

Abstract
Pre-mRNA splicing involves recognition of a consensus sequence at the 5′ splice site (SS). However, only some of the many potential sites that conform to the consensus are true ones, whereas the majority remain silent and are not normally used for splicing. We noticed that in most cases the utilization of such a latent intronic 5′ SS for splicing would introduce an in-frame stop codon into the resultant mRNA. This finding suggested a link between SS selection and maintenance of an ORF within the mRNA. Here we tested this idea by analyzing the splicing of pre-mRNAs in which in-frame stop codons upstream of a latent 5′ SS were mutated. We found that splicing with the latent site is indeed activated by such mutations. Our findings predict the existence of a checking mechanism, as a component of the nuclear pre-mRNA splicing machine, to ensure the maintenance of an ORF. This notion is highly important for accurate gene expression, as perturbations that would lead to splicing at these latent sites are expected to introduce in-frame stop codons into the majority of mRNAs.
Most eukaryotic genes contain introns whose precise removal by the pre-mRNA splicing machine is an essential step in gene expression. The accuracy and efficiency of pre-mRNA splicing is attributed to a number of transacting factors, which include the spliceosomal U small nuclear ribonucleoproteins (snRNPs) and several non-snRNP protein splicing factors, as well as to cis-acting sequence elements. The latter include 5′ and 3′ splice sites (SSs), a branch point, a polypyrimidine tract, and splicing enhancer and silencer sequence elements. A key step in pre-mRNA splicing involves the recognition and selection of a consensus sequence AG/GTRAGT (in mammals, where R denotes purine and/denotes the splice junction) at the 5′ SS (1–3). Frequently, however, sequences that comply with the consensus are not selected for splicing (4). We refer to such 5′ consensus sequences as latent 5′ SSs, to splicing events in which they are used as latent splicing, and to the resulting mRNA as latent RNA.
Latent 5′ SSs are highly abundant in pre-mRNAs. In a survey of a database consisting of 446 human protein-coding genes (http://www.fruitfly.org/seq_tools/datasets/Human/) we identified 10,626 latent 5′ SSs located within introns. Thus, the splicing machine must frequently discriminate between normal and latent 5′ SSs. How this is achieved is not yet understood. A clue for a possible mechanism arises from the fact that the intron sequences upstream to 94.5% of the intronic latent sites contain at least one stop codon in the reading frame determined by the bona fide upstream exons. This general finding conforms with the idea that the cell's nucleus harbors a checking mechanism that is capable of recognizing premature stop codons in pre-mRNAs, and consequently aborting splicing at downstream latent 5′ SSs (5).
A straightforward prediction of this model is that the removal of in-frame stop codons would render downstream latent 5′ SSs active in splicing. As a test case we chose the gene encoding the constitutively expressed multifunctional CAD (carbamoyl-phosphate synthetase, aspartate transcarbamylase, dihydroorotase) protein in Syrian hamster cells (6). Our previous analysis of the splicing pattern of CAD pre-mRNA revealed an intronic latent 5′ SS, which conforms to the consensus better than the normal one (latent: AG/GTGGGT vs. normal: AG/GTGACA). Nonetheless, its involvement in splicing never occurred under normal growth conditions. It could, however, be activated for splicing under stress conditions (5), indicating its capability to serve as a splice donor. The presence of in-frame stop codons upstream of the latent SS raised the possibility that splicing at this latent site under normal growth conditions was aborted, thus eliminating the formation of an mRNA that contains premature translation termination codons (PTCs).
A second system tested here is the human enzyme α-l iduronidase (IDUA) that participates in degradation of glycosaminoglycanes in lysosomes. Mutations in the IDUA gene are the cause of a group of lysosomal disorders, the most severe being Hurler syndrome (7). A latent 5′ SS is present within the first intron of IDUA, which is preceded by an in-frame stop codon. Our hypothesis predicted that mutating this stop codon to a missense codon would result in activation of the latent SS.
Here we verify the predictions made by our model, in both gene systems, by examining the splicing of pre-mRNAs transcribed from mini-gene constructs that harbor one or more in-frame stop codons upstream to an intronic latent site, and from mutant constructs in which part or all of these stop codons were mutated.
Materials and Methods
Plasmids.
The wild-type CAD chimeric mini-gene was inserted into pcDNAI/Amp expression vector (Invitrogen). It contains the first two exons and the first intron of the CAD gene (Ex1–Ex2) fused in-frame to a downstream CAD subgenomic region composed of four exons and three introns. Because the complete sequence of the CAD gene is not yet available, the latter genomic fragment is designated ExN–ExN+3. Two constructs, CAD1 and CAD2, were cloned. DNA sequencing confirmed an ORF in ExN of CAD1 and revealed four in-frame stop codons between the normal and the latent 5′ SSs in the downstream intron (IntN). CAD2 had a single nucleotide deletion in the Ex2–ExN junction. DNA sequencing confirmed that ExN in this construct maintained an ORF and revealed two in-frame stop codons in the same region of IntN. Mutants 1–4 were prepared by using the Power-Cloning technology developed by Gesher Advanced Biotecs (WO98/38297, WO98/38298 and WO98/38299, patent pending) by simultaneous assembly of three fragments.
The remaining mutants were prepared by the QuickChange PCR-based mutagenesis method (Stratagene). CAD Ter was generated by a CAG to TAG mutation at position 10 in exon N+1 of CAD1. For further details on plasmid construction see Appended Methods, which are published as supporting information on the PNAS web site, www.pnas.org.
A plasmid carrying a 9-kb fragment from the 5′ end of the IDUA gene (8), was kindly given to us by Elizabeth Neufeld, University of California, Los Angeles. The wild-type IDUA minigene was constructed by cloning into pcDNAI/Amp an 893-bp PCR fragment comprising the first IDUA intron flanked by the first two exons. The mutant IDUA mini-genes were prepared by the Power-Cloning technology as described above. All constructs were confirmed by DNA sequence analyses.
A normal β-globin construct (pmCMV-Gl-Normal) and a construct expressing a PTC-containing β-globin mRNA [pmCMV-Gl-39Ter (9)] were kindly provided by L. E. Maquat, Univ. of Rochester, Rochester, NY.
Transfections.
Syrian hamster SV28 fibroblast cells and human 293T cells were grown to 50% confluence in tissue culture plates as described (5) and transiently transfected with the appropriate DNA constructs (10 μg or 2 μg per 5 × 106 SV28 or 293T cells, respectively) by using the calcium phosphate method (10). Cells were harvested 24 or 48 h after transfection, and total cellular RNA was extracted with guanidinium thiocyanate as described (5). Treatment with antibiotics was as described in Fig. 4.
Figure 4.

Abrogation of NMD does not reveal latent splicing. (a) Human 293T cells (lanes 1–13) were transfected with CAD mini-gene constructs as indicated (lanes 1–4 are cotransfections with pEGFP-N3). Twenty-four hours posttransfection the cultures were treated with CHX (20 μg/ml) for the indicated lengths of time. RNA was analyzed by RT-PCR as in Fig. 1b (lettering and symbols are as in Fig. 1). (b) Human 293T cells were cotransfected as indicated (β-globin WT, wild-type β-globin; β-globin Ter39, a mutant construct expressing β-globin mRNA having a PTC at position 39; CAD Ter, a mutant construct expressing CAD mRNA having a PTC in exon N+1). Treatment with CHX was as in a. β-globin mRNA was revealed by RT-PCR. Mature CAD 1 and CAD Ter mRNAs were revealed as in Fig. 1b.
Reverse Transcription–PCR (RT-PCR).
Total RNA was treated with RNase-free DNase I (50 units/ml; Promega), and cDNA was synthesized from 1 μg of RNA with dT15 primer by using Moloney murine leukemia virus reverse transcriptase (GIBCO/BRL). PCRs (20 μl) contained cDNA synthesized from 0.2 μg of total RNA, 10 pmol each of the indicated primer pairs (listed in Appended Methods on the PNAS web site), and 1.0 units of TaqDNA polymerase (Roche Molecular Biochemicals). Amplification was carried out in a DNA Programmable Thermal Controller (PTC-100; MJ Research, Cambridge, MA) for 30 cycles, at annealing temperature of 63°C (for primer pairs a+b) or 58°C (for primer pair c+b). The amplified products were analyzed by electrophoresis in 2% agarose gels. The assignment of the bands observed was confirmed by sequencing of the DNA extracted from a gel and purified with a commercial DNA purification kit (Qiagen, Chatsworth, CA).
Analysis of IDUA RNA was carried out on total RNA, prepared 48 h after transfection, by RT-PCR. For the preparation of IDUA cDNA a primer complementary to the fusion region of IDUA with the vector was used. PCR was done as described for CAD but with the addition of 5% DMSO, at an annealing temperature of 63°C (primers b+c) or 59°C (primers d+c).
Results
Latent Splicing in CAD Pre-mRNA Is Activated on Removal of In-Frame Stop Codons by Point Mutations.
The CAD constructs fused the first two exons and intron 1 of CAD DNA in-frame to a wild-type or mutated genomic fragment starting at the exon that precedes the intron with the latent SS and ending at the fourth downstream exon (Fig. 1a). The various mini-gene constructs were transfected into Syrian hamster or 293T cells, and the transiently expressed CAD RNAs were analyzed by RT-PCR. Fig. 1b shows the amplified PCR fragments produced from primer a, which spans the junction between exon 2 and exon N, and primer b, which is complementary to a sequence within the downstream exon N+1. Analysis of RNA from control (untransfected) cells indicates that endogenous CAD RNA is not amplified (Fig. 1b, lane C). Transfection with a wild-type CAD mini-gene construct (CAD1), which contains four in-frame stop codons upstream of the latent site in intron N (Fig. 1a Upper), gave rise to a 428-bp DNA fragment representing the pre-mRNA (and/or remnants of the mini-gene DNA) and a 203-bp fragment representing normally spliced CAD RNA (normal RNA) (Fig. 1b, CAD1). Another wild-type construct (CAD2), which contains two in-frame stop codons upstream of the latent site in intron N (Fig. 1a Lower), gave similar results (Fig. 1b, CAD2). Notably, neither constructs gave rise to latent splicing. However, eliminating all stop codons from both of them allowed latent splicing in the respective mutants, as predicted by the model and detailed below.
Figure 1.
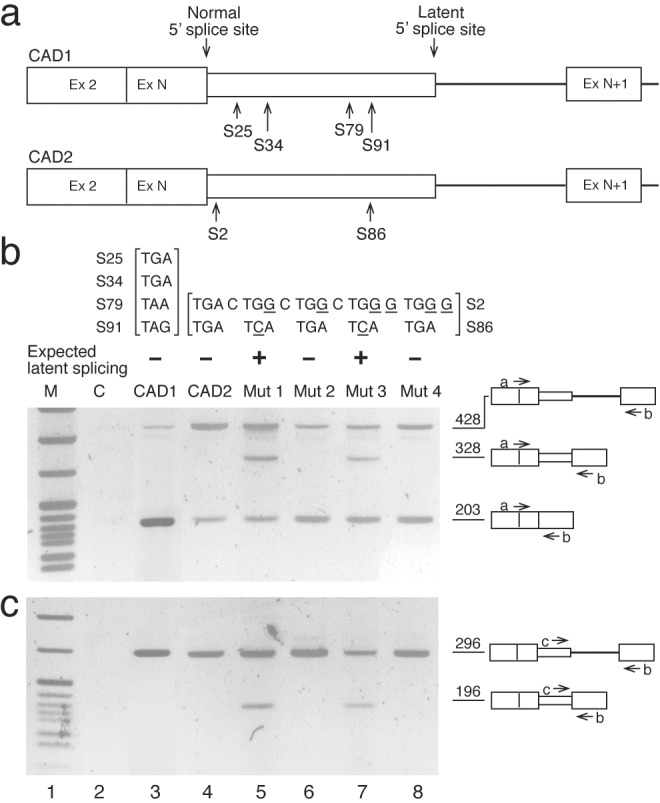
An intronic latent 5′ SS is activated in CAD mutant mini-genes devoid of upstream stop codons—RT-PCR analysis. (a) Schematic drawings of wild-type CAD mini-genes (open boxes, exons; heavy line, intron; narrow box, intronic sequence included as part of the exon in the latent RNA). The normal and latent 5′ SSs and the in-frame stop codons between them are indicated (S# designates the T nucleotide in a stop codon and its respective distance from the normal 5′ SS). (b and c) Gel electrophoretic analysis of RT-PCR DNA fragments obtained from CAD mini-genes. Sequences of stop codons and their mutations (underlined nucleotides) in each construct are indicated above each of the respective lanes. Also indicated (−/+) is the expected occurrence of latent splicing. Bands corresponding to precursor and mature (normal and latent) CAD fragments amplified with primers a+b (b) and primers c+b (c) are indicated by schematic drawings on the right. These assignments were confirmed by sequence analyses of the DNA fragments extracted from the gel. An additional minor band, which is assigned to a heteroduplex between precursor and mature PCR-amplified DNAs, occasionally appeared just below the 428-nt band in Figs. 1b, 2a, 3a, and 4a, as confirmed by sequence analyses of the DNA extracted from the gel, and by rerunning it on a second gel. Lane 2, control with untransfected cells. Lane 1, size markers, pBR322 cut with MspI.
Thus, a construct in which both stop codons in CAD2 were mutated [S2: TGA to TGG, S86: TGA to TCA (Fig. 1b, Mut 1)] gave rise to a fragment of 328 bp, which corresponds to an RNA species produced by a splicing event at the latent 5′ SS. Mutating the upstream stop codon (S2) in CAD2, which is located within the normal 5′ SS, increases the number of mismatches with the consensus at this site from two to three. However, the alteration in the splicing pattern cannot be attributed to this change because a construct in which both stop codons were mutated, but an additional C to G mutation restored the number of mismatches with the consensus to two (Mut 3; Fig. 1b, lane 7), gave rise to splicing at both the normal and latent sites. As a control we show that the double mutation in the normal 5′ SS alone, which did not seem to perturb splicing from the normal SS, was not sufficient to elicit latent splicing (Fig. 1b, Mut 4). Furthermore, a construct in which only the stop codon included in the normal 5′ SS (S2) was mutated, leaving the downstream stop codon (S86) unchanged, did not give rise to latent RNA but normal splicing was observed (Fig. 1b, Mut 2).
It can be argued that latent splicing occurs in transcripts containing in-frame stop codons, but its amplification by PCR is suppressed by the high concentration of normally spliced RNA (see ref. 11). To exclude this possibility, PCR was performed with an intronic primer (primer c) and primer b (Fig. 1c). These primers are not expected to amplify the normally spliced RNA, so that suppression of amplification of the latent RNA is not expected to take place. Yet, no PCR product arising from the latent RNA was observed in cells expressing the wild-type constructs (Fig. 1c, lanes 3 and 4), or in cells expressing constructs in which only S2 was mutated to a sense codon (Fig. 1c, lanes 6 and 8). Mutating both stop codons to sense codons, however, gave rise to the latent RNA (Fig. 1c, lanes 5 and 7), thus confirming the results obtained in Fig. 1b. To further demonstrate the null phenotype of latent splicing in stop codon-containing constructs, we used quantitative RT-PCR analyses (see Appended Methods on the PNAS web site). These experiments, in which we used 30-fold serial dilutions of the input RNA and 15–30 cycles of amplification (total amplification range of more than 4 orders of magnitude, assuming an average amplification of 1.5-fold per cycle), revealed latent splicing only in mutants lacking stop codons but not in wild type or mutants containing stop codons (data not shown). Thus, latent nonsense RNA in quantities larger than 0.0076% of normally spliced RNA should have been detected by this analysis. The RT-PCR results were independently confirmed by S1 nuclease mapping (see Fig. 6, which is published as supporting information on the PNAS web site).
Activation of Latent Splicing Cannot Be Attributed to Interference with Splicing Control Elements.
The relative efficiencies of alternative splicing events often may be influenced by short enhancer sequences (12, 13), as well as other splicing control elements that suppress or enhance splicing (1, 14–16). We therefore have asked whether activation of the latent 5′ SS by the point mutations could have been attributed to the suppression of an element that silences splicing or to the activation of an element that enhances splicing.
We can exclude this interpretation because it predicts that mutating either S2 (Mut 2) or S86 (Mut 5) alone would have activated latent splicing. As shown in Fig. 2 these constructs did not give rise to latent splicing (Mut 2 and Mut 5; Fig. 2, lanes 3 and 4, respectively), whereas latent splicing was elicited only when both S2 and S86 were mutated (Mut 1; Fig. 2, lane 2). Furthermore, latent splicing was suppressed when the TGA stop codon in S86 of Mut 2 was mutated to either of the remaining stop codons TAG or TAA (Mut 10 and Mut 11; Fig. 2, lanes 5 and 6, respectively), whereas mutating S86 to the missense codon TGG elicited latent splicing (Mut 12; Fig. 2, lane 7). Thus, the phenotypic behavior of mutants 10–12 supported our conclusion that the effect on latent splicing should be attributed to the reading context of the mutation in S86 rather than the mutation per se.
Figure 2.
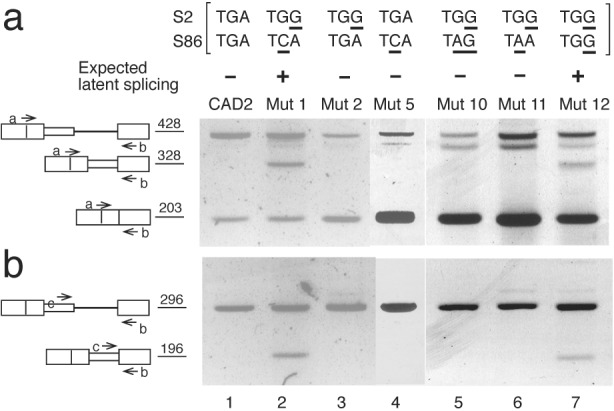
Splicing at a latent 5′ SS in CAD pre-mRNA is suppressed by all three in-frame stop codon variants. RT-PCR analyses of CAD constructs were performed with primers a and b (a) and primers c and b (b). Lettering and symbols are as in Fig. 1.
The fact that splicing enhancer and silencer sequence elements are degenerate and their location is not precisely defined with respect to the splice junctions (15, 17) makes it difficult to rely only on point mutations to exclude the attribution of our results to alterations in these elements. Thus, we eliminated the in-frame stop codons by frame-shift mutations. We inserted the dinucleotide AT 53 nt downstream of the normal 5′ SS, thereby frame-shifting the S86 TGA located 33 nt further downstream. Frame-shifting of S86 alone, leaving S2 unchanged, did not give rise to latent RNA but gave rise to normal RNA (Mut 6; Fig. 3, lane 3). On the other hand, a frame-shift mutation of S86 together with a point mutation in S2 resulted in latent splicing (Mut 7; Fig. 3, lane 4). Likewise, a frame-shift mutation in CAD1, eliminating all four stop codons upstream of the latent site, also gave rise to latent splicing (Mut 9; Fig. 3, lane 5). These results reinforce our model by showing the importance of the reading context of putative stop codons, rather than the sequence in which they are embedded, in rendering a downstream 5′ SS latent.
Figure 3.
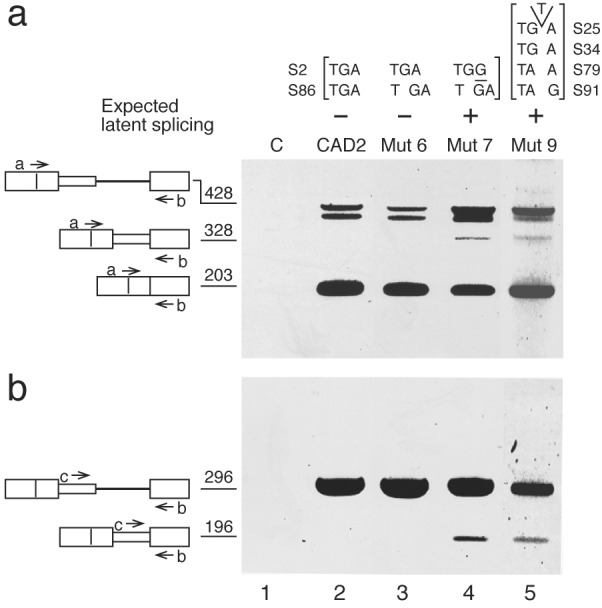
An intronic latent 5′ SS is activated in frame-shifted CAD mutants that eliminate in-frame stop codons. S86 in CAD2 was frame-shifted by an AT insertion at a distance of 53 nt downstream of the normal 5′ SS (Mut 6 and Mut 7). All four stop codons in CAD1 (see Fig. 1b, CAD1) were mutated by a T insertion into S25, thus frame-shifting the remaining three (Mut 9). C, Control RNA from untransfected cells. RT-PCR analyses were performed with primers a and b (a) and primers c and b (b). Lettering and symbols are as in Fig. 1.
Suppression of Latent Splicing Is Not Alleviated by Drugs that Abrogate Nonsense-Mediated mRNA Decay (NMD).
Messenger RNAs that contain PTCs lead to truncated proteins, which can be toxic to cells if they function in a dominant-negative or gain-of-function fashion (18–20). Numerous studies have shown that the nuclear and cytoplasmic abundance of many nonsense mRNAs expressed in mammalian cells is reduced. This reduction was attributed to NMD, whereby the occurrence of PTCs activates a mechanism that rapidly degrades such aberrant mRNAs (18, 19, 21, 22).
In the reported cases of NMD, the levels of nonsense mRNAs were substantially reduced but remained detectable [1–30% of the normal message (18, 21)]. In our case, however, no PTC-containing CAD RNA was detected in cells expressing constructs harboring intronic in-frame stop codons upstream of the latent 5′ SS, whether analyzed by RT-PCR or nuclease S1 mapping. To exclude the possibility that latent splicing had occurred, but the resulting nonsense RNAs were subjected to rapid degradation by NMD to an extent that rendered them undetectable, we tested the splicing pattern of CAD RNAs, expressed from several constructs, in cells treated with drugs known to abrogate NMD.
Cycloheximide (CHX), as well as other protein synthesis inhibitors, was shown to efficiently reverse the down-regulatory effects of PTCs in exons of the T cell receptor β and the β-globin mRNAs (23) and in the HEXA mRNA (24). Likewise, the aminoglycoside antibiotic G-418 was shown to abrogate NMD (25). Here we show that treatment with CHX does not alter CAD splicing patterns. Notably, latent splicing was not detected in cells expressing constructs harboring in-frame stop codons upstream of the latent 5′ SS [CAD1, four stop codons (Fig. 4a, lanes 1–4); CAD2, two stop codons (Fig. 4a, lanes 5–7); Mut 2, one stop codon (Fig. 4a, lanes 8–10)], whether or not treated with CHX. Latent splicing was, however, observed only when stop codons were absent (Mut 1) and was not affected by CHX (Fig. 4a, lanes 11–13).
The CHX experiments were performed under conditions in which NMD is abrogated in 293T cells, as shown in Fig. 4b. For this experiment 293T cells were cotransfected either with a wild-type β-globin construct (Fig. 4b, lanes 1–3), or with a PTC-containing β-globin construct (Ter 39) together with CAD1 as reference (Fig. 4b, lanes 4–6). Fig. 4b (lanes 1–3) shows that the expression of wild-type β-globin RNA did not change on CHX treatment. However, the level of Ter 39 β-globin RNA was reduced significantly (Fig. 4b, lane 4), presumably because of NMD, and treatment with CHX reversed this effect (Fig. 4b, lanes 5 and 6). To further exclude the possibility that an unusual rapid degradation by NMD may have accounted for the absence of transcripts spliced at the latent 5′ SS in wild-type CAD RNA, we analyzed CAD RNA that contains a PTC in a bona fide exon (CAD Ter). If this interpretation was valid, the transcript expressed from CAD Ter should have been undetectable as well. In contrast, Fig. 4b Upper (lane 7) shows that CAD Ter RNA was detected. Furthermore, the stability of this nonsense RNA increased by about 40% on treatment with CHX (Fig. 4b Upper, lanes 8 and 9), as estimated by comparing to the coexpressed green fluorescent protein RNA reference (Fig. 4b Lower). Taken together, these results lead us to conclude that the absence of a signal for nonsense RNA should be attributed to the suppression of the latent 5′ SS by the presence of upstream in-frame stop codons.
Latent Splicing Is Activated in the IDUA Gene by Mutating an In-Frame Stop Codon Upstream of the Latent 5′ SS.
To extend the generality of our model we analyzed RNA expressed from the human IDUA gene (7). Sequence examination of this gene (26) revealed a latent site within the first intron, which is preceded by an in-frame stop codon (Fig. 5a). Our hypothesis predicted that mutating this stop codon to a missense codon would result in activation of the latent SS.
Figure 5.

An intronic latent 5′ SS is activated in IDUA mutant mini-genes devoid of upstream in-frame stop codons. (a) A schematic drawing of wild-type IDUA mini-genes (symbols are as in Fig. 1; S, intronic in-frame stop codon). (b and c) Gel electrophoretic analysis of RT-PCR DNA fragments obtained from IDUA mini-genes. The sequences of the normal 5′ SSs in the wild-type and the mutant mini-genes (mutations underlined) are indicated above each of the respective lanes. Bands corresponding to precursor and mature (normal and latent) IDUA fragments amplified with primers b+c (b) and primers d+c (c) are indicated by schematic drawings on the right (other symbols are as in Fig. 1). These assignments were confirmed by sequence analyses of the DNA fragments extracted from the gel. The additional minor band that appears just below the 537-nt band in lanes 4–6 of c was assigned to a heteroduplex between precursor and spliced PCR-amplified DNAs, as confirmed by sequence analyses of the DNA extracted from the gel, and by rerunning it on a second gel. Lane 2, control with RNA from untransfected cells. Lane 1, size markers, pBR322 cut with MspI.
Chinese hamster ovary cells were transiently transfected with the wild-type and mutant IDUA mini-genes, and the IDUA RNAs were analyzed by RT-PCR. Total RNA from control (untransfected) cells gave no PCR signal corresponding to IDUA RNA with either combination of primers (Fig. 5 b and c, lane 2). PCR amplification with primers b and c of the RNA obtained from cells transfected with the wild-type construct gave rise to the expected fragments representing IDUA pre-mRNA and normally spliced RNA (778 and 212 bp, respectively; Fig. 5b, lane 3). Amplification of the same RNA with primers d and c gave rise only to a PCR product of 537 bp representing the pre-mRNA (Fig. 5c, lane 3). Amplification with primers b and c of total RNA from cells transfected with the Mut 1 construct (Fig. 5b, lane 6) gave rise to the expected 778-bp pre-mRNA PCR product and a 447-bp PCR product representing latent RNA. This result was confirmed by the nested PCR experiment (Fig. 5c, lane 6), showing PCR products of 537 and 206 bp, representing the pre-mRNA and latent RNA, respectively.
Notably, the 212-bp fragment, which represents normally spliced IDUA RNA, was not detected in the Mut 1 IDUA RNA (Fig. 5b, lane 6), although trace amounts of this band were detected by radioactive PCR (not shown), suggesting that the TGA to TGG mutation in Mut 1 rendered the normal 5′ SS almost inactive in splicing. We attributed this observation to the fact that the number of mismatches with the consensus of the normal 5′ SS increased from two in the wild type to three in the mutant. We therefore carried out analogous experiments with constructs in which further mutations that decreased the number of mismatches with the consensus were introduced (Mut 2, two mismatches; Mut 3, one mismatch). Fig. 5b (lanes 4 and 5) shows that each one of these mutants gave rise to PCR products resulting from splicing at the normal 5′ SS without impairing splicing from the latent 5′ SS.
Finally, as observed in the CAD case, the null phenotype of latent splicing in IDUA gene constructs having an in-frame stop codon upstream of the latent 5′ SS was maintained even after treatment with CHX and G-418 (not shown). We may thus conclude that the absence of a signal for nonsense RNA should be attributed to the suppression of the latent 5′ SS by the presence of upstream in-frame stop codons.
Discussion
In this article, we address the question of why highly abundant intronic sequences that conform with the consensus sequence of a 5′ SS are not selected for splicing. We have previously shown that such 5′ SSs have the potential to function in splicing because they can be activated under stress conditions (5) and thus may be regarded as latent 5′ SSs. This issue is of great relevance to accurate gene expression, as more than 90% of the protein-coding transcripts of RNA polymerase II have at least one latent 5′ SS, and perturbations that would lead to splicing at such latent sites are expected to introduce PTCs into the expressed mRNAs. Using two gene systems, we demonstrate that splicing involving latent intronic 5′ SSs can be activated by mutations that remove in-frame stop codons present between a normal and a latent 5′ SS. This study implies that the necessity to maintain an ORF may cause the suppression of splicing at 5′ SSs whose selection for splicing could result in the inclusion of a PTC into the resultant mRNAs.
Two alternative interpretations of our results should be considered. One possibility is that the effects of the mutations we made on splicing could have been attributed to the modulation of splicing control sequences (12, 13). We have used three different strategies to test against this possibility. First, by showing that eliminating only one of each of the two in-frame stop codons in CAD2 was not sufficient to elicit latent splicing; second, by showing that mutating a wild-type in-frame stop codon (TGA) to the remaining two (TAG and TAA) did not activate latent splicing, yet mutating it to a missense codon (TCA or TGG) did; and third, by eliciting latent splicing in mutants in which the in-frame stop codons were mutated by frame shifting. The second possibility is that the absence of mRNAs arising from splicing at latent 5′ SSs could have been attributed to their rapid and complete degradation by the NMD pathway. Our results clearly show that this was not the case, because two cell types expressing four different CAD constructs having an in-frame stop codon upstream of the latent 5′ SS did not show latent splicing even when treated with two drugs known to abrogate NMD (23–25).
We therefore favor the interpretation that the suppression of latent 5′ SSs may be attributed, among other factors, to the presence of upstream in-frame stop codons. This notion implies the existence of a nuclear surveillance mechanism that is functionally coupled to the splicing machine. By scanning the pre-mRNA, this mechanism detects the presence of in-frame stop codons and suppresses splicing at downstream potential 5′ SSs, which could have led to the inclusion of PTCs in the mRNA. Our results are consistent with studies that suggested a nuclear scanning mechanism. These include mutations in the dihydrofolate reductase gene (27) and several cases of human genetic diseases in which exons harboring PTCs were skipped (8, 21, 28, 29). In the fibrillin gene, associated with Marfan syndrome, nonsense, but not missense, mutations were associated with skipping of exon 51 (29). These studies also include cases of intron retention (30–34). In particular, nonsense mutations in the Igκ gene cause defective splicing of this pre-mRNA in a B cell in vitro system (32). While the above studies dealt with the effect of mutations that generate exonic in-frame stop codons, the present study addresses the issue of nonsense RNA from the viewpoint of SS selection and the role endogenous intronic stop codons play in this process.
Although the data presented here, and the extensive sequence analysis we have done, indicate that our model may be of a general nature, it still remains to be asked why a large number of protein-coding transcripts that contain nonsense mutations in their exons are spliced. This is an important issue; as such nonsense transcripts are characteristic of many genetic diseases (35). A clue to a possible answer lies in the sequence environment in which the stop codons are embedded. In the cases reported here, the intronic in-frame stop codons are flanked by 5′ SSs on both sides. In the absence of stop codons, two alternative exons can be defined by the exon definition process (14) and independently, or in a regulated fashion, enter the splicing pathway. The presence of an in-frame stop codon between the alternative 5′ SSs apparently interferes with the exon definition involving the downstream one, thereby suppressing its participation in splicing. A different situation arises when a stop codon is located in a bona fide exon, where it is flanked by an upstream 3′ SS and a downstream 5′ SS, as is the case for most nonsense transcripts that are spliced and contain PTCs in their exons. It is possible that interference with the exon definition process has the alternative of choosing a further downstream 3′ SS, thereby skipping the PTC-containing exon, alternatively it may result in intron retention. However, if no alternative splice choices that could eliminate the in-frame stop codon are available, such “ill-defined exons” might escape the surveillance mechanism and enter the splicing pathway. This would result in the production of PTC-containing mRNAs that can be subjected to the NMD pathway.
Such a surveillance mechanism is difficult to conceive mainly because it implies that the reading frame of mRNAs can be recognized at the level of pre-mRNA before splicing commences. The machine performing this role could involve intranuclear ribosomes. Support for such a view is the occurrence in the nucleus of charging of tRNA (36), translation initiation factors (37), and coupled transcription and translation (38). Alternatively, a yet-unknown nuclear machine might perform this reading frame task. But even if a nuclear “ribosome-like” machine, capable of reading nucleotide triplets was invoked (18, 20), it would be difficult to envisage its function on a pre-mRNA because the presence of introns whose length is not an integer multiple of three alter the reading frame. We propose the following speculative model to account for this circumstance. We have previously shown that nuclear pre-mRNA transcripts are packaged with all known splicing components in multisubunit large nuclear ribonucleoprotein particles that function as supraspliceosome complexes (39–41). We proposed that the supraspliceosome complex serves as a frame onto which the pre-mRNA is folded to align exons about to be spliced, while introns are looped out of each of the respective spliceosomes (see figure 5 in ref. 41). This configuration allows exon sequences to be scanned consecutively even when introns are still part of the pre-mRNA, suggesting that the decision whether to splice or not is made at the level of the complex. Namely, splicing complexes in which a latent 5′ SS is aligned with a 3′ SS may be formed, but splicing does not take place because an in-frame stop codon is recognized upstream of that site in the context of the reading frame of the not-yet-formed mRNA. Such unproductive complexes may reassociate to form productive splicing complexes. When the stop codon is removed, the homologous complex is stable and is capable of entering the splicing pathway. Further studies are required to explore this model and to identify the components involved.
In summary, the suppression of splicing (SOS) mechanism proposed here acts to prevent the inclusion of PTCs in mRNAs rather than dealing with them after splicing by activating NMD. Despite this conceptual difference between NMD and SOS, it is plausible that independent or complementary nuclear and cytoplasmic mechanisms are required to alleviate the potentially toxic effect of in-frame stop codons. All of these pathways, which share an underlying RNA surveillance mechanism, may be related or integrated into a common model.
Supplementary Material
Acknowledgments
We thank Drs. James E. Dahlberg and Nissim Benvenisty for useful discussions and advice, Mrs. Aviva Pecho for excellent technical assistance, and Ms. Tamar Golan for help with the CAD mutants. This research was supported by grants (to J.S.) from the Israeli Ministry of Health and the Leo and Julia Forchheimer Center for Molecular Genetics at the Weizmann Institute and (to R.S.) from the Israel Science Foundation.
Abbreviations
- SS
splice site
- CAD
carbamoyl-phosphate synthetase, aspartate transcarbamylase, dihydroorotase
- PTC
premature translation termination codon
- IDUA
α-l iduronidase
- RT-PCR
reverse transcription–PCR
- NMD
nonsense-mediated mRNA decay
- CHX
cycloheximide
References
- 1.Black D L. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 2.Krämer A. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- 3.Burge C B, Tuschl T H, Sharp P A. In: The RNA World. 2nd Ed. Gesteland R F, Cech T R, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 525–560. [Google Scholar]
- 4.Green M R. Annu Rev Cell Biol. 1991;7:559–599. doi: 10.1146/annurev.cb.07.110191.003015. [DOI] [PubMed] [Google Scholar]
- 5.Miriami E, Sperling J, Sperling R. Nucleic Acids Res. 1994;22:3084–3091. doi: 10.1093/nar/22.15.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Padgett R A, Wahl G M, Stark G R. Mol Cell Biol. 1982;2:293–301. doi: 10.1128/mcb.2.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neufeld E F. Annu Rev Biochem. 1991;60:257–280. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- 8.Bach G, Moskowitz S M, Tieu P T, Matynia A, Neufeld E F. Am J Hum Genet. 1993;53:330–338. [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Sun X, Qian Y, Maquat L E. RNA. 1998;4:801–815. doi: 10.1017/s1355838298971849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kingston R E. In: Short Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. New York: Wiley; 1992. pp. 9.1–9.16. [Google Scholar]
- 11.Valentine C R, Heflich R H. RNA. 1997;3:660–676. [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H X, Chew S L, Cartegni L, Zhang M Q, Krainer A R. Mol Cell Biol. 2000;20:1063–1071. doi: 10.1128/mcb.20.3.1063-1071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaal T D, Maniatis T. Mol Cell Biol. 1999;19:261–273. doi: 10.1128/mcb.19.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berget S M. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 15.Blencowe B J. Trends Biochem Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Manley J L. Curr Opin Genet Dev. 1997;7:205–211. doi: 10.1016/s0959-437x(97)80130-x. [DOI] [PubMed] [Google Scholar]
- 17.Liu H X, Cartegni L, Zhang M Q, Krainer A R. Nat Genet. 2001;27:55–58. doi: 10.1038/83762. [DOI] [PubMed] [Google Scholar]
- 18.Li S, Wilkinson M F. Immunity. 1998;8:135–141. doi: 10.1016/s1074-7613(00)80466-5. [DOI] [PubMed] [Google Scholar]
- 19.Hentze M W, Kulozik A E. Cell. 1999;96:307–310. doi: 10.1016/s0092-8674(00)80542-5. [DOI] [PubMed] [Google Scholar]
- 20.Frischmeyer P A, Dietz H C. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 21.Maquat L E. RNA. 1995;1:453–465. [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson A, Peltz S W. Annu Rev Biochem. 1996;65:693–739. doi: 10.1146/annurev.bi.65.070196.003401. [DOI] [PubMed] [Google Scholar]
- 23.Carter M S, Doskow J, Morris P, Li S, Nhim R P, Sandstedt S, Wilkinson M F. J Biol Chem. 1995;270:28995–29003. doi: 10.1074/jbc.270.48.28995. [DOI] [PubMed] [Google Scholar]
- 24.Rajavel K S, Neufeld E F. Mol Cell Biol. 2001;21:5512–5519. doi: 10.1128/MCB.21.16.5512-5519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedwell D M, Kaenjak A, Benos D J, Bebok Z, Bubien J K, Hong J, Tousson A, Clancy J P, Sorscher E J. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 26.Scott H S, Guo X H, Hopwood J J, Morris C P. Genomics. 1992;13:1311–1313. doi: 10.1016/0888-7543(92)90053-u. [DOI] [PubMed] [Google Scholar]
- 27.Urlaub G, Mitchell P J, Ciudad C J, Chasin L A. Mol Cell Biol. 1989;9:2868–2880. doi: 10.1128/mcb.9.7.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dietz H C, Valle D, Francomano C A, Kendzior R J, Pyeritz R E, Cutting G R. Science. 1993;259:680–683. doi: 10.1126/science.8430317. [DOI] [PubMed] [Google Scholar]
- 29.Dietz H C, Kendzior R J. Nat Genet. 1994;8:183–188. doi: 10.1038/ng1094-183. [DOI] [PubMed] [Google Scholar]
- 30.Naeger L K, Schoborg R V, Zhao Q, Tullis G E, Pintel D J. Genes Dev. 1992;6:1107–1119. doi: 10.1101/gad.6.6.1107. [DOI] [PubMed] [Google Scholar]
- 31.Lozano F, Maertzdorf B, Pannell R, Milstein C. EMBO J. 1994;13:4617–4622. doi: 10.1002/j.1460-2075.1994.tb06783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aoufouchi S, Yélamos J, Milstein C. Cell. 1996;85:415–422. doi: 10.1016/s0092-8674(00)81119-8. [DOI] [PubMed] [Google Scholar]
- 33.Gersappe A, Burger L, Pintel D J. J Biol Chem. 1999;274:22452–22458. doi: 10.1074/jbc.274.32.22452. [DOI] [PubMed] [Google Scholar]
- 34.Mühlemann O, Mock-Casagrande C S, Wang J, Li S, Custodio N, Carmo-Fonseca M, Wilkinson M F, Moore M J. Mol Cell. 2001;8:33–44. doi: 10.1016/s1097-2765(01)00288-x. [DOI] [PubMed] [Google Scholar]
- 35.Culbertson M R. Trends Genet. 1999;15:74–80. doi: 10.1016/s0168-9525(98)01658-8. [DOI] [PubMed] [Google Scholar]
- 36.Lund E, Dahlberg J E. Science. 1998;282:2082–2085. doi: 10.1126/science.282.5396.2082. [DOI] [PubMed] [Google Scholar]
- 37.Dostie J, Lejbkowicz F, Sonenberg N. J Cell Biol. 2000;148:239–247. doi: 10.1083/jcb.148.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iborra F J, Jackson D A, Cook P R. Science. 2001;293:1139–1142. doi: 10.1126/science.1061216. [DOI] [PubMed] [Google Scholar]
- 39.Yitzhaki S, Miriami E, Sperling J, Sperling R. Proc Natl Acad Sci USA. 1996;93:8830–8835. doi: 10.1073/pnas.93.17.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sperling R, Koster A J, Melamed-Bessudo C, Rubinstein A, Angenitzki M, Berkovitch-Yellin Z, Sperling J. J Mol Biol. 1997;267:570–583. doi: 10.1006/jmbi.1997.0898. [DOI] [PubMed] [Google Scholar]
- 41.Müller S, Wolpensinger B, Angenitzki M, Engel A, Sperling J, Sperling R. J Mol Biol. 1998;283:383–394. doi: 10.1006/jmbi.1998.2078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}