

Abstract
Minimally invasive molecular biomarkers have been applied to the early detection of multiple cancers in large scale case-control and cohort studies. These demonstrations of feasibility herald the potential for permanent transformation of current cancer screening paradigms. This commentary discusses the major opportunities and challenges facing the preclinical development and clinical validation of multicancer early detection test strategies. From a diverse set of early detection research perspectives, the authors recommend specific approaches and highlight important questions for future investigation.
Keywords: biomarkers, early detection of cancer, liquid biopsy, translational medical research, tumor
BRIEF BACKGROUND
Globally, cancer is the second leading cause of death.1 Although presymptomatic early detection improves clinical outcomes and mortality,2 only a few of the most common cancer types, including colorectal, cervical, and breast are currently targeted for general population screening in the United States.3 Most cancers go unscreened because of low-population prevalence,2 high costs of single-organ screening,4,5 suboptimal screening-test performance, and disparities in access and uptake of the approved tests at the population level. Accordingly, cancers such as pancreatic, lung, ovarian, and esophageal cancers are typically diagnosed after symptom onset, which invariably indicates advanced-stage disease and poor prognosis.6 This enormous gap in cancer control can be filled in large part through effective presymptomatic, population-wide early-detection strategies that shift the burden of disease to an earlier stage, where treatment may be more effective or potentially curative.
This goal could be achieved by development and wide use of tests that detect multiple cancers in a screened population. A multicancer early detection (MCED) test has strong epidemiologic rationale7 as the aggregate prevalence of the most lethal unscreened cancers exceeds that of any single-organ cancer.2 Encouragingly, emerging clinical-translational data corroborate the biological expectations that currently unscreened cancers harbor tissue-specific molecular profiles8,9 and can be detected by a minimally invasive “liquid biopsy” of nucleic acid and protein markers assayed from bodily fluids,10,11 such as blood.12 Most compellingly, early data now strongly indicate that this liquid-biopsy approach can identify the location of a clinically silent primary tumor,13,14 with the expectation of rapid diagnosis and treatment. Recently published modeling analyses are also encouraging, suggesting that 26% of all cancer-related deaths could be avoided through using an MCED strategy to intercept cancers among the 50- to 79-year-old population in the United States.15
As industry and academia actively pursue this approach toward a goal of more universal cancer screening, the field is faced with critically important and previously unaddressed questions. How can technical and biological limits be pushed to inform the community on which biomarkers should be developed to optimally distinguish clinically progressive or lethal cancers from other neoplastic or incidental findings that could lead to unnecessary diagnostic testing or harmful treatment? As the interest in this strategy grows there has been a rapid proliferation of early-phase research and publications on cancer biomarkers; however, few of these will be validated, commercialized, or gain regulatory approval—why? How can the fundamental process of biomarker development,16 unchanged for almost 2 decades, and designed around early detection of single-organ cancers be adapted to MCEDs in preclinical research and prospective human trials?
Here we begin to systematically address these questions to propose a framework for MCED development, validation, and clinical utility. New questions raised herein should form the basis for targeted and collaborative research toward a long-term yet urgent goal to reduce the global burden and human suffering from cancer. Beginning with the current and suboptimal state of single-organ cancer screening, a vision for the desired future clinical research state is presented. The opportunity assessment for multicancer testing is supported by discussion of emerging clinical research on this approach. Barriers to implementation are manifold: For example, engaged stakeholders must work from a common lexicon, anticipate the needs of a changing population, and anticipate downstream implications on diagnosis, treatment, and surveillance after a “test positive” is defined.
Because of these challenges, emphasis on rigor and reproducibility must be maintained. We argue that the general principles of the discovery and assay development pathway in human biospecimens can and should be adapted in novel ways to rapidly facilitate the development and validation of MCED tests with a long-term goal of widespread adoption, if proven to be as clinically impactful as anticipated. To demonstrate the clinical utility of multicancer testing, biologic and epidemiologic principles are anticipated to guide the cancers targeted, the inclusion or exclusion of precancerous targets, and the intended target populations. These will in turn drive considerations of trial designs, end points, and sample-size requirements for the evaluation of MCEDs. A clinical research model and questions for new investigation are proposed to speed the translational development of multicancer testing into evidence-based practice.
CURRENT STATUS
Before assessing the opportunities and barriers to clinical research on multicancer testing, the current state of cancer screening should be considered. Because the design and delivery of cancer-screening programs across global regions are markedly different, the emphasis of this article is from the US perspective, with discussion of key topics related to other low-middle-high–income countries addressed in the companion international perspectives commentary.17 Broadly speaking, screening of asymptomatic average-risk adults is recommended for cancers of high incidence, by tests proven to reduce mortality from those cancers. Although a detailed review of each disease and screening modality is beyond our present scope, an overview is provided to highlight common themes on the successes and substantial challenges of the status quo.
As of 2020, the American Cancer Society screening guidelines endorse mammography for early detection and mortality reduction from breast cancer, Pap smear with or without human papillomavirus testing for the prevention of cervical cancer incidence and mortality, and structural (colonoscopy or sigmoidoscopy), molecular (fecal immunochemical hemoglobin, guaiac occult blood, or multitarget stool DNA) or imaging-based (computed tomography [CT] colonography) screening for the prevention of incident and mortal colon cancer.3 Cervical cancer incidence is the lowest of these 3 at <14,000 new cases in 2020,2 and steadily falling since the widespread use of the Pap smear began in the mid-1960s.6 Although successes have been seen in the rates of death from both breast cancer and colon cancer, the annual incidence of both diseases remains high, at >276,000 and >147,000 new cases, respectively, leading to >95,000 attributable deaths in 2020 alone.2
There are numerous unresolved questions about our current approach to cancer screening. Rates of participation in colorectal cancer screening have slowly improved, but remain suboptimal; breast and cervical cancer-screening–adherence rates are either stable or decreasing.3 Because each organ is screened by a different disease-specific test, the rate of completion for screens of colon, breast, and cervical cancer is only 42% by self-report and likely overestimated.18 Screening is expensive: Although considered cost-effective by “willingness to pay” thresholds, there are few instances in which cancer screening pays for itself.19,20 Screening strategies must respond to the impact of emerging therapies, vaccine prevention,21 and changing population trends in cancer incidence22 and mortality,23 as well as screening’s attendant benefits or harms.24 In part, these considerations account for the differences in recommendations regarding the age at which breast or colorectal cancer screening should begin.3,25,26 Emerging molecular screening tests, notably for colorectal cancer, must account for potential trends in the biochemical hallmarks of cancer in association with age or prevalent exposures,27 variation in the prevalence of predisposing germline mutations,28 and clonal mutations of indeterminate potential.29
Some of these risks are sufficient to drive single-organ surveillance testing for special populations. Examples such as liver cancer surveillance among those with cirrhosis or noncirrhotic chronic hepatitis B infection30 or lung cancer screening by CT among those with significant smoking history31 have compelling evidence for effectiveness on cancer mortality, but will not detect the majority of these cancers,32 which most often occur de novo in the general population or in persons in whom these high-risk states are unrecognized. Accordingly, this review will maintain focus on an intended target population of asymptomatic average-risk adults.
In this average-risk population, cancers with high prevalence result in a low number needed to screen (NNS) to detect 1 invasive cancer (Fig. 1A). The NNS for less-common cancers rises rapidly, when considered under the current single-organ paradigm. In contrast, when a multi-organ perspective is considered, the aggregate prevalence of the less-common cancers overtakes even the most common single-organ cancers, dramatically lowering the NNS for the group of cancers. When tests with reasonably high specificity are applied to the aggregate prevalence of all cancers, the positive-predictive value (PPV) is anticipated to fall well within currently accepted performance ranges (Fig. 1B).7 This approach will require tools (ie, screening/early detection tests) with high sensitivity and specificity for curable-stage disease across multiple currently lethal cancer types, with the capability of efficiently directing diagnostic evaluation and curative treatments. This reimagined paradigm creates a new vision for the future state of cancer screening, expected to be transformational in advancing cancer control as a compelling complement to sociobehavioral interventions that promote healthy lifestyles at the individual and population levels.
Figure 1.
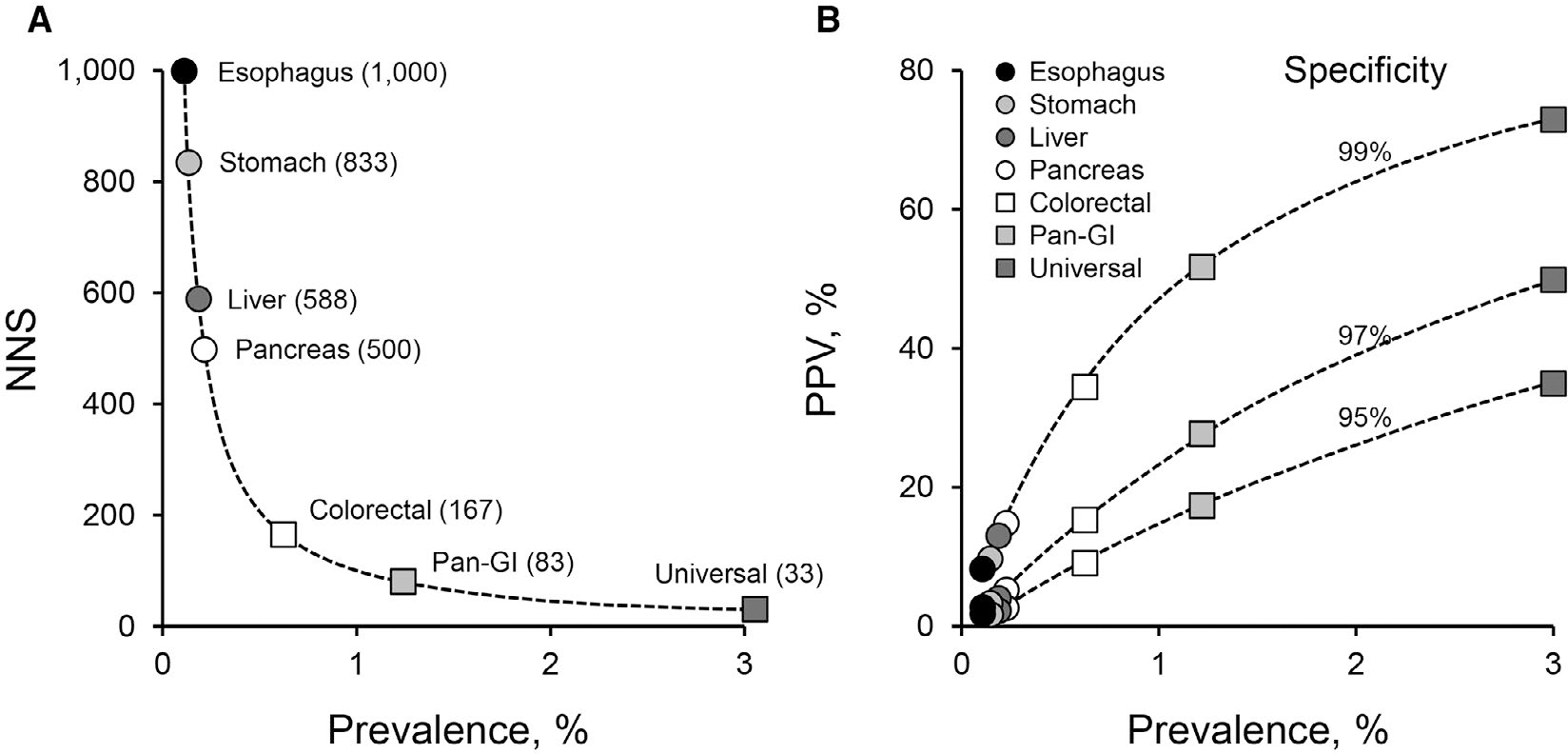
Impact of cancer prevalence on screening efficiencies. (A) Exponential relationship between cancer prevalence and the number of patients needed to be screened to detect a single cancer (NNS). Estimated NNS is plotted for cancers at individual gastrointestinal organs (only colorectal screening is currently practiced), for combined gastrointestinal cancers (Pan-GI), and for all cancer types in aggregate (Universal). For this illustration, detection sensitivities of 100% were assumed in calculations of NNS. (B) Influence of cancer prevalence on positive predictive value (PPV) at various specificities. For illustrative purposes, estimated PPVs are plotted for same spectrum of single and combined cancer-screening approaches as in A (from Ahlquist DA7 used with permission).
FRAMING THE FUTURE
Maturation of Biomarker-Based Early-Cancer Detection
The ability to detect cancers in blood and other biofluids is 1 of the most exciting advances in cancer diagnostics; it meets the many criteria of a potentially disruptive innovation. Easily accessible, minimally invasive media sampled for a multicancer test could reach a larger patient demographic than structural examinations of individual organs requiring specialized equipment, may be comparably accurate and potentially much less costly. This increasingly likely future state builds upon decades of oncology research that was harnessed in the identification and development of biomarkers to distinguish cancer from noncancer. Technical advances using these biomarkers resulted in sensitive assays capable of detecting cancer-specific analytes in the presence of a vast excess of analytes derived from sources not specific to the presence of cancer. These developments propelled our ability to evaluate tumor-derived material in the circulation and other biofluids.14,33–36 Although a description of the numerous assays for cancer biomarkers falls outside of this article’s scope, the analytical sensitivities and specificities of assay technologies have vastly improved over the years.
Clinical research indicates that most cancer biomarkers are not cancer-type specific and can be used to detect multiple cancers in a group of patients. Such a biomarker might also detect different cancer types with different sensitivity.14,36,37 To overcome some of these challenges, biomarker research has recently focused on the identification of marker combinations that might be more accurate for early-stage cancers and validation of biomarkers that can increase the sensitivity for certain tumor types or indicate their tissue of origin.
A cornucopia of analytes exists, each with its own advantages and disadvantages. DNA is the most common analyte used in liquid biopsies. DNA can be highly informative in that genetic changes, such as point mutations, small insertions and deletions, copy number changes, and large rearrangements, fragment size, and epigenetic changes, such as methylation, can be measured and quantified. Such changes also take place in nonneoplastic cells; if no measures are used to eliminate them, they may result in false positives. Benign mutations in blood cells, as in clonal hematopoiesis of indeterminate potential must be accounted for in liquid-biopsy analysis of mutation signatures and correctly identified as nonpathogenic.38 In fact, there are reports of mutation frequencies in normal tissues within the limits of detection of new liquid-biopsy technologies.39–41 Such issues affect the specificity of the test; they are common among biomarkers, necessitating the careful development of algorithms for their interpretation. Even for biomarkers extensively used in liquid biopsies for the detection of cancer, such as circulating tumor DNA (ctDNA), an understanding of the pathophysiology of their release is lagging. Despite a good correlation between the amount of ctDNA, tumor size, and tumor stage, there is a lack of predictability because of large variations. In early-stage cancers, another confounding issue is the low and variable amount of an analyte at any given point in time, resulting in stochastic events where 2 different blood draws from the same individual might yield different test results.
One approach to try to alleviate these challenges is the development of MCED tests that rely on a combination of analytes to increase the sensitivity of the detection of early-stage cancers. This approach increases the chances of providing a positive result based on the possibilities that 1) at least 1 will be positive in a sample and 2) each has a different distribution of positivity. For example, the multitarget stool DNA test for colon cancer screening takes advantage of the presence of blood, methylation, and mutations in stool.42 Studies have indicated that protein biomarkers in combination with ctDNA also help in the detection of cancer when applied with criteria different than their more common use as markers for tumor burden.43 The authors also anticipate that academic and industry scientists will identify new candidates as biomarkers for the detection of cancer and explore other avenues, such as metabolites and novel biomarkers that are associated with, rather than causative for cancer, such as markers of host immune response.
Although multianalyte tests could result in increased sensitivity, performing multiple assays within a single test can compromise specificity and raise costs. The specificity of a test could be also affected by age. Many of the biomarkers currently considered for the early detection of cancer are known to increase in prevalence with age in individuals without cancer or with comorbidities; therefore, the effects of age must be controlled and deliberately studied during development, validation, and clinical testing.44
Opportunities and Needs to Move Forward Quickly
The majority of prototype MCED assays have been developed and evaluated in case-control studies that include individuals who are known to have cancer at the time of testing as cases. A test, CancerSEEK (now licensed by Thrive Earlier Detection), made positive or negative calls by a logistic regression algorithm from multiplexed (polymerase chain reaction [PCR]) detection of mutations in regions of 16 commonly mutated genes and measurements of validated protein biomarkers.14 In contrast, Galleri (GRAIL) is a test that uses a targeted methylation bisulfite sequencing assay coupled with machine learning.36 Other approaches in development include genome-wide bisulfite-free plasma DNA methylation profiling,45 targeted methylation bisulfite sequencing followed by semitargeted PCR of differentially methylated regions,46 and whole-genome sequencing for genome-wide DNA fragmentation patterns.35
Cancers in these studies are likely to be larger and more advanced because most were diagnosed based on symptoms, and presumably can be identified with higher sensitivity compared with undiagnosed cancers in the average-risk population. Such studies, although necessary to evaluate the candidate assay, will overestimate its sensitivity. In addition, retrospective studies usually have control arms that do not represent the target population. These factors have to be addressed in large clinical studies at later phases of biomarker development as detailed below (see the subsection What Are the Required Phases of Biomarker Development?). The ability of a test to identify cancers in individuals with previously undetected disease can be evaluated only in a prospective study in the target population, like DETECT-A (Detecting Cancers Earlier Through Elective Mutation-Based Blood Collection and Testing), which used a version of the CancerSEEK test.48
The performance characteristics of a test need to be thought of in the context of the prevalence of the disease in the target population and the test’s intended clinical use. The same test, for example, detecting mutations in plasma, may require different specificity and sensitivity to be effective depending on its clinical application. A screening test targeting average-risk individuals without a known cancer requires high specificity to avoid the detection of false positives that will lead to extensive diagnostic testing and possible overtreatment. A disease prevalence of 1% and a test with a sensitivity of 50% and a specificity of 99% will result in twice as many false positives as true positives. For the same test applied to a high-risk population of individuals already under the usual clinical surveillance, a lesser specificity might be acceptable to favor an increase in sensitivity. In a screening program in which individuals are tested at predefined intervals, a test could have different PPVs and negative-predictive values (NPVs) as the prevalence of the disease changes with time, especially if the program is successful in identifying cancers.
However, a screening test is not a diagnostic test. What then is the diagnostic cascade after a positive result? One approach is to provide information about the potential location of the cancer based on biomarkers; this is referred to as site prediction or tissue-of-origin modeling.14,35,36,47 Such information would be valuable and funnels the individual toward a streamlined diagnostic pathway. In practice, a test incorporating appropriate biomarkers for the detection of the cancer type will require exquisite specificity because general tissue of origin signals will be misleading in terms of the false–positive results. The approach also requires exquisite specificity for localizing the cancer—or the number of false negatives will increase.
Another approach is to use diagnostic positron emission tomography (PET)-CT for the accurate localization of the cancer in the individuals with a positive blood test, as in the DETECT-A study, a prospective, interventional study of 10,000 women between 65 and 75 years of age without a previous diagnosis or family history of cancer.48 The radiographic results provided much more than an accurate localization of the lesion. First, the results excluded half of the individuals for any further follow-up procedure increasing the PPV. For the remaining individuals, PET-CT also provided data used for cancer staging, required for the immediate management of the patient.
Observational studies do not report results to the patients and their physicians. Interventional studies will be required to evaluate the impact of a test on the management of individuals. DETECT-A started exploring such an impact of a multianalyte test where participants with positive blood-test results underwent diagnostic evaluation and intervention with intent to cure depending on the stage and tumor type identified. We anticipate that the next steps in this continuum of work will include the development and definition of clinical pathways from the MCED test result to a definitive diagnosis for each target cancer. One example of this work is the PATHFINDER study (Assessment of the Implementation of an Investigational Multi-Cancer Early Detection Test Into Clinical Practice; ClinicalTrials.gov Identifier: NCT04241796), which will enumerate the number and types of diagnostic tests required to achieve diagnostic resolution following a “signal detected” MCED test result, for each of 6200 participants. These studies will also help inform numerous stakeholders, beginning with patients, primary care providers, disease specialists, payers, regulators, and guidance committees, on the potential benefit:harm ratio of test implementation.
OPPORTUNITIES AND CHALLENGES
Changing Landscape of Cancer-Risk Factors
It is estimated that nearly half of all cancer cases are attributable to lifestyle (eg, tobacco and alcohol use) and environmental/occupational exposures (eg, ultraviolet radiation and air pollution).49 Consequently, it will be critical that any multicancer screening test includes markers reflective of these exposures. However, these exposures vary significantly between populations and within a single population over time. These variations in exposures, combined with variations in cancer treatments within and between populations, determine the set of cancers that can be screened for in a particular population, taking into consideration well-established criteria for a screening test.50 Therefore, the development pathway, and ultimately the clinical application, of a MCED test must take into account the current exposures and cancer landscape of the population in which it is to be applied.
It is highly likely that no single MCED test will be the sole solution to the challenge of early detection. Different intended circumstances of use will affect the performance characteristics that are needed from a test (eg, a primary screening test vs a rule-out triage test). Furthermore, the epidemiology of particular cancers will affect how the test performs for that particular cancer. The differences in prevalence between cancers will affect the PPVs and NPVs of the test for each cancer type: As the prevalence increases, the PPV also increases but the NPV decreases. Similarly, as the prevalence decreases, the PPV decreases while the NPV increases.51 This will also apply to the performance of a test in particular subpopulations, as stratified by cancer-causing exposures (eg, behavioral factors) or germline characteristics (eg, ethnicity, germline genomic variants), who may have a different prevalence of cancer compared with the general population. Indeed, even within a particular population and organ-of-origin cancer type, there will likely be molecular subtypes that are more or less susceptible to a given screening test because of the particular markers included in the test. All these factors must be borne in mind by test developers when considering their tests’ markers or performance characteristics, their intended populations or circumstances of use, and when establishing their cutpoint values or guidance regarding the interpretation of test results.
As 1 example, if the United States is considered the population of intended use, development of a MCED test would need to consider a cancer landscape characterized by declining mortality, shifting risk factors, therapeutic improvements including vaccines and immunotherapies, the availability of and unequal access to evidence-based single-organ screening tests (ie, breast, cervical, colon, and lung for certain patients) and, of course, by the numerous molecular paths to cancer that are known to exist. Overall cancer mortality in the United States is down 29% since its peak in 1991, driven largely by declines in tobacco use and lung cancer mortality, along with improvements in early detection and treatment. The prevalence of leading cancer-risk factors in the population has changed markedly. Implementation of evidence-based tobacco control measures over the last 50 years has reduced the prevalence of tobacco use from 42% to 20% of the US population;52 whereas the prevalence of obesity has steadily increased from 15% to 42% of Americans over the same period.53 These changes in the epidemiology of cancer-risk factors will shape the future epidemiology of cancer incidence and mortality in the United States. Indeed, the incidence of tobacco-related cancers is declining, whereas the incidence of obesity-related cancers is rising. And although tobacco use still accounts for the highest proportion of cancer cases in the United States, it is followed closely by excess body weight. It is expected that at some point in the future, excess body weight will overtake tobacco use as the leading cause of preventable cancers. Any MCED test will need to be responsive to changes in lifestyle and environmental/occupational exposures to ensure that it detects cancers of maximum public health importance.
Therapeutic enhancements and their availability and uptake among populations must also be considered in the design and implementation of a MCED test. For example, an effective cancer-prevention vaccine was approved in 2006 to prevent or reduce the risk of several types of human papillomavirus (HPV)–associated cancers, including cervical, genital, and recently, oropharyngeal cancers. Australia was the first country to implement a national HPV vaccination program in 2007; 10 years later, they transitioned their organized cytology-based screening program to primary HPV testing. They are now on track to eliminate cervical cancer by 2028.54 Yet, in the United States, HPV vaccine uptake lags far behind that of Australia and lacks an organized cervical screening program, making elimination of cervical cancer in the near future in the United States much less likely. Although screening for HPV-related cancers/markers as part of a MCED test would likely not be required in Australia in the near future, it would definitely be needed in the United States if vaccination rates remain low. Additionally, immunotherapies are resulting in significant improvements in survival for highly lethal cancers like melanoma55 and non–small cell lung cancer (NSCLC).56 Significant mortality reductions in these (and other cancers) attributable to immunotherapies may make these cancers less of an important public health problem necessitating early-detection strategies by MCEDs, particularly in the context of other favorable improvements in relevant risk factors, such as greater awareness of sun-protective behaviors in the case of melanoma and reduced tobacco use in the case of NSCLC.
In addition, evidence-based single-organ screening examinations already exist in the United States for the most common cancers. These tests have been shown to result in significant mortality reductions in their associated cancers, yet unequal access to these examinations persists. For example, the prevalence of the receipt of mammography, and cervical and colorectal cancer screening among the uninsured is substantially less than what it is among the insured.57 Moreover, there are technical and logistical aspects of these screening tests and their associated screening programs that challenge their widespread implementation in low-resource settings. Where and when these tests are accessible, we anticipate that MCED testing should be complementary to existing options. It is also unclear if a multicancer screening test would be subject to the same barriers to access as single-organ screens; these disparities must be kept in mind as such tests are developed and marketed.
Finally, as alluded to earlier, there are many possible molecular paths to cancer. Although these paths are organized around the acquisition of at least 6 “hallmarks” or phenotypic traits,58 relatively little is known about the order, timing, and frequency in which these hallmarks, and their underlying genotypic variations, occur in the neoplastic process. Additionally, there are enabling characteristics crucial to the acquisition of these 6 hallmark capabilities, as well as the tumor microenvironment acting on neoplastic progression.59 These features of early cancer development and its dynamic evolution present a significant challenge to the proper clinical interpretation of a MCED test based on a snapshot of various markers at a single point in time, especially if precursor lesions can be detected. A related issue is the existence of extensive subclinical genomic clonal mosaicism that has been revealed in normal tissue.60 High rates of mosaicism could complicate the interpretation of a multicancer screening test.
In summary, within- and between-population variations in cancer-risk factors, cancer incidence and mortality, and treatment patterns will need to be considered in the design and implementation of a multicancer screening test to ensure that it captures the relevant/dominant exposures and germline susceptibilities occurring within the intended-use population at a particular point in time.
Lack of a Shared Lexicon for Critical Terminology in Early Detection
The early detection of cancer is an inherently multidisciplinary exercise: To be fully informed about the appropriate purpose, population, application, and interpretation of an MCED test, integrated expertise is needed from across biology, biomarker research, the physical sciences, engineering, data science, epidemiology, clinical research, behavioral science, and other areas. Each of these fields has its own language and terminology; across the disciplines (and indeed even within them), multiple terms may be used to describe the same concept, and the same words may be used to convey different concepts. This ambiguity is a significant barrier to interdisciplinary understanding, scientific progress, and crucially to communicating effectively with the public, health care payers/providers, and government agencies—all of whom are key stakeholders in the implementation, uptake, and reimbursement of any multicancer screening approach. We propose that the establishment of a shared lexicon and understanding of terms concerning cancer screening would be of great value in accelerating progress in this field. Our suggestions for simple, standard definitions are provided in Table 1.
TABLE 1.
A Cancer-Screening Lexicon
| True positive | The screening test gives a positive result, which is subsequently confirmed to be malignant cancer or a precancerous lesion that requires active surveillance, if that is the intended purpose of the test. |
| False positive | The screening test gives a positive result, which is subsequently confirmed to not be malignant cancer or a precancerous lesion that requires active surveillance, if that is the intended purpose of the test. |
| True negative | The screening test gives a negative result when no malignant cancer or precancerous lesion requires active surveillance, if that is the intended purpose of the test, is indeed present. |
| False negative | The screening test gives a negative result, but a malignant cancer or a precancerous lesion that requires active surveillance, if that is the intended purpose of the test, is subsequently confirmed to be present. |
| Consequential disease | A condition that will cause mortality or significant morbidity within the individual’s expected remaining lifespan. |
| Overdiagnosis | Making a person into a patient unnecessarily; a screen-positive result that detects an inconsequential condition, which does not require treatment or monitoring. |
| Sensitivity | The ability of a test to correctly identify those with the disease; a test with high sensitivity will miss fewer cancers (ie, fewer false negatives). |
| Specificity | The ability of a test to correctly identify those without the disease; a test with high specificity will have fewer instances of saying a cancer is present in error (false positives). |
| Positive predictive value | The probability that individuals with a positive test result actually have the disease increases with increasing disease prevalence. |
| Negative predictive value | The probability that individuals with a negative test result do not actually have the disease. |
| Precancerous lesion | A cellular or gross deviation away from normal that has an elevated risk of, or is on a trajectory toward, transformation into cancer. |
| indeterminate (lesion/nodule) | An abnormality that has a risk of progressing to become consequential cancer, the extent of which is unknown. |
| Detection | Identification, by means of a test, of a previously undiagnosed condition. |
| Diagnosis | Identification and characterization of a specific disease/condition following detection/symptomatic presentation. |
| Cancer screening | Testing asymptomatic, apparently healthy people for signs of cancer. |
| Targeted cancer screening | Testing asymptomatic individuals for cancer, within a subset of the population who are at higher risk of cancer (based on genomic risk, family history, behavioral factors, etc, beyond a simple age cutoff that is applied in all screening programs); therefore, finding a higher incidence of disease (synonymous with “surveillance”). |
| Risk stratification | Using genotypic, phenotypic, environmental, or behavioral information to identify individuals or populations who are at higher risk for developing cancers; can also identify those who are at lower risk and tailor interventions appropriately. |
Further to adoption of the above-referenced common terminology standards to improve knowledge exchange, there are other common areas of miscommunication and uncertainty that have hampered progress toward the early detection of cancer. Researchers in both academia and industry need to think carefully and express themselves clearly around the parameters used to evaluate test performance, result interpretation, and recommended action(s). As touched on in the lexicon above, test developers must carefully consider how to define a positive/negative result: Does the test aim only to identify a curable-stage cancer versus any cancer versus any cancer or precancerous lesions? In some cases (usually where intervention is simple and low risk), identification of a precancerous lesion can be beneficial, so a test developer might classify these findings as true positives. However, in other cases (where risk of progression is low and/or intervention is complex or risky), one might not wish to identify precancerous lesions, so would define the same category of abnormality as true negatives. Test developers must be mindful of this contextual distinction and clearly set test-performance expectations accordingly. Thus, even though many MCED test prototypes do not yet appear reliable for the detection of precancerous lesions or have not been reported for patients known to harbor such lesions, these should be included in clinical testing to verify sensitivity and specificity estimates, as appropriate. For the majority of biomarker classes, state of the art assays report a marker signal as continuous, and defining test positive/negative becomes a matter of establishing a clinically consequential cut point (see the subsection What Are the Required Phases of Biomarker Development? below). In tests based on continuous variables, then a “positive” result might actually indicate a spectrum of cancer risk (with further workup and confirmatory diagnosis required) rather than a simple dichotomous answer. Again, test developers must be mindful of these issues, and look to quantify and express the uncertainty in the range of values in positive and negative cases.
Determining Appropriate Specificity of First-Line Test/Intervention
For any proposed MCED approach, definition of the clinical pathways, from first-line test to diagnosis for each target cancer, is essential. These pathways will need to establish minimum benefit:harm ratios at diagnosis, with input from all stakeholders. One approach might be to back-calculate the minimum required sensitivity, specificity, and/or other performance metrics that a first-line test must exhibit to be clinically, ethically, and economically workable. These metrics would be dependent, at least in part, on the downstream consequences of the test and the invasiveness of a diagnostic evaluation and treatment strategy. One noteworthy example is ovarian cancer. Biomarkers for this disease are required to have very high specificity because the subsequent diagnostic cascade includes expensive and/or invasive follow-up, such as CT or ultrasound imaging, as well as laparoscopic or even open surgery for definitive diagnosis.
Integration With Existing Screening Paradigms and Risk of Overdiagnosis
At present, there is a paucity of rigorously reproduced, independently validated data to suggest that liquid-biopsy tests of single- or multicancer targets can detect clinically significant precancers, such as colorectal advanced adenomas. A further potential pitfall is that a negative result from a MCED test might dissuade individuals from participating in standard-of-care (SOC) screening (mammography, fecal testing, etc) or delay a symptomatic presentation because the test result provided false reassurance of no underlying disease. We thus anticipate that MCED testing should be additive and complementary to already SOC–screening programs, in settings where these services are available, at least until the full implications of the additional MCED testing are better clarified. This was the case in the DETECT-A study, where the blood test doubled the number of cancers identified by SOC screening. Importantly, participation in the study did not dissuade individuals from following-up to receive SOC–screening tests.48
In the event that a screen–positive result identifies an increased risk of cancer or an indeterminate precancerous lesion, rather than detecting cancer itself, there will be significant implications for clinical decision-making; clinical research must be cognizant of and plan for this outcome. Increasing screening-test sensitivity and usage levels will increasingly provide results that 1) indicate that a cancer may be present, but which cannot be visualized at the time of testing, with the attendant need for continued careful surveillance over time; or 2) lead to identification of a lesion of indeterminate significance/unclear prognosis based on follow-up data from existing diagnostic tests. Tumor-localization measures such as PET-CT or tissue-of-origin biomarkers described previously aim to reduce overdiagnosis; the detection of a lesion that is indolent and will never result in death may lead to unnecessary invasive testing and morbid treatment effects. Although the overdiagnosis bell cannot be “unrung,” overdiagnosis does not necessarily require overtreatment; observation or surveillance may be an option. Thus, fear of overdiagnosis should not make us deaf to the bell of underdiagnosis, which is being rung approximately 60,000 times a year in the United States alone.61 Clear clinical pathways are needed to deal with each of these scenarios; further research will need to address remaining knowledge gaps.
Determining the Appropriate Target Population for a MCED Test
Ideally, a MCED test would be applicable universally. For the test to be economically practical and to minimize potential harms from false positives/overdiagnosis/invasive follow-up, it is likely that risk stratification by age would be a prerequisite. Further risk stratification might be appropriate. When designing clinical studies, the use of high-risk groups will increase the incidence rate, but will come with a trade-off: The high-risk group will not be representative of the general population, so the performance characteristics of the test will differ. Usually, the high-risk group designates a limited number of higher incidence cancers (eg, lung cancer in smokers, or breast and ovarian cancer in BRCA1/2 carriers), so that other target cancers included in the MCED test will not have higher incidence. Indeed, it is possible that the markers themselves (which the test measures) could differ between such a high-risk group and the general population.
High-risk patients represent an important, yet distinct, target population because they typically require different test performance characteristics than average-risk groups, including higher sensitivity and lower specificity, because the criteria to recommend potentially invasive follow-up testing are often more aggressive. High-risk populations might require different markers starting from discovery and preclinical work, and a separate evidence-generation plan and regulatory-approval pathway. Thus, definition of the appropriate target population(s) is critical to optimizing the benefit:risk ratio of a MCED strategy and requires careful consideration of the integrated approach used to establish criteria for defining average versus increased risk across the evaluated organ sites. Target populations could vary by geographic region based on cultural, economic, or other factors; and consideration should be given to pediatric cancers as well.
KEY CONSIDERATIONS TO ACCELERATE PROGRESS
Which Cancers Should Be Targeted?
Another major consideration for MCED test developers is which cancer types should be targeted. It is a reasonable working assumption that, for the foreseeable future, no single MCED test will have universal coverage of all cancer types. Even the broadest MCED tests currently under development claim to be able to identify only a subset of all cancers. There are various criteria by which a test developer might choose to prioritize cancer types for detection, each with a legitimate rationale. Targeting the most prevalent cancers would give the test the best chance of a high PPV and would potentially shorten the development time, as research studies may be shorter to complete because the cases of interest are more common. Alternatively, a test developer could prioritize the most lethal cancer types, where the societal and ethical imperative is greatest, and where there may be a greater willingness in payers to reimburse and of patients to undergo testing with the potential need for subsequent diagnostic evaluations. Another strategy might be to prioritize the cancers that are most amenable to early treatment with curative intent, such as those occurring in readily accessible luminal organs such as the gastrointestinal tract or upper airway. Yet another approach would be to prioritize detection of those cancers that have the worst current detection rates at an early-disease stage, and where commen-surately survival rates are poorest, such as pancreas, ovary, or lung. Test developers must be very clear as to which of these criteria they would prioritize, why they are doing so, and acknowledge the projected consequences (favorable and unfavorable) of that prioritization.
What Are the Required Phases of Biomarker Development?
To begin to address the crucial questions raised above, any biomarker-based MCED test will need to follow a rigorous pathway to ultimately demonstrate clinical benefit with acceptable harms in an asymptomatic screening population. The National Cancer Institute Early Detection Research Network (EDRN) uses a “go/no go” decision-point schema to determine which biomarkers identified through discovery (phase 1) can progress to verification (phase 2) and then to validation (phases 3–5). This framework was developed in part to clearly distinguish biomarker validation steps from those of therapy-directed clinical trials. It is based on a 5-step approach developed by the EDRN Data Management and Coordinating Center as follows: phase 1—discovery, phase 2—clinical assay and validation, phase 3—retrospective longitudinal testing, phase 4—prospective screening, and phase 5—cancer control. The criteria to determine whether to move a biomarker from 1 phase to the next include both its analytical (accuracy and reproducibility) and clinical (sensitivity, specificity, area under the curve, PPV, NPV) performance. To move a biomarker(s) from phase 1 to phase 2, there must be data showing that it has sufficient sensitivity and specificity to meet its anticipated clinical use and, if applicable, the biomarker outperforms the currently used methodology or adds significant complementary value. If the biomarker(s) fulfills these criteria, it progresses to phase 2 to show that it can accurately detect established disease (samples from case-control or cross-sectional studies) using randomized, blinded samples. If verified, the biomarker(s) moves to phase 3 to show that it can detect preclinical disease using samples from a retrospective longitudinal cohort (eg, from the Prostate, Lung, Colon, and Ovarian Cancer Screening Trial or Women’s Health Initiative) or samples collected by EDRN. If the biomarker has the sensitivity to accurately detect preclinical cancer and has an acceptable false–positive rate, it can progress to phase 4, a prospective screening trial to determine the extent and characteristics of the detected disease and determine the false-referral rate. To date, the EDRN has not undertaken any phase 4 trials with novel candidate biomarkers, although several of the Food and Drug Administration- or Clinical Laboratory Improvement Amendments-approved biomarkers could be evaluated in a phase 4 trial.
The proposed assay development phases may not all be necessary/appropriate in a given circumstance and may not be mutually exclusive. If the developmental pathway for biomarkers is shortened by novel/alternative designs, the principals underlying these outlined phases must still be deliberately considered and addressed as summarized below:
Phase 1: Phase 1 is also known as the discovery phase; during this phase, potential leads or biomarkers derived from genomic, proteomic, epigenomic, and other “-omic” analyses of biospecimens are identified. Candidate discovery biomarkers are prioritized based on the 1) degree of differentiation between cancer and noncancer, 2) potential clinical application, 3) sample source (convenience sample or high-quality specimens), 4) mechanistic or other biological evidence, and 5) applied study design. The prioritized biomarkers are then advanced to phase 2.
Phase 2: In this phase, a clinical-grade assay based on specimens that can be obtained noninvasively is developed. The true–positive rate, false–positive rate, or receiver operating characteristics curve for the clinical biomarker assay are determined and assessed for ability to distinguish subjects with cancer from subjects without cancer. The assay is optimized, verified for accuracy, and evaluated with respect to the association between biomarker measurement and disease state. Associations with relevant demographic (ie, age, gender), environmental (ie, smoking behavior), and other potentially influencing factors are further established. The biomarker moves to phase 3 only when it demonstrates superior or complementary diagnostic performance in comparison with an existing modality, if an appropriate comparator exists. By completion of phase 2, most—if not all—of the preclinical steps in the biospecimen-based assessment modality pathway should be completed (Fig. 2).62
Phase 3: In this phase, biomarkers are evaluated retrospectively, typically using previously collected specimens from a longitudinal cohort study, to assess whether the biomarker detects the disease before it has become clinically evident. Typically, a screen-positives rule is defined as a function of time before clinical diagnosis to establish capacity of the biomarker to detect preclinical disease. Criteria for a positive–screening test (sensitivity, specificity, NPV, PPV) are also defined, in preparation for phase 4 studies.
Phase 4: In a prospective-screening study, the candidate assay is applied to healthy, asymptomatic individuals, and definitive diagnostic procedures are applied at that time to those who screen positive. Thus, the number and nature of cases detected with the screening tool are determined in phase 4, as are the numbers of subjects who screen positive, but have no relevant findings identified during follow-up evaluation.
Phase 5: The final biomarker assay development phase addresses whether screening reduces the burden of cancer in the population. During this phase, estimation on the reductions in cancer mortality afforded by the screening test is made. This has historically required a prospective trial in the target population, with end points based on reductions in cancer-related mortality or late-stage cancer incidence (late-stage reduction).
Figure 2.
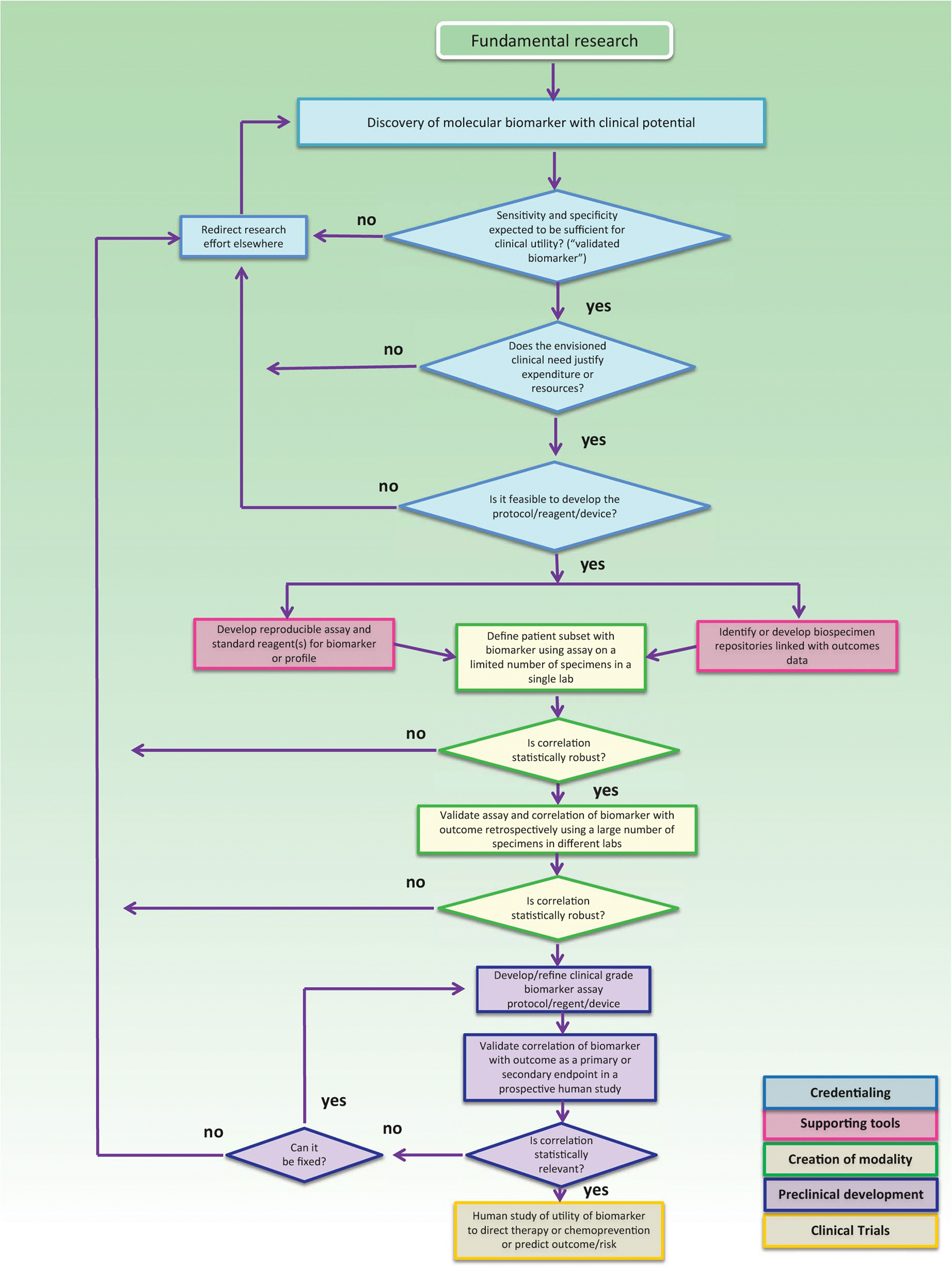
Biospecimen-based assessment modality (BM) pathway. The BM pathway is depicted as a flowchart, a schematic process representation widely used in engineering. Rounded rectangle at the top indicates the origin of the process, square-cornered rectangles, activity steps; diamonds, conditional tests or decision steps; unidirectional arrows, the direction of the activity sequence, and the direction of transfer of supporting tools from their parallel development paths to the main path of modality development. The 3 diamonds in the initial steps of the pathway (blue) are decisions required to proceed through the pathway and represent the credentialing step. Subsequent steps include the development of supporting tools (red), the creation of the modality (green), preclinical development (purple), and early-stage clinical trials (yellow). For each activity or decision point, it is understood that there are many more variations that can occur, and that not all steps may occur in each instance. The pathway does not address the ways in which insights gained from late-stage clinical trials can influence the development process. Biospecimen-based assessment devices can be used for screening, early detection, diagnosis, prediction, prognosis, or response assessment. The pathways are conceived not as comprehensive descriptions of the corresponding real-world processes, but as tools designed to serve specific purposes, including research program and project management, coordination of research efforts, and professional and lay education and communication (adapted from Srivastava et al62 used with permission provided by the American Association for Cancer Research (publisher) on May 28, 2021, under license number 5077790591401).
Defining Clinical Utility End Points
The reduction in cancer-associated mortality end points in the setting of a randomized controlled trial is often required before new screening approaches are broadly recommended or implemented at the population level. Given the relatively low incidence of cancer in screening populations, these mortality-powered trials require tens of thousands of patients to be followed over 1 or more decades, at the cost of millions of dollars.63,64 Relying on this approach as the only definitive evidence-generation model means that implementation of any novel multicancer-screening approach would be delayed by decades. Although we anticipate that MCED tests will ultimately have to demonstrate measurable and meaningful effects on cancer outcomes, intermediate indicators of clinical utility can be derived in the interim. Encouragingly, large observational and interventional studies of emerging MCED tests in addressing the intended-use population are (or will shortly be) underway as demonstration projects. For example, GRAIL recently announced a prospective evaluation of their Galleri test in 165,000 persons through a collaboration with the United Kingdom National Health System,65 and may commercially release the Galleri test as a laboratory-developed test before study completion. When available, it is likely that “real-world” postmarketing studies will be conducted based on the experience of early-adopter patients and health systems.
If acceptable to regulatory agencies, guideline developers, and payers, assessing the reduction in absolute incidence of late-stage diagnosis for targeted cancers (rather than stage shift expressed as a fraction of all cases) may represent a compelling, earlier end point to expedite prospective study execution, completion, and results reporting. This approach would likely need to be coupled to a post-implementation, real-world study of longer-term outcomes such as the demonstration of significant reductions in target cancer mortality once the screening test is in clinical use on a larger scale. This integrated evidence-generation strategy could then accumulate cancer mortality data over a longer term, although potentially benefitting many patients through more rapid MCED test access in the nearer term.
SUMMARY AND NEXT STEPS
Although many uncertainties remain with respect to research priorities, study design standards, trial-execution efficiencies, and other key topics in this rapidly evolving field, we highlight below those areas where available data have minimized controversy, as well as remaining knowledge gaps that must be urgently addressed through further investigation:
Apply the Pepe framework for preclinical and clinical development of novel cancer-associated biomarkers, to streamline interdisciplinary MCED-assay evidence-generation activities involving academic, industry, and other collaborators.
Include well-phenotyped, archival specimens in MCED-test marker-development studies. Marker candidates should be tested for validity before advancing to subsequent steps (see Fig. 2). Later phase-marker development should take advantage of samples collected at an asymptomatic stage, ideally in the context of prospective studies in the target population. Although not available for all cancers that might be considered in a prototype MCED test, the EDRN is a valuable resource for such samples, with capabilities and resources using prospective sample collections with retrospective blinded evaluations.
Verify that prespecified, standardized definitions of test performance attributes and interactions of those attributes with the population under study are available during clinical- and prospective-validation studies.
Prioritize the description of MCED prototype-test sensitivity, specificity, predictive values, numbers of cancers detected (beyond those identified by current standards of care), the specific diagnostic tests required to make or exclude a definitive cancer diagnosis, and any physical, emotional, or financial harms and benefits generated, in comparison with existing SOC options.
Incorporate interim analyses into the data analysis plans for prospective clinical trials, including safety and efficacy assessments so that accrual targets and performance goals can be appropriately confirmed or modified based on the emerging ratio of benefits:harms at diagnosis.
Achieve consensus agreement from stakeholder groups regarding how best to account for changing population trends (disease patterns, environmental exposures, etc.) in longer-term studies.
Determine a clear pathway for MCED test results of indeterminate clinical significance. As MCED tests progress with respect to both performance sensitivity and clinical application, it is possible that an increasing number of early lesions will be detected, which could represent relatively indolent malignancies (unlikely to cause death or significant morbidity within the individual’s expected remaining lifespan) or precancerous lesions of uncertain prognosis.66 Interdisciplinary input is critical to mitigate the risks associated with potential overtreatment.
Clarify the acceptability of reducing the absolute incidence of late-stage cancers as a surrogate end point for cancer-related mortality reduction in later-phase MCED clinical trials. Consideration should also be given to study design innovations that could be more feasible and efficient to establish clinical utility.
FUNDING SUPPORT
No specific funding was disclosed.
We are grateful for the conceptual and organizational input of Steven J. Skates, PhD. We thank Amanda Bedard for her administrative assistance in the preparation of this work.
The authors thank the Summit Participants, Planning Committee, and Host Organizations for defining a collaborative roadmap for the development, implementation, and adoption of a multicancer early detection (MCED) assay.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
John B. Kisiel and the Mayo Clinic have rights to receive royalties paid to the Mayo Clinic through a contracted services agreement with Exact Sciences. Nickolas Papadopoulos is a founder of PGDx and Thrive an Exact Sciences company. He holds equity in PGDx, Exact Sciences, CAGE Pharam, NeoPhore, and ManaTbio. He is a scientific consultant to Thrive, CAGE, NeoPhore, and ManaTbio. He is entitled to royalties from technologies that have been licensed by the above and other companies. The terms of all these arrangements are being managed by Johns Hopkins University in accordance with its conflict of interest policies.
REFERENCES
- 1.Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev. 2016;25:16–27. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Smith RA, Andrews KS, Brooks D, et al. Cancer screening in the United States, 2019: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin. 2019;69:184–210. [DOI] [PubMed] [Google Scholar]
- 4.Yeh JM, Ho W, Hur C. Cost-effectiveness of endoscopic surveillance of gastric ulcers to improve survival. Gastrointest Endosc. 2010;72:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rulyak SJ, Kimmey MB, Veenstra DL, Brentnall TA. Cost-effectiveness of pancreatic cancer screening in familial pancreatic cancer kindreds. Gastrointest Endosc. 2003;57:23–29. [DOI] [PubMed] [Google Scholar]
- 6.Howlader NNA, Krapcho M, Miller D, et al. SEER cancer statistics review, 1975–2017. Accessed September 30, 2020. https://seer.cancer.gov/csr/1975_2017/
- 7.Ahlquist DA. Universal cancer screening: revolutionary, rational, and realizable. NPJ Precis Oncol. 2018;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304, e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang S, Li Q, Chen Q, et al. CancerLocator: non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017;18:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majumder S, Raimondo M, Taylor WR, et al. Methylated DNA in pancreatic juice distinguishes patients with pancreatic cancer from controls. Clin Gastroenterol Hepatol. 2020;18:676–683, e673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med. 2018;379:1754–1765. [DOI] [PubMed] [Google Scholar]
- 12.Chalasani NP, Ramasubramanian TS, Bhattacharya A, et al. A novel blood-based panel of methylated DNA and protein markers for detection of early-stage hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2020;S1542–3565(20)31224–6. [DOI] [PubMed] [Google Scholar]
- 13.Sun K, Jiang P, Chan KC, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A. 2015;112:E5503–E5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubbell E, Clarke CA, Aravanis AM, Berg CD. Modeled reductions in late-stage cancer with a multi-cancer early detection test. Cancer Epidemiol Biomarkers Prev. 2021;30:460–468. [DOI] [PubMed] [Google Scholar]
- 16.Pepe MS, Etzioni R, Feng Z, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93:1054–1061. [DOI] [PubMed] [Google Scholar]
- 17.Johnson SE, Zurn SJ, Anderson BO, et al. International perspectives on the development, application, and evaluation of a multicancer early detection strategy. Cancer. 2022;128(suppl 4):875–882. [DOI] [PubMed] [Google Scholar]
- 18.Sauer AG, Liu B, Siegel RL, Jemal A, Fedewa SA. Comparing cancer screening estimates: Behavioral Risk Factor Surveillance System and National Health Interview Survey. Prev Med. 2018;106:94–100. [DOI] [PubMed] [Google Scholar]
- 19.Ladabaum U Cost-effectiveness of current colorectal cancer screening tests. Gastrointest Endosc Clin N Am. 2020;30:479–497. [DOI] [PubMed] [Google Scholar]
- 20.Rim SH, Allaire BT, Ekwueme DU, et al. Cost-effectiveness of breast cancer screening in the National Breast and Cervical Cancer Early Detection Program. Cancer Causes Control. 2019;30:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crowe E, Pandeya N, Brotherton JM, et al. Effectiveness of quadrivalent human papillomavirus vaccine for the prevention of cervical abnormalities: case-control study nested within a population based screening programme in Australia. BMJ. 2014;348:g1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf AMD, Fontham ETH, Church TR, et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J Clin. 2018;68:250–281. [DOI] [PubMed] [Google Scholar]
- 23.Siegel RL, Miller KD, Jemal A. Colorectal cancer mortality rates in adults aged 20 to 54 years in the United States, 1970–2014. JAMA. 2017;318:572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peterse EFP, Meester RGS, Siegel RL, et al. The impact of the rising colorectal cancer incidence in young adults on the optimal age to start screening: microsimulation analysis I to inform the American Cancer Society colorectal cancer screening guideline. Cancer. 2018;124:2964–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for colorectal cancer: US Preventive Services Task Force Recommendation Statement. JAMA. 2016;315:2564–2575. [DOI] [PubMed] [Google Scholar]
- 26.Siu Albert L US Preventive Services Task Force. Screening for breast cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2016;164:279–296. [DOI] [PubMed] [Google Scholar]
- 27.Limburg PJ, Mahoney DW, Ahlquist DA, et al. Comparison of tissue-based molecular markers in younger versus older patients with colorectal neoplasia. Cancer Epidemiol Biomarkers Prev. 2020;29:1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heimbach JK, Kulik LM, Finn R, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67:358–380. [DOI] [PubMed] [Google Scholar]
- 31.Wood DE, Kazerooni EA, Baum SL, et al. Lung cancer screening, version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:412–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma J, Ward EM, Smith R, Jemal A. Annual number of lung cancer deaths potentially avertable by screening in the United States. Cancer. 2013;119:1381–1385. [DOI] [PubMed] [Google Scholar]
- 33.Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Douville C, Cohen JD, Ptak J, et al. Assessing aneuploidy with repetitive element sequencing. Proc Natl Acad Sci U S A. 2020;117:4858–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shumilov E, Flach J, Pabst T, et al. Genetic alterations crossing the borders of distinct hematopoetic lineages and solid tumors: Diagnostic challenges in the era of high-throughput sequencing in hematooncology. Crit Rev Oncol Hematol. 2018;126:64–79. [DOI] [PubMed] [Google Scholar]
- 39.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee-Six H, Øbro NF, Shepherd MS, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osorio FG, Rosendahl Huber A, Oka R, et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 2018;25:2308–2316.e2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;370:1287–1297. [DOI] [PubMed] [Google Scholar]
- 43.Cohen JD, Javed AA, Thoburn C, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci U S A. 2017;114:10202–10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahlquist DA, Taylor WR, Yab TC, et al. Aberrantly methylated gene marker levels in stool: effects of demographic, exposure, body mass, and other patient characteristics. J Mol Biomark Diagn. 2012;3:133. [Google Scholar]
- 45.Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–583. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Gole J, Gore A, et al. Non-invasive early detection of cancer 4 years before conventional diagnosis using a blood test. Nat Commun. 2020;11:3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lennon AM, Buchanan AH, Kinde I, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. 2020;369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Islami F, Goding Sauer A, Miller KD, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68:31–54. [DOI] [PubMed] [Google Scholar]
- 50.Wilson JMG, Jungner G, World Health Organization. Principles and Practice of Screening for Disease / Wilson JMG, Jungner G. World Health Organization; 1968. [Google Scholar]
- 51.Tenny S, Hoffman MR. Prevalence. StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- 52.Creamer MR, Wang TW, Babb S, et al. Tobacco product use and cessation indicators among adults—United States, 2018. MMWR Morb Mortal Wkly Rep. 2019;68:1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief. 2020:1–8. [PubMed] [Google Scholar]
- 54.Hall MT, Simms KT, Lew JB, et al. The projected timeframe until cervical cancer elimination in Australia: a modelling study. Lancet Public Health. 2019;4:e19–e27. [DOI] [PubMed] [Google Scholar]
- 55.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381:1535–1546. [DOI] [PubMed] [Google Scholar]
- 56.Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for pd-l1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. [DOI] [PubMed] [Google Scholar]
- 57.American Cancer Society. Cancer prevention & early detection facts & figures 2019–2020. Accessed February 18, 2021. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/cancer-prevention-and-early-detection-facts-and-figures/cancer-prevention-and-early-detection-facts-and-figures-2019-2020.pdf
- 58.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 59.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 60.Vattathil S, Scheet P. Extensive hidden genomic mosaicism revealed in normal tissue. Am J Hum Genet. 2016;98:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viale PH. The American Cancer Society’s facts & figures: 2020 edition. J Adv Pract Oncol. 2020;11:135–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srivastava S, Gray JW, Reid BJ, Grad O, Greenwood A, Hawk ET. Translational Research Working Group developmental pathway for biospecimen-based assessment modalities. Clin Cancer Res. 2008;14:5672–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prorok PC, Andriole GL, Bresalier RS, et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials. 2000;21:273s–309s. [DOI] [PubMed] [Google Scholar]
- 64.Mandel JS, Bond JH, Church TR, et al. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota Colon Cancer Control Study. N Engl J Med. 1993;328:1365–1371. [DOI] [PubMed] [Google Scholar]
- 65.GRAIL and UK government to make galleri multi-cancer early detection blood test available to patients. Accessed February 18, 2021. https://grail.com/press-releases/grail-and-uk-government-to-make-galleri-multi-cancer-early-detection-blood-test-available-to-patients/
- 66.Srivastava S, Koay EJ, Borowsky AD, et al. Cancer overdiagnosis: a biological challenge and clinical dilemma. Nat Rev Cancer. 2019;19:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]