

Abstract
The elucidation of physical and molecular mechanisms by which a membrane tube is generated from a membrane reservoir is central to the understanding of the structure and dynamics of intracellular organelles and of transport intermediates in eukaryotic cells. Compelling evidence exists that molecular motors of the dynein and kinesin families are involved in the tubulation of organelles. Here, we show that lipid giant unilamellar vesicles (GUVs), to which kinesin molecules have been attached by means of small polystyrene beads, give rise to membrane tubes and to complex tubular networks when incubated in vitro with microtubules and ATP. Similar tubes and networks are obtained with GUVs made of purified Golgi lipids, as well as with Golgi membranes. No tube formation was observed when kinesins were directly bound to the GUV membrane, suggesting that it is critical to distribute the load on both lipids and motors by means of beads. A kinetic analysis shows that network growth occurs in two phases: a phase in which membrane-bound beads move at the same velocity than free beads, followed by a phase in which the tube growth rate decreases and strongly fluctuates. Our work demonstrates that the action of motors bound to a lipid bilayer is sufficient to generate membrane tubes and opens the way to well controlled experiments aimed at the understanding of basic mechanisms in intracellular transport.
Biological membranes, such as the endoplasmic reticulum (ER), the Golgi apparatus, and endosomes, form elaborate and highly dynamic tubular networks (1, 2). Recently, microscopy of living cells has illustrated that membrane tubes also participate in transport events between cellular compartments. For instance, long-range tubular transport intermediates have been observed between Golgi and ER and between Golgi and the plasma membrane, challenging the classical notion of small spherical vesicles as the sole transport intermediates (3–8).
The formation and movement of membrane tubes in animal cells is thought to involve an interaction of the membranes with the cytoskeleton, especially the microtubule network (9). Numerous experiments, in vitro and in vivo, suggest a direct role of microtubules in the formation of the endoplasmic reticulum and Golgi membrane networks (9–11). Microtubule-dependent tubulation of Golgi and endosomal membranes is amplified after the treatment of cultured cells with the fungal metabolite brefeldin A (BFA) (12, 13). The interactions of membranes with microtubules are mediated by several classes of proteins, notably motor proteins of the dynein and kinesin families (14). The involvement of these motors in the movement of various organelles, including transport intermediates and cellular structures, along microtubules is now well established. Their role in the formation of membrane tubes is less clear, but it has been reported that BFA-induced tubulation of Golgi membranes requires both in vivo and in vitro microtubule-based motor activity (11, 15). It should be pointed out that several proteins involved in membrane fission events, such as endophilin, amphiphysin, and the GTPase dynamin, have been shown to induce the formation of tubules from liposomes in vitro (16–18). These proteins can directly bind to membranes by means of lipid binding domains. Whether these proteins are necessary for allowing motors to pull tubes in vivo is however still unclear.
Membrane tubes can also be pulled out from membranes of controlled lipid composition by hydrodynamic flow (19) and by direct manipulation, using either micropipettes (20, 21) or optical tweezers (22). These systems have been useful in understanding the physics of membrane tube formation (19, 20, 23, 24). However, the growth rates of the membrane tubes in these systems are in the range of a few tens to a few hundred micrometers per second, values that are significantly higher than the growth rates of tubes generated by motor proteins pulling on biological membranes in vivo [in the range of 1–2 μm/s (5–8)]. This rate of in vivo membrane movement corresponds well to the rates of in vitro motility of kinesin motors (the fastest—Neurospora kinesin—has a velocity value of about 2.6 μm/s; see ref. 25).
The goal of this study was to determine whether binding a motor protein to a lipid bilayer would be sufficient to generate membrane tubes. We show that a lipid reservoir, kinesin-coated beads, microtubules, and ATP provide a minimal system for generating tubular structures that resemble tubes observed in vitro with complex biological membranes (9–11).
Materials and Methods
Reagents.
Egg phosphatidylcholine (EPC), cholesterol, and N-biotinyl-dioleyl-phosphoethanolamine (Biot-DOPE) were purchased from Avanti Polar Lipids. β-BODIPY 530/550 C5-hexadecanoyl phosphatidylcholine, β-BODIPY 581/591 C5-hexadecanoyl phosphatidylcholine, cholesteryl BODIPY 542/563 C11, and cholesteryl BODIPY Fl C12 were obtained from Molecular Probes. All chemicals were purchased from Sigma Aldrich except ATP, GTP, and adenosine 5′[β,γ-imido]triphosphate (AMP-PNP), which were purchased from Roche Molecular Biochemicals. Streptavidin beads (100 nm) were purchased from Bangs Laboratories (Carmel, IN). Biotinylated hemagglutinin-kinesin (a gift of F. Nédélec, European Molecular Biology Laboratory, Heidelberg) was purified as described (26). By dividing the number of kinesins by the number of beads, the number of kinesin molecules per bead was estimated at approximately 1,500.
Giant Unilamellar Vesicles (GUVs).
GUVs were prepared by the electroformation technique (27). Rat liver Golgi membranes were purified according to a standard procedure (28). Golgi lipids were then prepared as described (29).
Assay for Tube Formation.
Coverslips were washed for 5 min in sulfochromic acid, rinsed 4–6 times in milliQ water, once in 95% ethanol, and stored at 4°C in ethanol. A coverslip was then dried under a nitrogen flux and incubated for 1 to 2 min in a poly(L-lysine) solution (0.01% wt/vol). A flow chamber was built by intercalating two sheets of parafilm between this poly(L-lysine)-coated coverslip and a clean slide. The chamber was finalized by heating the stack for a few seconds at 100–150°C on a heater plate. Its volume was approximately 25 μl. Diluted microtubules in IMI buffer (50 mM imidazole, pH 6.7/50 mM NaCl/2 mM EGTA/1 mM MgCl2) were injected into the chamber and incubated for a few minutes. The chamber was rinsed with 50 μl of IMI buffer and rinsed with 50 μl of 5 mg/ml casein diluted in IMI buffer. After a 15-min incubation, the chamber was rinsed with 50 μl of MB buffer (IMI buffer plus 1 mM ATP and 10 μM Taxol). Streptavidin beads (5 μl) mixed with 2.5 μl of 5 mg/ml casein were sonicated for 15 min on ice. IMI buffer (30 μl) was added to the beads, and 1 μl of this mixture was mixed and incubated for a few minutes with 5 μl of 1 μM biotinylated kinesins. Then, 44 μl of MB buffer was added to the kinesin-coated beads, and the chamber was filled with this mixture. GUVs (1–5 μl) were injected into the chamber and allowed to sediment on the coated coverslip.
A similar assay was used for biotinylated Golgi membranes except that a smaller chamber volume (12 μl) and twice as many kinesins per bead were used for pulling out tubes.
Tube Imaging.
Tubes were visualized by fluorescence confocal microscopy, differential interference contrast (DIC) microscopy, and reflection interference contrast microscopy (RICM) (30).
The fluorescence intensity profiles of tubes were measured on digital confocal images. The maximum was taken as the value of the fluorescence intensity of one tube. To avoid any saturation or nonlinear effect, highly fluorescent tubes were omitted. On each network of tubes, all intensity values were normalized by dividing by the smallest intensity value. This operation was repeated on many images to obtain an accurate estimation of the fluorescence distribution.
Electron Microscopy.
We built a chamber made of four 300-μm mesh grids aligned in the middle of a coverslip, covered with a collodion film, and dried at 60°C. The coverslip was incubated for 1 min with poly(L-lysine), washed in water, and dried under a nitrogen flux. A slide was then prepared with 2 spacers made of 2 parafilm layers each and fixed by melting with a heater. After solidification of the spacers, the coverslip was assembled on them, keeping the grids aligned between the spacers and fixed with vacuum grease. The assay was then performed as described above. To avoid nonspecific adsorption onto the glass, 10 μg/ml of bacitracin in IMI buffer was used instead of casein. Negative staining was achieved by rinsing the chamber with 100 μl of 1% (wt/vol) uranyl acetate in water and drying with a Whatman filter paper. The chamber was opened, and the grids were removed from the coverslip. Carbon was then evaporated onto the grids. Grids were observed in a Philips (Eindhoven, The Netherlands) EM 12 electron microscope fitted with a LaB 6 filament and operating at 80 kV. Micrographs were taken on Kodak electron microscope films.
Results
Biotinylated kinesins (consisting of the motor domain of Drosophila melanogaster kinesin) were attached to the membrane of biotinylated GUVs (diameter ranging from 5 to 50 μm) by means of 100-nm polystyrene beads coated with streptavidin. The assay was first developed by using lipid bilayers consisting of 95% EPC and 5% biotinylated dioleyl-phosphoethanolamine (Biot-DOPE). GUVs and the kinesin-coated beads were injected into a chamber coated with Taxol-polymerized microtubules, and the chamber was filled with a buffer containing 1 mM ATP. Approximately 10 min after injection, membrane tubes started to form (Fig. 1 a and b). Once generated, tubes continued to grow along the microtubules. Side branching events at the intersections of the underlying microtubule network were subsequently observed (Fig. 1 a and b), leading to the formation of a network of membrane tubes (Fig. 2a) often extending over 50 μm from the GUVs. No tubes were observed in the absence of either kinesin molecules or ATP (data not shown), indicating that tubes form through the action of kinesin. Fig. 1c illustrates a tube with two beads at its tip, with additional beads distributed along the tube. The beads are in fact required for tube formation, as demonstrated in the following way. To allow direct binding of kinesin to the membrane, we exploited the fact that kinesins are not released from microtubules in the absence of ATP (Fig. 1d Left). Kinesins and fluorescent Cy3-labeled streptavidin were sequentially injected into the chamber in the absence of ATP, leading to the formation of streptavidin/kinesin complexes bound to microtubules. After injection of GUVs and ATP, kinesins detached from microtubules and bound to the lipid bilayer, as demonstrated by the transfer of fluorescence from microtubules to the GUVs (Fig. 1d Center). Under these conditions, no tubes were observed up to 1 h after injection (Fig. 1d Right). By reducing 100-fold the amount of biotinylated lipids incorporated in the bilayer, we were able to show that the absence of tube formation was not because of a high membrane rigidity resulting from dense streptavidin grafting. On the other hand, tubes could be formed from these GUVs when kinesin-coated beads were added (data not shown).
Figure 1.

Formation of tubes and networks from EPC GUVs. (a) DIC images recorded with a charge-coupled device camera (time lapse, 12 s) showing several tubes growing simultaneously from GUVs (bright spherical objects visible at the bottom of the image). Branching events occur on images 1, 4, and 5, leading to the formation of a network (images 6–9). (b) DIC images (time lapse, 7 s) showing a tube growing along a microtubule that splits into 2 tubes at the intersection of 2 microtubules (image 7). A retraction event is visible on images 12 and 13, followed by further growth (images 14–18). Black arrows point to microtubules. (c) Tube growth observed by reflection interference contrast microscopy. Beads appear black or white depending on their distance to the substrate. Two beads (gray arrow) are visible at the tip of a growing tube. Beads are also present along the tube (white arrow) growing on a microtubule (black arrow). (d) Tubes do not form in the absence of beads. From Left to Right: without ATP, biotinylated kinesins-fluorescent Cy3 streptavidin complexes were fixed on the microtubule network. In the presence of 1 mM ATP, the complexes were transferred onto the GUV (Center). Absence of tubes growing from a GUV as shown by DIC microscopy (Right). [Bar = 5 μm.]
Figure 2.

Tube networks. (a and b) Fluorescence confocal microscopy of EPC tubes containing 1% (no./no.) fluorescent BODIPY 530/550 C5-HPC. A single representative confocal section is shown in a (the GUV appears out of focus). (c) Sequence showing the growth of a fluorescent tube along another tube (time lapse, 30 s). (d) Histogram of the normalized fluorescence intensities (NI), showing the discrete distribution of the fluorescence intensities of approximately 150 tubes. (e–g) Transmission electron microscopy images of EPC tubes (N); (e) Network of membrane tubes (white arrow) growing on a microtubule network (black arrow). The GUVs are no longer visible, probably because of washing out during the staining process. (f) A single tube aligned along a microtubule. The estimated diameter of this tube is 47 nm. (g) Bundles of 2 tubes on a microtubule. [Bars = 5 μm (for a, b, and e) and 0.5 μm (f and g)].
Membrane tubes can be readily observed by confocal fluorescence microscopy if fluorescent lipids are incorporated into the vesicles. As shown in Fig. 2 a and b, the membrane tubes showed a broad distribution in fluorescence intensity. The histogram of the normalized intensities is discontinuous, and only multiples of a unitary value are seen (Fig. 2d). Such a distribution implies that some tubes are either multilamellar or composed of bundles of unitary tubes. To define the precise structure of the network, we analyzed it by transmission electron microscopy (Fig. 2 e–g). Single tubes had a constant diameter (40 ± 10 nm), a value close to that estimated for membrane tubes in vivo. Interestingly, two or more tubes were frequently observed aligned along a single microtubule (Fig. 2g), suggesting that tubes of high fluorescent intensity (see Fig. 2 a and b) in fact represent bundles of several tubes. This hypothesis is supported by the fact that the tubes with the highest fluorescence intensity often split into several tubes of lower intensity (Fig. 2b). In addition, growth or retraction of a tube along another tube was frequently observed (Fig. 2c).
Based on growth velocity measurements, we were able to distinguish two phases in the formation of a tubular network. At the first phase (I), the average growth velocity was 340 ± 40 nm/s, a value close to that measured for kinesin-coated beads moving along microtubules in the absence of membranes (Fig. 3a). During this phase, the length of a tube increases essentially linearly with time (Fig. 3c). Phase I corresponds to the time following the emergence of tubes from a given vesicle, up to the beginning of network formation (see Fig. 1a). Network growth then enters gradually in a second phase (phase II) in which the average velocity decreased by a factor of two as compared with phase I (Fig. 3g, EPC I and II). The observation of single tubes shows a slowing of growth, and even a complete stop, after a growth period at constant velocity (data not shown). Moreover, the instantaneous velocity of the tip of the tube (Fig. 3f) fluctuated to a much greater extent than in phase I (Fig. 3d), where the fluctuations were similar to those of a kinesin-coated bead not bound to GUVs moving along a microtubule (Fig. 3b). Phase II corresponds to a progressive densification of the network, and the number of growing tubes gradually decreased. No significant, further growth activity was detected 30–60 min after the beginning of the experiment, which was not a result of ATP exhaustion (data not shown).
Figure 3.

Kinetics of tube growth. Typical length (L) vs. time (t) and corresponding instantaneous velocity (V) vs. time (t) plots, for, respectively: (a and b) a free bead in a standard motility assay performed under the same conditions (L corresponding to the distance covered by the bead); (c and d) an EPC tube in phase I; (e and f) an EPC tube in phase II. (g) Average velocities: free bead (Bd), EPC lipid tubes in phases I and II (EPC), Golgi-purified lipid tubes (GPL) in phases I and II, biotinylated purified Golgi membranes in phase II (Gol), and EPC lipids with the addition of 50% (no./no.) cholesterol (Chl) in phase II.
We next tested the influence of the lipid composition of the membrane on the generation of tubes. The addition of cholesterol (from 16 to 50% no./no., in the initial lipid solution) to the EPC bilayer did not significantly change either the growth velocity (Fig. 3g, Chl) or the diameter of the fluorescent tubes (data not shown). To obtain a more complex lipid composition, GUVs were made of lipids prepared from purified rat liver Golgi membranes (GPL, Golgi-purified lipid). Golgi lipids are composed of 50% (no./no.) phosphatidylcholine, 20% phosphoethanolamine, 6% phosphatidylserine, 12% phosphatidylinositol, 8% sphingomyelin, and cholesterol (lipid ratio 0.16) (29, 31). Kinetic parameters comparable to the ones found with EPC GUVs were obtained, in particular for velocity values during phases I and II (Fig. 3g, GPL I and II). In addition, Golgi lipid GUVs also gave rise to networks consisting of bundles of membrane tubes.
Finally, we wished to determine whether tubes could be obtained by binding the kinesin-coated beads to membrane proteins instead of lipids. For this purpose, purified rat liver Golgi membranes were incubated with N-hydroxysuccinimide-biotin, a reagent that biotinylates amino groups in proteins. The addition of kinesin-coated beads led to the formation of tube networks resembling those described above (Fig. 4). However, a phase I dynamic was not observable. Rather, the growth velocity of the tubes resembled that of phase II in GUVs (Fig. 3g, Gol). Bundles of tubes were also present in the preparations. In control experiments, tube formation was not observed when biotinylated Golgi membranes were incubated in buffer alone.
Figure 4.
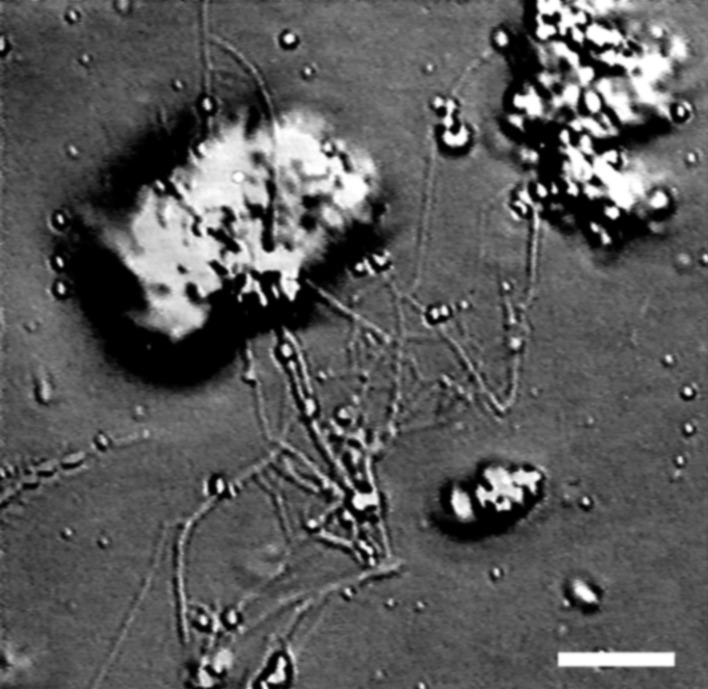
DIC microscopy images of the network extracted from biotinylated rat liver Golgi stacks. [Bar = 5 μm.]
Discussion
The minimal system described above has two dynamic regimes. When the growth velocity of tubes is nearly identical to the velocity of a free bead (phase I in our assay), this implies that the force experienced by individual motors is smaller than 1 pN (32). In our assay, the velocity is such that the force necessary for pulling a tube can be estimated from static arguments (20): f = πrσ = 2πκ/r, in which σ is the membrane tension, and r = (κ/2σ)1/2 is the tube radius (κ is the membrane-bending rigidity modulus). Based on values of κ ≈ 4 × 10−20 J for pure phospholipids and r ≈ 20 nm from electron microscopy measurement, we obtain f ≈ 12 pN. From geometrical arguments based on motor steric exclusion, bead curvature, and microtubule diameter (33), the number of motors likely to interact simultaneously with a microtubule at any given time can be estimated to be between 10 to 20. Therefore, the load per motor, assuming that pulling is carried out by a single bead, is of the order of or smaller than 1 pN. After a few minutes, the growth regime gradually reaches a second stage, in which tube growth velocity is reduced by a factor of two. This observation indicates that each individual motor experiences a force about half stall force (≈2–3 pN) (32). Such an increase in force may result from an increase in membrane tension, likely caused by membrane extraction from GUVs (34). A high initial tension of the purified Golgi membrane could explain why phase I is not observed in this system. The velocity fluctuations in phase II (Fig. 3f) could reflect either fluctuations in tension (35, 36) or fluctuations in the number of motors working at a given time in a high-tension condition.
A striking finding is that beads linking motor proteins to the membrane bilayer seem to be a key element in our assay. Under a load of a few pN, single phospholipid molecules are extracted from a membrane in a few seconds (36), and motors detach from the microtubule at even higher rates (37). On the other hand, it takes more than a minute to pull out a 30-μm-long tube (36). Therefore, the role of beads could be to distribute the load among a large number of lipids and motors, thus avoiding lipid extraction or motor detachment. It is also important to emphasize that the binding of several motors to a bead increases its processivity. How motors bind to biological membranes is still poorly understood, but it has been reported that a kinesin (KIF13A) directly interacts with coat proteins (38), large cytosolic complexes that are recruited onto membranes and play an essential role in the formation of transport intermediates in cells (39, 40). A tentative extrapolation of our results is that coat protein complexes, by forming patches on the membranes, fulfil a role similar to that of the beads in our assay. Lipid subdomains, such as detergent-resistant membranes (rafts), could also have the same function, if motors can directly bind to them. Interestingly, a kinesin (KIFC3) that associates with triton-insoluble membranes has been recently characterized (41).
Another interesting observation is the existence of bundles of tubes in the network, in apparent contradiction with elastic energy minimization, which predicts that tubes elongated from the same GUV should coalesce once generated [I. Derényi, personal communication (21)]. One possible explanation is that tubes originating from different points on the GUV membrane form bundles because of the topology of the underlying microtubules network, allowing bundles to form far enough from the base of the vesicle (Fig. 5a). However, most of the bundles that we observed are localized very close to the GUV (see Fig. 2a). In this case, bundles of tubes could be formed if tubes connect at different points to the same microtubule (Fig. 5b). Attractive interactions between tubes and microtubules should then stabilize the first points of contact, preventing tube coalescence.
Figure 5.
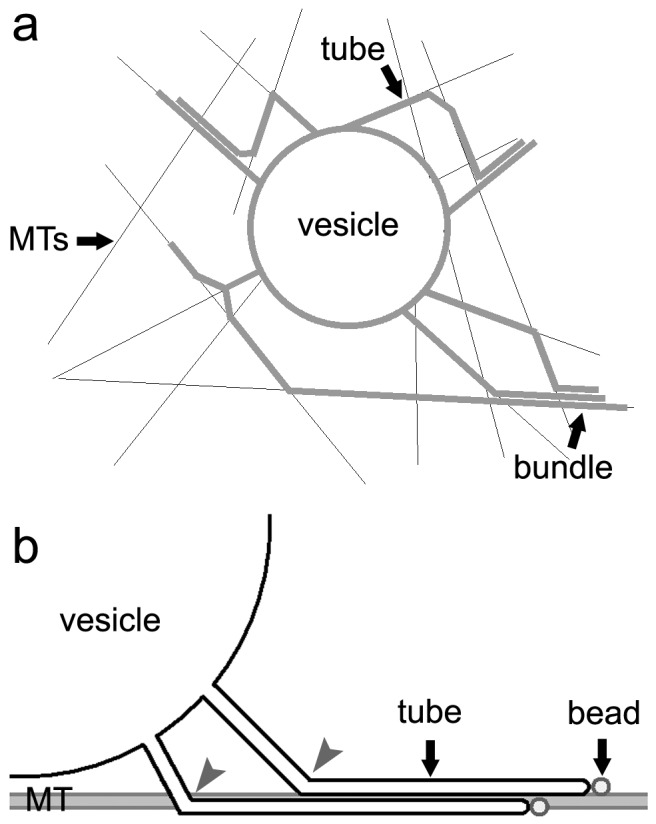
Possible mechanisms for the formation of bundles in the network. (a) Single tubes, originating from different points of the same vesicle and moving along different microtubules, converge at a distance from the vesicle on a single microtubule and form bundles. (b) Single tubes can also grow along the same microtubule and form bundles near the GUV membrane if weak interactions exist between tubes and microtubules. The contact points (gray arrows) of the two tubes with the microtubule should be distant enough to prevent tubes from coalescence.
In conclusion, our work demonstrates that the action of motor proteins is sufficient to produce membrane tubules from a lipid bilayer. That tubes have been obtained with GUVs of complex lipid composition, as well as with purified Golgi membranes, indicates that this assay will be worthwhile for determining the pertinent biophysical parameters and for addressing the role of lipid domains and protein coats in the events underlying the tubulation of biological membranes. It will be also interesting to investigate how growing tubules can detach (fission event) from the membrane reservoir.
Acknowledgments
We thank F. Nédélec for kindly providing the kinesin plasmid; F. Jülicher, I. Derényi, and M. Dogterom for stimulating discussions; J. Pécréaux and L. Legoff for their assistance in data processing; and B. Antonny, M. Bornens, D. Cassel, D. Chatenay, P. Chavrier, B. Hoflack, L. Johannes, R. McDermott, F. Nédélec, F. Perez, and T. Surrey for critical reading of the manuscript. This work was supported by grants from the Centre National de la Recherche Scientifique (Program Physique et Chimie du Vivant), the Institut Curie, and the Association pour la Recherche Contre le Cancer.
Abbreviations
- GUV
giant unilamellar vesicle
- EPC
egg phosphatidylcholine
- IMI
imidazole
- DIC
differential interference contrast
References
- 1.Lee C, Chen L B. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- 2.Mollenhauer H H, Morré D J. Histochem Cell Biol. 1998;109:533–543. doi: 10.1007/s004180050253. [DOI] [PubMed] [Google Scholar]
- 3.Hirschberg K, Miller C M, Ellenberg J, Presley J F, Siggia E D, Phair R D, Lippincott-Schwartz J. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez-Menarguez J A, Geuze H J, Slot J W, Klumperman J. Cell. 1999;98:81–90. doi: 10.1016/S0092-8674(00)80608-X. [DOI] [PubMed] [Google Scholar]
- 5.Polishchuk R S, Polishchuk E V, Marra P, Saverio A, Roberto B, Luini A, Mironov A A. J Cell Biol. 2000;148:45–58. doi: 10.1083/jcb.148.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White J, Johannes L, Mallard F, Girod A, Grill S, Reinsch S, Keller P, Tzschaschel B, Echard A, Goud B, Stelzer E H K. J Cell Biol. 1999;147:743–760. doi: 10.1083/jcb.147.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller P, Toomre D, Diaz E, White J, Simons K. Nat Cell Biol. 2001;3:140–149. doi: 10.1038/35055042. [DOI] [PubMed] [Google Scholar]
- 8.Stephens D J, Pepperkok R. J Cell Sci. 2001;114:1053–1059. doi: 10.1242/jcs.114.6.1053. [DOI] [PubMed] [Google Scholar]
- 9.Allan V J, Schroer T A. Curr Opin Cell Biol. 1999;11:476–482. doi: 10.1016/s0955-0674(99)80068-4. [DOI] [PubMed] [Google Scholar]
- 10.Dabora S L, Sheetz M P. Cell. 1988;54:27–35. doi: 10.1016/0092-8674(88)90176-6. [DOI] [PubMed] [Google Scholar]
- 11.Robertson A M, Allan V J. Mol Biol Cell. 2000;11:941–955. doi: 10.1091/mbc.11.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lippincott-Schwartz J, Donaldson J G, Schweizer A, Berger E G, Hauri H P, Yuan L C, Klausner R D. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- 13.Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner R D. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- 14.Hirokawa N. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 15.Lippincott-Schwartz J, Cole N B, Marotta A, Conrad P A, Bloom G S. J Cell Biol. 1995;128:293–308. doi: 10.1083/jcb.128.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farsad K, Ringstad N, Takei K, Floyd S R, Rose K, De Camilli P. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takei K, McPherson P S, Schmid S L, De Camilli P. Nature (London) 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- 18.Takei K, Haucke V, Slepnev V, Farsad K, Salazar M, Chen H, De Camilli P. Cell. 1998;94:131–141. doi: 10.1016/s0092-8674(00)81228-3. [DOI] [PubMed] [Google Scholar]
- 19.Waugh R E. Biophys J. 1982;38:19–37. doi: 10.1016/S0006-3495(82)84526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans E, Yeung A. Chem Phys Lipids. 1994;73:39–56. [Google Scholar]
- 21.Evans E, Bowman H, Leung A, Needham D, Tirrell D. Science. 1996;173:933–935. doi: 10.1126/science.273.5277.933. [DOI] [PubMed] [Google Scholar]
- 22.Raucher D, Sheetz M P. Biophys J. 1999;77:1992–2002. doi: 10.1016/S0006-3495(99)77040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bukman D J, Yao J H, Wortis M. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Top. 1996;54:5463–5468. doi: 10.1103/physreve.54.5463. [DOI] [PubMed] [Google Scholar]
- 24.Heinrich V, Bozic B, Svetina S, Zeks B. Biophys J. 1999;76:2056–2071. doi: 10.1016/S0006-3495(99)77362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinberg G, Schliwa M. J Biol Chem. 1996;271:7516–7521. doi: 10.1074/jbc.271.13.7516. [DOI] [PubMed] [Google Scholar]
- 26.Surrey T, Elowitz M B, Wolf P E, Yang F, Nedelec F, Shokat K, Leibler S. Proc Natl Acad Sci USA. 1998;95:4293–4298. doi: 10.1073/pnas.95.8.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angelova M I, Soléau S, Méléard P, Faucon J F, Bothorel P. Prog Colloid Polym Sci. 1992;89:127–131. [Google Scholar]
- 28.Slusarewicz P, Hui N, Warren G. Cell Biology: A Laboratory Handbook. Vol. 1. San Diego: Academic; 1994. pp. 509–516. [Google Scholar]
- 29.Brugger B, Sandhoff R, Wegehingel S, Gorgas K, Malsam J, Helms J B, Lehmann W D, Nickel W, Wieland F T. J Cell Biol. 2000;151:507–518. doi: 10.1083/jcb.151.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rädler J, Sackmann E. J Phys II [French] 1993;3:727–748. [Google Scholar]
- 31.van Meer G. Trends Cell Biol. 1998;8:29–33. doi: 10.1016/s0962-8924(97)01196-3. [DOI] [PubMed] [Google Scholar]
- 32.Visscher K, Schnitzer M J, Block S M. Nature (London) 1999;400:184–189. doi: 10.1038/22146. [DOI] [PubMed] [Google Scholar]
- 33.Badoual M. Ph.D. thesis. Paris: Université Pierre et Marie Curie; 2001. [Google Scholar]
- 34.Helfrich W, Servuss R-M. Nuovo Cimento. 1984;3 D:137–161. [Google Scholar]
- 35.Sandre O, Moreaux L, Brochard-Wyart F. Proc Natl Acad Sci USA. 1999;96:10591–10596. doi: 10.1073/pnas.96.19.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans E, Ludwig F. J Phys Condens Matter. 2000;12:315–320. [Google Scholar]
- 37.Coppin C M, Pierce D W, Hsu L, Vale R D. Proc Natl Acad Sci USA. 1997;94:8539–8544. doi: 10.1073/pnas.94.16.8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakagawa T, Setou M, Seog D, Ogasawara K, Dohmae N, Takio K, Hirokawa N. Cell. 2000;103:569–581. doi: 10.1016/s0092-8674(00)00161-6. [DOI] [PubMed] [Google Scholar]
- 39.Schekman R, Orci L. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- 40.Wieland F, Harter C. Curr Opin Cell Biol. 1999;11:440–446. doi: 10.1016/s0955-0674(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 41.Noda Y, Okada Y, Saito N, Setou M, Xu Y, Zhang Z, Hirokawa N. J Cell Biol. 2001;155:77–88. doi: 10.1083/jcb.200108042. [DOI] [PMC free article] [PubMed] [Google Scholar]