

Abstract
The processing of precursor tRNAs at their 5′ and 3′ termini is a fundamental event in the biosynthesis of tRNA. RNase P is generally responsible for endonucleolytic removal of a leader sequence of precursor tRNA to generate the mature 5′ terminus. However, much less is known about the RNase P counterparts or other proteins that are active at the tRNA 3′ terminus. Here we show that one of the human RNase P subunits, Rpp14, together with one of its interacting protein partners, OIP2, is a 3′→5′ exoribonuclease with a phosphorolytic activity that processes the 3′ terminus of precursor tRNA. Immunoprecipitates of a crude human RNase P complex can process both ends of precursor tRNA by hydrolysis, but purified RNase P has no exonuclease activity. Rpp14 and OIP2 may be part of an exosome activity.
Keywords: tRNA processing‖phosphorolytic enzymes‖exosome
Functional tRNAs are generated from their precursor RNAs. Precursor tRNAs must undergo a complex series of processing steps that includes removal of both extra sequences at their 5′ and 3′ termini. Final maturation of the 5′ terminus of tRNA is undertaken by RNase P (1), whereas the 3′ end processing differs in various genetic systems and is thought to be somewhat more complicated. In bacteria, the 3′-trimming reaction can be catalyzed by a variety of exonucleases (2). In the eukaryotic nucleus, 3′-terminal processing of a tRNA precursor seems to be either exonucleolytic or endonucleolytic, depending on the organism analyzed (3).
Unlike 3′-terminal processing, the generation of the 5′ terminus of mature tRNA is well understood and involves only one enzyme, RNase P (1). The chemical composition of this enzyme differs in various organisms. Its functionality remains almost the same from bacteria to yeast to humans. In human HeLa cells, the nuclear RNase P consists of one RNA molecule, and at least ten protein subunits (4–10). In addition to a possible function in RNase P catalysis, the protein subunits may find other roles in vivo. For example, Rpp20 is suggested to have an ABC transporter-like ATPase activity (11). Here we report the characterization of Rpp14 and its non-RNase P interacting partner, OIP2, as 3′→5′ exoribonucleases. Moreover, we found that the processing of the 5′ and 3′ termini of tRNA precursors can be performed simultaneously with a crude RNase P complex that has been immunoprecipitated from HeLa cell crude extracts.
Materials and Methods
Substrates.
The tRNA substrates used were precursor tRNATyr (Escherichia coli) containing 5′ and 3′ extra sequences (denoted as 5′-Tyr-3′), precursor tRNATyr that contains 5′ extra sequences (denoted as 5′-Tyr) or 3′ extra sequences (Tyr-3′), precursor to the suppressor tRNASer (Saccharomyces cerevisiae), SupS1, with both 5′ and 3′ extra sequences (denoted as 5′-SupS1-3′), precursor SupS1 with 5′ extra sequences (denoted as 5′-SupS1), and precursor SupS1 with 3′ extra sequences (denoted as SupS1-3′). Internally labeled 5′-Tyr-3′ and 5′-Tyr were synthesized in vitro in the presence of [α-32P]GTP. Internally labeled 5′-SupS1-3′ and 5′-SupS1 were synthesized in vitro in the presence of [α-32P]UTP. Internally labeled SupS1-3′ was prepared from internally labeled 5′-SupS1-3′ by removing the 5′ leader sequence by using purified human RNase P. For the synthesis of 5′-32P-labeled 5′-Tyr-3′, transcription reactions were performed with nonlabeled NTPs, the RNA was purified and then was treated with calf intestinal phosphatase, extracted two times with phenol/chloroform and one time with chloroform. Recovery was by ethanol precipitation. 5′-Tyr-3′ was labeled with [γ-32P]ATP by using polynucleotide kinase.
Adult enhancer factor-1 (AEF) RNA, a single-stranded RNA from Drosophila melanogaster (12), was labeled as indicated by the same methods used for 5′-32P-labeled 5′-Tyr-3′.
All of the substrates used in the reaction were purified on a 5% polyacrylamide gel.
Protein Expression and Immunoprecipitation.
DNA sequence encoding OIP2 (kindly provided by R. F. Rest, Hahnemann University, Philadelphia; ref. 13) was subcloned in-frame with the six histidine residues of pHTT7K. Overexpressions of recombinant 6XHis-OIP2, 6XHis-Rpp14, and 6XHis-Rpp20 were performed in strain E. coli BL21(DE3) as described in Jarrous et al. (6). The cells (100 ml) were collected and resuspended in lysis buffer (4 ml) consisting of 20 mM Tris⋅HC1 (pH 8.0)/500 mM NaC1/10 mM DTT/0.l% Nonidet P-40. After sonication followed by centrifugation at 16,000 × g for 10 min, the lysates were clarified by centrifugation at 100,000 × g for 2 h. The S100 aqueous phases (12.6 μg/ml) were recovered and stored at −80°C.
To prepare 6XHis-OIP2, 6XHis-Rpp14, and 6XHis-Rpp20 for exoribonuclease assay in vitro, each preparation of the expressed proteins of 6XHis-OIP2, 6XHis-Rpp14, and 6XHis-Rpp20 was immunoprecipitated by mixing 200 μl of each lysate, 10 μl of antipolyhistidine monoclonal antibody (Sigma), and 15 mg of protein A-Sepharose beads at 4°C for 3–4 h, and by washing the beads thoroughly with buffer (10 mM Tris⋅HC1, pH 8.0/50 mM KCl/10 mM DTT) at least five times. The pellets were resuspended in 1 ml of the exo buffer (see below) and divided into six aliquots. Before use of each aliquot for an activity assay, the aliquot was centrifuged, the aqueous phase removed, and the resulting material in the test tube added to the substrate and the reaction buffer (see below).
Immunoprecipitation from HeLa Cell Crude Extracts.
HeLa cell crude extracts (S100) were prepared as described (5, 14) and were about 58.8 μg/ml of protein. Immunoprecipitates were first cleaned with preimmune sera coupled to protein A-Sepharose CL-4B beads; polyclonal rabbit antibodies (80 μl of sera) were mixed with 10 mg of washed protein A-Sepharose CL-4B in 250 μl NET-2 buffer (50 mM Tris⋅HC1, pH7.5/150 mM NaC1/0.05% Nonidet P-40). Coupling was made by nutating beads 2 h at 4°C, followed by four brief washes of beads with NET-2 buffer. HeLa cell crude extracts (100 μl) were added to the beads in a final volume of 250 μl by supplementing 1× exo buffer, and nutated about 3 h at 4°C. Pellets were then collected by a short centrifugation, followed by five to six washes with 1× exo buffer without RNasin. Pellets were resuspended in 1 ml of exo buffer and distributed in three 330-μl aliquots. After removal of the aqueous phases, the aliquots were assayed for exoribonuclease or RNase P activity.
Exoribonuclease Assay in Vitro.
Assays in vitro were performed directly on immunoprecipitates of 6Xhis-Rpp14, 6XHis-Rpp14, and 6XHis-OIP2 or of HeLa cell extracts (see above). Each aliquot of immunoprecipitate, prepared as described in Materials and Methods, was incubated at 37°C with 5 μl of 32P-labeled RNA substrate (about 3,000 cpm) in 1× exo buffer (10 mM Tris⋅HC1, pH 8.0/5 mM MgC12/15 mM Na2HPO4/50 mM KCl/0.8 units/μl of RNasin). The amount of substrate per reaction is about 0.04–0.1 nmol for ptRNA precursors and about half as much for AEF RNA. The time of incubation for each immunoprecipitate is indicated in the figure legends. The reactions were stopped by the addition of 2–3 μl of phenol and 1 volume of formamide gel-loading buffer, and the reaction products were resolved in polyacrylamide gels indicated in the figure legends. The reaction products were quantitated by autoradiography and quantitated with a phosphoimager. Nucleoside 5′-monophosphate and nucleoside 5′-diphosphate reaction products were identified by thin-layer chromatography.
Reactions with internally labeled 5′-supS1-3′ ([α-32P]UTP) as substrate were stopped by addition of 3–5 μl of phenol and vortexing. After brief centrifugation, 5 μl of supernatant were mixed with 1 μl of a UTP, UDP, and UMP mixture (20 mM each), spotted onto polyethyleneimine-cellulose plates and developed in 1 M LiCl/1 M HOAc. The standards were visualized under UV light, and the reaction products visualized by autoradiography.
RNase P Activity Assay.
RNase P activity assays have been described (14), except that the reactions in this study were performed in 1× exo buffer with different substrates as indicated in the figure legends.
Results
Rpp14 and OIP2 Are 3′→5′ Exoribonucleases with a Processing Activity on the 3′ Terminus of Precursor tRNA.
Rpp14 and OIP2 were identified as interacting partners in a yeast two-hybrid assay in which screening was performed on a HeLa cell cDNA library. In this assay, Rpp14 was used as bait (14). A search of databases had indicated that both Rpp14 and OIP2 contained sequences that were related to those found in the E. coli RNase PH family, i.e., 3′→5′ exonucleases (15–17). The fusion proteins of 6XHis-Rpp14, 6XHis-OIP2, and control 6XHis-Rpp20 were isolated from crude extracts of E. coli that were immunoprecipitated by monoclonal antibodies against the polyhistidine bound to protein A-Sepharose beads (Fig. 1A), and assayed for ribonuclease activity. In Fig. 1A, one aliquot of each recombinant protein resolves into a single band with no other protein species present, except for the major bands found in the serum medium, which indicates the high purity of the immunoprecipitates. Incubation of 5′-32P-labeled AEF RNA (a small RNA of 52 nt encoded by D. melanogaster; ref. 12) with either immunoprecipitated 6XHis-Rpp14 or immunoprecipitated 6XHis-OIP2 generated a series of smaller fragments (Fig. 1B). The final, processed products are four nucleotides in size after analysis on sequencing gels (data not shown). We also found that the exonuclease activities of 6XHis-Rpp14 and 6XHis-OIP2 are dramatically decreased without phosphate to a low-level background (Fig. 1 C and D; see below). A rough calculation of structure in AEF RNA revealed no stable secondary structure, which indicates that 6XHis-Rpp14 and 6XHis-OIP2 are probably phosphorolytic exoribonucleases (see below). We note also that 6XHis-Rpp20 showed no significant exonuclease activity in the presence of phosphate, as expected from the negative control.
Figure 1.

Recombinant Rpp 14 and OIP2 exhibit 3′→5′ exoribonuclease activity in vitro. Assays in vitro were performed with 6XHis-Rpp 14, 6XHis-Rpp 20, or 6XHis-OIP2 immunoprecipitated by antipolyhistidine monoclonal antibody (Sigma), and the reaction products were analyzed by PAGE (see Materials and Methods). (A) 6XHis-Rpp 14, 6XHis-Rpp 20, or 6XHis-OIP2 were expressed in E. coli and then immunoprecipitated with antipolyhistidine monoclonal antibody bound to protein A-Sepharose CL-4B beads. The measurement of an aliquot of each immunoprecipitate is defined in Materials and Methods. One aliquot of each sample was boiled in 1× SDS-polyacrylamide gel-loading buffer, separated on a SDS-12% polyacrylamide gel, and visualized by Coomassie brilliant blue staining. Lane 1, anti-polyhistidine antibody bound to protein A-Sepharose CL-4B bead; lane 2, 6XHis-Rpp 20 precipitated with antipolyhistidine antibody bound to protein A-Sepharose CL-4B bead (the arrow indicates 6XHis-Rpp 20); lane 3, 6XHis-Rpp 14 precipitated with anti-polyhistidine antibody bound to protein A-Sepharose CL-4B bead (the arrow indicates 6XHis-Rpp 14); lane 4, 6XHis-OIP2 with anti-polyhistidine antibody bound to protein A-Sepharose CL-4B bead (the arrow indicates 6XHis-OIP2). For reference purposes, the approximate molecular weight of the OIP2 protein is 32–36 kDa and that of ribonuclease PH is 22–25 kDa. (B) Assay of 6XHis-Rpp 14, 6XHis-Rpp 20, and 6XHis-OIP2 with 5′-32P-labeled AEF RNA. One aliquot of each immunoprecipitate was used for each assay shown in B–D. Substrate was approximately 0.05 nmol per reaction. Reaction products were resolved in a 12% polyacrylamide gel. Time course with substrate alone (lanes 1–4), 6XHis-Rpp 20 (lanes 5–7), 6XHis-Rpp 14 (lanes 8–10), or 6XHis-OIP2 (lanes 11–13). Time (minutes) indicated in the figure. (C) Time course of processing of 5′-32P-labeled AEF RNA by 6XHis-Rpp 14 in the absence of exogenously added 15 mM Na2HPO4 (pH 8.0) or 5 mM MgCl2. Lane 1, substrate; lanes 2–5, 6XHis-Rpp 20 in the presence of Na2HPO4 and MgCl2; lanes 6–17, 6XHis-Rpp 14 in presence of both Na2HPO4 and MgCl2(lanes 6–9), only MgCl2 (lanes 10–13) or only Na2HPO4 (lanes 14–17), respectively. (D) Time course of processing of 5′-32P-labeled AEF RNA by 6XHis-OIP2 in absence of exogenously added 15 mM Na2HPO4 (pH 8.0) or 5 mM MgCl2. Lane 1, substrate; lanes 2–5, substrate incubated in the presence of Na2HPO4 and MgCl2; lanes 6–17, 6XHis-OIP2 in presence of both phosphate and MgCl2 (lanes 6–9), only MgCl2 (10–13), or only phosphate (lanes 14–17), respectively.
To investigate the action of 6XHis-Rpp14 and 6XHis-OIP2 on precursor tRNA, precursor tRNATyr, with both a 5′ leader and a 3′ trailer sequence (designated as 5′-Tyr-3′; 43 nucleotides as the 5′ leader sequence, and 46 nt at the 3′ end) and precursor tRNATyr with only a 5′ leader (denoted as 5′-Tyr) were used as substrates. When 6XHis-Rpp14 (Fig. 2), or 6XHis-OIP2 (data not shown), was incubated with 5′-32P-labeled 5′-Tyr-3′, only one large product was accumulated (Fig. 2, lanes 13 and 14). No fragments of other lengths were observed. Incubation of internally 32P-labeled 5′-Tyr-3′ with 6XHis-Rpp14 (Fig. 2, lanes 5 and 6; the product is the same size as that seen in lanes 13 and 14), in addition to the observation of the previously identified large product, also showed a smaller product. Thin-layer chromatography analysis of the small products released from the internally labeled substrate showed that nucleoside 5′ diphosphates were generated with 6XHis-Rpp14 or 6XHis-OIP2 (Fig. 3, lanes 1–3). No UDP was generated by 6XHis-Rpp20. We conclude that either 6Xhis-Rpp14 or 6XHis-OIP2 can process the 3′ end of precursor tRNA.
Figure 2.

Processing in vitro of precursor tRNATyr with 6XHis-Rpp 14. Assay in vitro of 3′ processing of precursor tRNATyr containing 5′ and 3′ extra sequences (denoted as 5′-Tyr-3′) by 6XHis-Rpp 14. Aliquots of each immunoprecipitate and substrate concentration used in each reaction are given in the legend of Fig. 1. The reaction products were separated on a 12% sequencing gel. Lanes 1–8, internally 32P-labeled 5′-Tyr-3′ as substrates; lanes 1 and 2, substrates incubated for 0 or 45 min as controls; lanes 3 and 4, time course with immunoprecipitated 6XHis-Rpp 20 for 20 or 45 min; lanes 5 and 6, immunoprecipitated 6XHis-Rpp 14 for 20 or 45 min; lanes 7 and 8, immunoprecipitated human RNase P from HeLa cells crude extracts by anti-Rpp 29 polyclonal antibody for 20 or 45 min. Several faint bands appear in the lane below Tyr-3′, an indication of processing at the 3′ terminus. Lanes 9–16, 5′-32P-labeled 5′-Tyr-3′ as substrates; lanes 9 and 10, substrate incubation for 0 or 45 min as controls; lanes 11 and 12, time course with immunoprecipitated 6XHis-Rpp 20 for 20 or 45 min; lanes 13 and 14, with immunoprecipitated 6XHis-Rpp 14 for 20 or 45 min; lanes 15 and 16, with immunoprecipitated human RNase P from HeLa cells crude extracts by anti-Rpp 29 polyclonal antibody for 20 or 45 min. Major products are indicated on both sides of the figure. Tyr-3′ was the precursor of tRNATyr, of which 5′ leader sequence was removed; 5′-Tyr was a processed precursor of tRNATyr from which the trailing 3′ sequence was removed; 5′ is the 5′ leader sequence.
Figure 3.
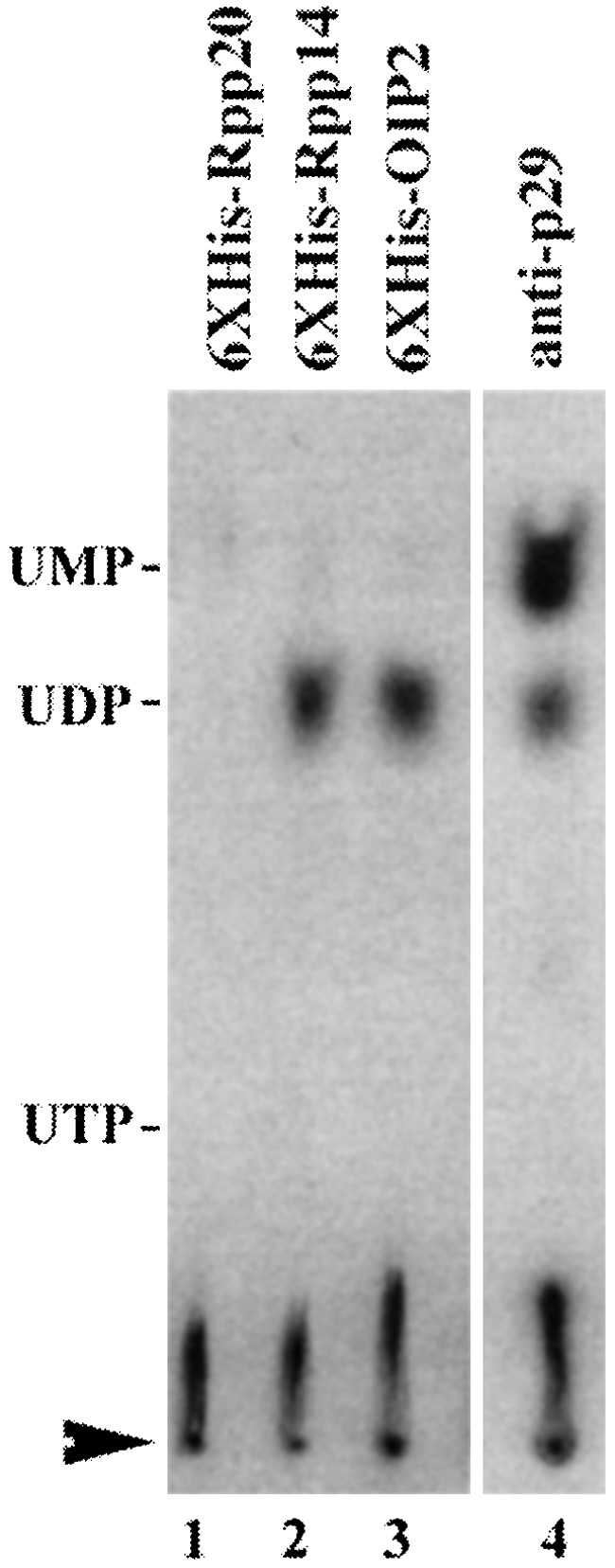
Thin-layer chromatographic analysis of the products released by the 6XHis-Rpp 20, 6XHis-Rpp 14, 6XHis-OIP2, and human RNase P-associated exoribonuclease activity. Aliquots for each immunoprecipitate and substrate concentration are given in the legend to Fig. 1. The reactions were performed as described in Materials and Methods with 5′-SupS1-3′ transcribed in the presence of [α-32P]UTP. The standards are UTP, UDP, and UMP. The reaction products were mixed with the standards and were resolved by chromatography on polyethyleneimine-cellulose thin-layer plates. Lanes 1–4, 5′-SupS1-3′ incubated for 10 min at 37°C with 6XHis-Rpp 20, 6XHis-Rpp 14, 6XHis-OIP2, and immunoprecipitated human RNase P by anti-Rpp 29 polyclonal antibody (anti-p29), respectively. The standards were visualized with UV light and their positions are shown at the left side of the figure. The sample application points are indicated by an arrowhead, the direction of chromatography was from bottom to top.
Requirements for the 3′→5′ Exonuclease Activity.
Requirements for 3′-terminal processing of precursor tRNA by Rpp14 or OIP2 were further investigated with internally 32P-labeled precursor SupS1 with a 3′ trailer sequence (designated as SupS1-3′) but no 5′ leader sequence. The 6XHis-Rpp14 and 6XHis-OIP2 from E. coli require a divalent cation, Mg2+, for specific activity (Fig. 4A). In the absence of added Mg2+, the substrate SupS1-3′ was almost completely degraded in a nonspecific manner to small products. This phenomenon was also observed with AEF RNA as a substrate (Fig. 1 C and D), and we do not know at present if it represents an additional enzymatic activity. The optimal Mg2+ concentration for both proteins was 5–20 mM, with relatively little change in activity up to at least 40 mM, whereas in a low concentration of Mg2+ (1 mM), the substrates were intact.
Figure 4.

Requirements of the reactions governed by recombinant Rpp 14 and OIP2. Assays in vitro were performed with 6XHis-Rpp 14 or 6XHis-OIP2 immunoprecipitated by anti-polyhistidine monoclonal antibody. Aliquots as defined earlier, as was substrate concentration (Fig. 1). Time of incubation was 10 min at 37°C. (A) Effect of Mg2+ on 3′ processing of internally labeled (with [α-32P]UTP) SupS1-3′ by 6XHis-Rpp 14 and 6XHis-OIP2. Lane 1, substrate; lane 2, substrate incubation in the presence of 5 mM MgCl2 as control; lanes 3–9, 3′ end processing with 6XHis-OIP2 in the presence of 0, 1, 2.5, 5, 10, 20, and 40 mM MgCl2, respectively; lanes 10–16, 3′ end processing with 6XHis-Rpp 14 in the presence of 0, 1, 2.5, 5, 10, 20, and 40 mM MgCl2, respectively. (B) Phosphate-dependent processing of SupS1-3′ by 6XHis-Rpp 14 and 6XHis-OIP2. Lane 1, substrate; lane 2, substrate incubation in the presence of 15 mM phosphate as control; lanes 3–11, 3′ end processing with 6XHis-Rpp 14 in the presence of 0, 2.5, 5, 10, 20, 40, 80, 160, and 320 mM of phosphate, respectively; lanes 12–20, 3′ end processing with 6XHis-OIP2 in the presence of 0, 2.5, 5, 10, 20, 40, 80, 160, and 320 mM phosphate, respectively. The reaction products were separated on an 8% polyacrylamide gel. Mononucleotides are not shown on the gel.
The 3′→5′-phosphorolytic exonuclease activities of 6XHis-Rpp14 and 6XHis-OIP2 are also largely dependent on phosphate (Fig. 4B) and produce mononucleotide diphosphates. In the absence of phosphate, 6XHis-Rpp14 activity was virtually undetectable and 6XHis-OIP2 activity was much lower than in the presence of phosphate. The large, processed products were of a greater size than those found in the presence of phosphate, but a thin-layer analysis of the mononucleotide products made with and without phosphate present indicated that they were UDP in the presence of phosphate and UMP in the absence of phosphate (data not shown). Because 6XHis-Rpp20, prepared under the same conditions, yielded about the same low level of UMP in the absence or presence of phosphate (and no other product in the presence of phosphate), we conclude that the activity seen in the absence of phosphate is likely a contaminant from E. coli.
The optimal phosphate concentration for both the phosphorolytic exoribonucleolytic activity of Rpp14 and OIP2 was 2.5–80 mM. Optimal pH for Rpp14 and OIP2 3′ end-processing activity was in the range of 7.5–9.0 (data not shown).
A 3′→5′ Exoribonucleolytic Processing Activity Is Also Associated with an RNase P Complex Immunoprecipitated from Crude Cell Extracts.
One of the human RNase P subunits, Rpp14, together with its interacting partner OIP2, has 3′-5′ exoribonuclease activity. Does RNase P in crude enzyme complexes also exhibit this exonucleolytic activity?
Human RNase P was immunoprecipitated from crude HeLa cell extracts by using polyclonal rabbit antisera against Rpp14, Rpp20, and Rpp29. The immunoprecipitates were assayed for exoribonuclease activity with a 5′-32P-labeled AEF RNA substrate. The immunoprecipitates obtained with rabbit antisera against Rpp14, Rpp20, and Rpp29 exhibited exoribonuclease activity, with progressive disappearance of the RNA substrate and the accumulation of labeled end-products smaller than 10 nucleotides (Fig. 5).
Figure 5.

Exoribonuclease assay of immunoprecipitates from HeLa cells by anti-human RNase P subunits (Rpp 14, Rpp 20, and Rpp 29) polyclonal antibodies. HeLa cell crude extracts subject to immunoprecipitation assay were first cleaned with preimmune sera coupled to protein A-Sepharose beads. The immunoprecipitates were obtained by mixing the cleaned HeLa cell crude extracts with beads coupled to the preimmune rabbit sera as control, and with beads coupled to anti-Rpp 14, anti-Rpp 20, or anti-Rpp 29 as polyclonal antibody. The immunoprecipitates above, aliquots assayed as described in Materials and Methods, were assayed for exoribonuclease activity by using 5′-32P-labeled AEF RNA substrates. Lanes 1–3, substrate incubation; lanes 4–6, immunoprecipitates by preimmune sera; lanes 7–9, immunoprecipitates by anti-Rpp 20 polyclonal antibody; lanes 10–12, immunoprecipitates by anti-Rpp 14 polyclonal antibody; lanes 13–15, immunoprecipitates by anti-Rpp 29 polyclonal antibody.
The exoribonucleolytic activity associated with the immunoprecipitated enzyme produces 5′-UMP or 5′-AMP as its major degradation product (when either U or A are the only internally labeled nucleotide, respectively). This reaction is clearly different from the activities of the expressed, recombinant, immunoprecipitated, and purified proteins from E. coli, which produce 5′-UDP. Lower amounts of immunoprecipitated enzyme do not yield increased amounts of UDP. These data suggest that the RNase P-related exonucleolytic activity, if related to those of Rpp14 or OIP2, might contain a phosphatase activity. When pSupS-3′ labeled with [α-32P]ATP was used as a substrate, only AMP was found in the reaction products (data not shown). Furthermore, if [α-32P]ADP was purified and used as a substrate, no AMP was observed in the products of this reaction (data not shown).
Maturation of Precursor tRNA at Both Termini by the Human RNase P Complex and OIP2.
Human RNase P was immunoprecipitated by rabbit antisera against Rpp29. The immunoprecipitates were incubated with internally 32P-labeled 5′-Tyr-3′ and 5′-Tyr. Both ends of 5′-Tyr-3′ can be processed, with the accumulation of intermediate products with different truncated 3′ ends (Fig. 6A, lanes 10–12). To analyze the reaction products, two different precursor tRNAs were used as substrates, precursor SupS1 with both 5′ leader and 3′ trailer sequences (denoted as 5′-SupS1-3′), and precursor SupS1 without 5′ leader sequences (denoted as SupS1-3′). After incubation of internally 32P-labeled 5′-SupS1-3′ with RNase P immunoprecipitates (Fig. 6B, lane 11) or HeLa cell crude extracts (lane 12), the reaction products were resolved in a sequencing gel. In the reaction with RNase P immunoprecipitates, a small amount of mature SupS1 is produced. However, a series of processed, exonucleolytic products exists primarily with different truncated 3′ ends of about 90–82 nt. The same processing pattern is also observed when incubation of SupS1-3′ with RNase P immunoprecipitates (data not shown). In the incubation of internally 32P-labeled SupS1-3′ with immunoprecipitated 6XHis -Rpp20 (Fig. 6B, lane 6), 6XHis-Rpp14 (Fig. 6B, lane 5), and 6XHis-OIP2 (Fig. 6B, lane 4), only uniform products with six extra nucleotides following the mature 3′ end were observed. Analysis of the reaction products by thin-layer chromatography revealed that the accumulating end products are mainly nucleoside diphosphates (5′-UDP) for reactions with 6XHis-Rpp14 and 6XHis-OIP2, and both nucleoside diphosphates and nucleoside monophosphates, as the major product, are produced in reactions with RNase P immunoprecipitates (Fig. 3, lane 4). The actual source, therefore, of the activity that produces AMP or UMP is unknown at the moment. As a negative control (data not shown), preimmune serum beads were used and showed no enzymatic activity.
Figure 6.

5′ and 3′ processing of precursor tRNA by human RNase P and its associated exonucleolytic components: HeLa cell crude extracts subject to immunoprecipitation assay were precleaned with preimmune sera coupled to protein A-Sepharose beads. The immunoprecipitates were obtained by mixing the precleaned HeLa cell crude extracts with beads coupled to the preimmune rabbit sera as control, and with beads coupled to anti-OIP2 or anti-Rpp 29 polyclonal antibodies. The immunoprecipitate aliquots above (defined in Materials and Methods and amount of substrate used as in Fig. 1) were assayed for tRNA processing (times indicated in the figure) by using internally 32P-labeled precursor tRNATyr containing 5′ and 3′ extra sequences (denoted as 5′-Tyr-3′) or internally 32P-labeled precursor tRNATyr containing only 5′ extra sequences (denoted as 5′-Tyr). (A) Lanes 1–12, internally 32P-labeled 5′-Tyr-3′ as substrates. Lane 1, input substrate; lane 2, digestion with highly purified human RNase P (the positions of processed products pTyr-3′ and 5′ leader sequence were marked on the right side of the figure); lane 3, substrate incubation for 30 min; lanes 4–6, immunoprecipitates by preimmune sera; lanes 7–9, immunoprecipitates by anti-OIP2 antibody; lanes 10–12, immunoprecipitates by anti-Rpp 29 polyclonal antibody; lanes 13–19, internally 32P-labeled 5′-Tyr as substrates; lanes 13 and 15, substrate input; lane 14, digestion with purified RNase P for 10 min; lane 16, substrate incubation for 10 min; lane 17, digestion with immunoprecipitated human RNase P by anti-Rpp 29 polyclonal antibody for 10 min; lane 18, digestion with immunoprecipitated OIP2 by anti-OIP2 polyclonal antibody for 10 min; lane 19, digestion with immunoprecipitates by preimmune sera for 10 min. Major products are indicated on the left side of the figure. mTyr is mature tRNA; 5′ is the 5′ leader sequence. (B) Identification of products with internally 32P-labeled precursor SupS1 that contains only 5′ leader sequences (denoted as 5′-SupS1) or internally 32P-labeled precursor SupS1 containing only 3′ extra sequences (prepared by digestion of 5′-SupS1-3′ with human RNase P, denoted as SupS1-3′). After the reaction was finished, the processed products were analyzed in a 5% sequencing gel. Lane 1, 5′-SupS1 incubation; lane 2, 5′-SupS1 incubation with RNase P (produces a mature SupS1 of 82 nucleotides); lane 3, ladder made from mixture of four sequencing reactions of a DNA sample; lanes 4–6, SupS1-3′ incubation with immunoprecipitated 6XHis-OIP2, 6XHis-Rpp 14, and 6XHis-Rpp 20 for 30 min, respectively; lanes 7–9, 11, 12, 5′-SupS1-3′ incubation alone, with RNase P, immunoprecipitates by preimmune sera, immunoprecipitates by anti-Rpp 29 polyclonal antibody and HeLa cell extracts for 30 min, respectively; lanes 14 and 15, 5′-SupS1 incubation with buffer or with RNase P. The tRNAs and their size are labeled on both sides of the figure. The number 82 shows the size of mature SupS1 and is counted as a standard along the sequencing ladder. Therefore, number 88 is the size of partially 3′ end-processed products, indicating that the products have six extra nucleotides after the mature 3′ end.
Discussion
We have characterized Rpp14 and its yeast two-hybrid interacting partner, OIP2 (13), as 3′→5′ exoribonucleases. These enzymes are phosphorolytic and perform the 3′ terminus processing of tRNA precursors to yield mononucleotide diphosphate. Their end products on tRNA precursor molecules are limited to a few nucleotides in addition to the 3′ terminus of the tRNAs. RNase P, immunoprecipitated from crude cell extracts, also has a 3′→5′ exonucleolytic activity and produces mononucleotide monophosphate as its end product. The activity is absent in fractions of RNase P purified further.
The RNase P-associated exonuclease might be responsible for general exonucleolytic degradation of unwanted RNA molecules, for example, to remove 5′ leader products generated by RNase P (Fig. 6). The 3′ leader sequence is readily removed, whereas the 5′ leader is degraded after cleavage by RNase P, itself.
Other 3′→5′ exoribonucleases with sequences similar in part to the proteins we have just characterized, also exist in E. coli and in human cells (15–17). In E. coli, RNase PH is an important nuclease involved in several biochemical processes. In yeast and humans, sequences similar to RNase PH are found in several proteins, including the ones we characterized. In testing the aliquots of immunoprecipitates from E. coli that were made with monoclonal antibodies to the 6XHis tag protein, we observed that the proteins we isolated were pure, aside from the ones in the sera we used (Fig. 1A). One could argue that a small proportion of RNase PH copurified with the recombinant protein and that this copurification was the cause of the exoribonuclease function we observed. The same argument can be made with the immunoprecipitates made from human cell crude extracts with polyclonal antibodies to the required proteins. The amount of such crude contaminants seems unlikely; certainly less than 10% of the visible activity of the contaminant is shown in Fig. 1A. A rough estimate of the amount of protein that might account for the enzymatic function we measure is too low for the preparations we have made. Immunoprecipitates of E. coli designed to purify complexes of recombinant proteins (e.g., 6XHis-Rpp20) or HeLa cells made with preimmune sera show no such exoribonuclease activity and are, presumably, free of any contaminants.
We also purified the recombinant proteins separately from E. coli by affinity column chromatography using their nickel-binding properties. The proteins were treated more rigorously with respect to their exposure to solutions (300 mM imidazole) that might be denaturing. The purified proteins were then immunoprecipitated with antipolyhistidine monoclonal antibodies and then were tested for exoribonuclease activity. In each case, activity was recovered from the immunoprecipitates, giving further proof that the exoribonuclease activity was associated with the recombinant proteins (data not shown). Recombinant proteins other than Rpp 14 and OIP2 showed no such activity when subjected to the same procedure.
Although 5′ terminus processing of precursor tRNA in HeLa cells is generally performed by an endoribonuclease, human nuclear RNase P, little is known about human nuclear 3′ processing of precursor tRNA. One report indicated that the 3′ terminus processing of precursor tRNA in HeLa cells may be due to one step of endonucleolytic cleavage, because no stepwise degradation products had been detected (18). However, because of the exonuclease activity of Rpp14 and OIP2 on precursor tRNA, and especially the exonuclease activity associated with human RNase P, we conclude that a human RNase P-associated exonuclease activity that can generate mature 3′ termini is at least one of the mechanisms for 3′ end processing of precursor tRNA in HeLa cells. We do note, again, that purified RNase P does not contain the exonucleolytic activity. Accordingly, there are other factors associate with the RNase P complex in crude cell extracts, and are immunoprecipitated with them, and these factors are lost on further purification of the enzyme.
3′→5′ exoribonucleases form an exosome in eukaryotic cells (16, 17). The end products of this exosome are hydrolytic rather than phosphorolytic. Rpp14 and OIP2, as individual proteins, may be part of such an enzyme or at least contribute to its activity. OIP2, which is in weak association with RNase P (data not shown), may also explain the observations on exonucleolytic activity reported here. The arrangement of this protein and its exonuclease activity is quickly lost from the RNase P-associated complex during purification of this latter enzyme from crude extracts.
Acknowledgments
We thank our colleagues, Dr. C. Guerrier-Takada, Dr. Y. Li, and Donna Wesolowski, for helpful discussions. This research is supported by U.S. Public Health Service Grant GM19422 (to S.A.).
Abbreviation
- AEF
adult enhancer factor-1
References
- 1.Altman S, Gopalan V, Vioque A. RNA. 2000;6:1689–1694. doi: 10.1017/s1355838200001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deutscher M P, Li Z. Prog Nucleic Acids Res Mol Biol. 2001;66:67–105. doi: 10.1016/s0079-6603(00)66027-0. [DOI] [PubMed] [Google Scholar]
- 3.Mörl M, Marchfelder A. EMBO Rep. 2001;2:17–20. doi: 10.1093/embo-reports/kve006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lygerou Z, Pluk H, van Venrooij W J, Seraphin B. EMBO J. 1996;15:5936–5948. [PMC free article] [PubMed] [Google Scholar]
- 5.Eder P S, Kekuda R, Stolc V, Altman S. Proc Natl Acad Sci USA. 1997;94:1101–1106. doi: 10.1073/pnas.94.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarrous N, Eder P S, Guerrier-Takada C, Hoog C, Altman S. RNA. 1998;4:407–417. [PMC free article] [PubMed] [Google Scholar]
- 7.Jarrous N, Eder P S, Wesolowski D, Altman S. RNA. 1999;5:153–157. doi: 10.1017/s135583829800185x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jarrous N, Reiner R, Wesolowski D, Mann H, Guerrier-Takada C, Altman S. RNA. 2001;7:1153–1164. doi: 10.1017/s1355838201010469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Eenennaam H, Lugtenberg D, Vogelzangs J H, van Venrooij W J, Pruijn G J. J Biol Chem. 2001;276:31635–31641. doi: 10.1074/jbc.M103399200. [DOI] [PubMed] [Google Scholar]
- 10.Guerrier-Takada C, Eder P S, Gopalan V, Altman S. RNA. 2002;8:1–6. doi: 10.1017/s1355838202027954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Altman S. Proc Natl Acad Sci USA. 2001;98:441–444. doi: 10.1073/pnas.021555498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falb D, Maniatis T. Mol Cell Biol. 1992;12:4093–4103. doi: 10.1128/mcb.12.9.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams J M, Chen G-C, Zhu L, Rest R F. Mol Microbiol. 1998;27:171–186. doi: 10.1046/j.1365-2958.1998.00670.x. [DOI] [PubMed] [Google Scholar]
- 14.Jiang T, Altman S. Proc Natl Acad Sci USA. 2001;98:920–925. doi: 10.1073/pnas.021561498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly K O, Deutscher M P. J Biol Chem. 1992;267:17153–17158. [PubMed] [Google Scholar]
- 16.Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- 17.Allmang C, Petfalski E, Podtelejnikov A, Mann M, Tollervey D, Mitchell P. Genes Dev. 1999;13:2148–2158. doi: 10.1101/gad.13.16.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruissem W, Prescott D M, Greenberg B M, Hallick R B. Cell. 1982;30:81–92. doi: 10.1016/0092-8674(82)90014-9. [DOI] [PubMed] [Google Scholar]