectric focusing protocol that was used to generate CRCL. However, the complexity of this vaccine makes our path to the complete understanding of its mechanisms extremely difficult. We demonstrated that peptides are associated with CRCL and provide proof of principle that in this case BCR-ABL is among them and appears to play an important role in the immune response against CML. Eventually, understanding the biochemistry, biology, and immunology behind the vaccine would hopefully lead to an even better second-generation vaccine, crossing the bridge between basic and translational research.
Acknowledgments
The authors wish to thank Sara O. Dione for assistance with ELISPOT photography; Kerri L. Kislin and Gang Li for sharing unpublished data; and Xinchun Chen, Gang Li, and Angela Romanoski for technical assistance.
Footnotes
Supported in part by National Institutes of Health grant R01 CA104926, Department of Defense United States Army DAMD 17-03-1-0208, and the Tee Up for Tots Fund (Y.Z.).
References
- 1.Graner M, Zeng Y, Feng H, Katsanis E. Tumor-derived chaperone-rich cell lysates are effective therapeutic vaccines against a variety of cancers. Cancer Immunol Immunother. 2003;52:226–234. doi: 10.1007/s00262-002-0359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeng Y, Feng H, Graner MW, Katsanis E. Tumor-derived, chaperone-rich cell lysate activates den-dritic cells and elicits potent antitumor immunity. Blood. 2003;101:4485–4491. doi: 10.1182/blood-2002-10-3108. [DOI] [PubMed] [Google Scholar]
- 3.Graner M, Raymond A, Akporiaye E, Katsanis E. Tumor-derived multiple chaperone enrichment by free-solution isoelectric focusing yields potent antitumor vaccines. Cancer Immunol Immuno-therapy. 2000;49:476–484. doi: 10.1007/s002620000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng Y, Graner MW, Feng H, Li G, Katsanis E. Imatinib mesylate effectively combines with chaperone-rich cell lysate-loaded dendritic cells to treat bcr-abl+ murine leukemia. Intl J Cancer. 2004;110:251–259. doi: 10.1002/ijc.20115. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava PK, Maki RG. Stress-induced proteins in immune response to cancer. Curr Top Microbiol Immunol. 1991;167:109–123. doi: 10.1007/978-3-642-75875-1_7. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Ann Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 7.Reits E, Griekspoor A, Neijssen J, et al. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity. 2003;18:97–108. doi: 10.1016/s1074-7613(02)00511-3. [DOI] [PubMed] [Google Scholar]
- 8.Princiotta MF, Finzi D, Qian SB, et al. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 9.Baker-LePain JC, Sarzotti M, Fields TA, Li CY, Nicchitta CV. GRP94 (gp96) and GRP94 N-terminal geldanamycin binding domain elicit tissue nonrestricted tumor suppression. J Exp Med. 2002;196:1447–1459. doi: 10.1084/jem.20020436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicchitta CV. Re-evaluating the role of heat-shock protein-peptide interactions in tumour immunity. Nat Rev Immunol. 2003;3:427–432. doi: 10.1038/nri1089. [DOI] [PubMed] [Google Scholar]
- 11.Daley GQ. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 12.Matulonis UA. Role of B7–1 in mediating an immune response to myeloid leukemia cells. Cancer Genet Cytogenet. 2001;126:8–12. [Google Scholar]
- 13.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 14.Zeng Y, Feng H, Graner MW, Katsanis E. Tumor-derived, chaperone-rich cell lysates activate dendritic cells and elicit potent anti-tumor immunity. Blood. 2003;101:4485–4491. doi: 10.1182/blood-2002-10-3108. [DOI] [PubMed] [Google Scholar]
- 15.Graner M. Immunoprotective activities of multiple chaperone proteins isolated from murine B-cell leukemia/lymphoma. Clin Cancer Res. 2000;6:909–915. [PubMed] [Google Scholar]
- 16.Fields RC, Osterholzer JJ, Fuller JA, Thomas EK, Geraghty PJ, Mule JJ. Comparative analysis of murine dendritic cells derived from spleen and bone marrow. J Immunother. 1998;21:323–339. doi: 10.1097/00002371-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Rotzschke O, Falk K, Wallny HJ, Faath S, Rammensee HG. Characterization of naturally occurring minor histocompatibility peptides including H-4 and H-Y. Science. 1990;249:283–287. doi: 10.1126/science.1695760. [DOI] [PubMed] [Google Scholar]
- 18.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 20.Peto R, Peto J. Asymptotically efficient rank invariant procedures. J R Stat Soc. 1972;A135:185–207. [Google Scholar]
- 21.He L, Feng H, Raymond A, et al. Dendritic-cell-peptide immunization provides immunoprotection against bcr-abl-positive leukemia in mice. Cancer Immunol Immunother. 2001;50:31–40. doi: 10.1007/PL00006680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinilla-Ibarz J, Cathcart K, Korontsvit T, et al. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood. 2000;95:1781–1787. [PubMed] [Google Scholar]
- 23.Cathcart K, Pinilla-Ibarz J, Korontsvit T, et al. A multivalent bcr-abl fusion peptide vaccination trial in patients with chronic myeloid leukemia. Blood. 2004;103:1037–1042. doi: 10.1182/blood-2003-03-0954. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2002;2:185–194. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- 25.Janetzki S, Palla D, Rosenhauer V, Lochs H, Lewis JJ, Srivastava PK. Immunization of cancer patients with autologous cancer-derived heat shock protein gp96 preparations: a pilot study. Int J Cancer. 2000;88:232–238. doi: 10.1002/1097-0215(20001015)88:2<232::aid-ijc14>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 26.Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–1588. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- 27.Nieland TJ, Tan MC, Monne-van Muijen M, Koning F, Kruisbeek AM, van Bleek GM. Isolation of an immunodominant viral peptide that is endogenously bound to the stress protein GP96/GRP94. Proc Natl Acad Sci U S A. 1996;93:6135–6139. doi: 10.1073/pnas.93.12.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heikema A, Agsteribbe E, Wilschut J, Huckriede A. Generation of heat shock protein-based vaccines by intracellular loading of gp96 with antigenic peptides. Immunol Lett. 1997;57:69–74. doi: 10.1016/s0165-2478(97)00048-5. [DOI] [PubMed] [Google Scholar]
- 29.Meng SD, Gao T, Gao GF, Tien P. HBV-specific peptide associated with heat-shock protein gp96. Lancet. 2001;357:528–529. doi: 10.1016/S0140-6736(00)04050-2. [DOI] [PubMed] [Google Scholar]
- 30.Arnold D, Faath S, Rammensee H, Schild H. Cross-priming of minor histocompatibility antigen-specific cytotoxic T cells upon immunization with the heat shock protein gp96. J Exp Med. 1995;182:885–889. doi: 10.1084/jem.182.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breloer M, Marti T, Fleischer B, von Bonin A. Isolation of processed, H-2Kb-binding ovalbumin-derived peptides associated with the stress proteins HSP70 and gp96. Eur J Immunol. 1998;28:1016–1021. doi: 10.1002/(SICI)1521-4141(199803)28:03<1016::AID-IMMU1016>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 32.Nair S, Wearsch PA, Mitchell DA, Wassenberg JJ, Gilboa E, Nicchitta CV. Calreticulin displays in vivo peptide-binding activity and can elicit CTL responses against bound peptides. J Immunol. 1999;162:6426–6432. [PubMed] [Google Scholar]
- 33.Inoue K, Sugiyama H. [WT 1 and leukemia] Rinsho Ketsueki. 1995;36:552–558. [PubMed] [Google Scholar]
- 34.Nakao S. Identification of novel minor histocompatibility antigens responsible for graft-versus-leukemia (GVL) effect on chronic myeloid leukemia: usefulness of determining the clonotype of T cells associated with GVL effect after donor leukocyte infusion. Int J Hematol. 2002;76(suppl 1):274–276. doi: 10.1007/BF03165261. [DOI] [PubMed] [Google Scholar]
- 35.van der Harst D, Goulmy E, Falkenburg JH, et al. Recognition of minor histocompatibility antigens on lymphocytic and myeloid leukemic cells by cytotoxic T-cell clones. Blood. 1994;83:1060–1066. [PubMed] [Google Scholar]
- 36.Molldrem JJ, Clave E, Jiang YZ, et al. Cytotoxic T lymphocytes specific for a nonpolymorphic proteinase 3 peptide preferentially inhibit chronic myeloid leukemia colony-forming units. Blood. 1997;90:2529–2534. [PubMed] [Google Scholar]
- 37.Breloer M, Fleischer B, von Bonin A. In vivo and in vitro activation of T cells after administration of Ag-negative heat shock proteins. J Immunol. 1999;162:3141–3147. [PubMed] [Google Scholar]
- 38.Baker-LePain JC, Sarzotti M, Nicchitta CV. Glucose-regulated protein 94/glycoprotein 96 elicits bystander activation of CD4+ T cell Th1 cytokine production in vivo. J Immunol. 2004;172:4195–4203. doi: 10.4049/jimmunol.172.7.4195. [DOI] [PubMed] [Google Scholar]
- 39.Binder RJ, Blachere NE, Srivastava PK. Heat shock protein-chaperoned peptides but not free peptides introduced into the cytosol are presented efficiently by major histocompatibility complex I molecules. J Biol Chem. 2001;276:17163–17171. doi: 10.1074/jbc.M011547200. [DOI] [PubMed] [Google Scholar]
- 40.Menoret A, Peng P, Srivastava PK. Association of peptides with heat shock protein gp96 occurs in vivo and not after cell lysis. Biochem Biophys Res Commun. 1999;262:813–818. doi: 10.1006/bbrc.1999.1306. [DOI] [PubMed] [Google Scholar]
- 41.Arnold-Schild D, Hanau D, Spehner D, et al. Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol. 1999;162:3757–3760. [PubMed] [Google Scholar]
- 42.Ishii T, Udono H, Yamano T, et al. Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol. 1999;162:1303–1309. [PubMed] [Google Scholar]
- 43.Castellino F, Boucher PE, Eichelberg K, et al. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. J Exp Med. 2000;191:1957–1964. doi: 10.1084/jem.191.11.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh-Jasuja H, Toes RE, Spee P, et al. Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J Exp Med. 2000;191:1965–1974. doi: 10.1084/jem.191.11.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark RE. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood. 2001;98:2887–2893. doi: 10.1182/blood.v98.10.2887. [DOI] [PubMed] [Google Scholar]
- 46.Bocchia M, Wentworth PA, Southwood S, et al. Specific binding of leukemia oncogene fusion protein peptides to HLA class I molecules. Blood. 1995;85:2680–2684. [PubMed] [Google Scholar]
- 47.Yotnda P, Firat H, Garcia-Pons F, et al. Cytotoxic T-cell response against the chimeric p210 BCR-ABL protein in patients with chronic myelogenous leukemia. J Clin Invest. 1998;101:2290–2296. doi: 10.1172/JCI488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bosch GJ, Joosten AM, Kessler JH, Melief CJ, Leeksma OC. Recognition of BCR-ABL positive leukemic blasts by human CD4+ T cells elicited by primary in vitro immunization with a BCR-ABL breakpoint peptide. Blood. 1996;88:3522–3527. [PubMed] [Google Scholar]
- 49.Bocchia M, Korontsvit T, Xu Q, et al. Specific human cellular immunity to bcr-abl oncogene-derived peptides. Blood. 1996;87:3587–3592. [PubMed] [Google Scholar]
- 50.Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107:76–94. doi: 10.1159/000046636. [DOI] [PubMed] [Google Scholar]
- 51.Feng H, Zeng Y, Graner MW, Likhacheva A, Katsanis E. Exogenous stress proteins enhance the immunogenicity of apoptotic tumor cells and stimulate antitumor immunity. Blood. 2003;101:245–252. doi: 10.1182/blood-2002-05-1580. [DOI] [PubMed] [Google Scholar]
- 52.Buteau C, Markovic SN, Celis E. Challenges in the development of effective peptide vaccines for cancer. Mayo Clinic Proc. 2002;77:339–349. doi: 10.4065/77.4.339. [DOI] [PubMed] [Google Scholar]