

Abstract
In the absence of effective patient stratification approaches, tuberculosis (TB) treatment relies on overtreating most patients to ensure high cure rates. Shortening treatment duration without compromising efficacy is therefore high on the agenda of the global TB community. While new and better drugs are certainly needed, we argue that innovative but rational treatment strategies will help achieve this goal. There is growing recognition that patient stratification, based on host and pathogen factors, is key to delivering the right drug regimen for the right duration. In this Perspective, we review the current knowledge on the heterogeneity of TB disease and propose approaches to optimize treatment duration in distinct patient groups, taking into consideration the realities of TB control globally. We emphasize key insights that improve our understanding of bacterial vulnerabilities in easy- and hard-to-treat patients, helping to reduce diagnostic uncertainties. We explore how the TB research community can integrate disease biology, pathology and symptoms, to rethink therapeutic strategies and reduce TB treatment duration.
Introduction
The recommended 6-month regimen to treat tuberculosis (TB) is driven by the duration required to cure the most severe forms of pulmonary TB, while shorter durations appear sufficient to cure less extensive disease1,2. Thus, the current standard of care can be viewed as a compromise that relies on overtreating most for the benefit of few1. This is understandable from a health system and public health perspective in which regimen simplicity and high cure rates are necessary. However, given recent advances in biomarkers of disease heterogeneity, portable technologies to stratify patients and the healthy clinical development pipeline, it is time to revisit and better define the optimal TB treatment across the disease spectrum1.
Globally, the aim of TB research and development (R&D) organizations is to discover and design highly effective, markedly shorter (< 2 months) pan-TB treatment regimens, where ‘pan-TB’ is defined as a regimen that can be administered without prior knowledge of the patient’s drug resistance profile3,4 and co-administration of mechanistically novel agents is expected to slow the inevitable emergence of clinically meaningful drug resistance. In clinical trials, the efficacy of available treatments for drug susceptible and multidrug resistant TB is excellent, with favorable outcomes reported in 85–95% of patients. However, in real world practice, effectiveness is often as low as 65%5, with an average of 88% for first-line regimens6. This decreased real-world effectiveness is partly due to limitations in implementing drug susceptibility testing, loss to follow up, suboptimal drug availability and adherence, and differences in patient populations — for example, trial inclusion/exclusion criteria can limit patient heterogeneity and disease severity relative to real-world conditions.
Here, we provide an update on our understanding of disease heterogeneity4 and how this is informing strategies for patient stratification and TB treatment shortening approaches. We also discuss the formidable challenge of dealing with this most complex infectious disease while requiring standardized and simple treatment guidelines that can be implemented in all settings for effective disease control around the world.
Clinical TB is heterogeneous
Over the past 15 years, it has been increasingly recognized that TB presents as a continuum of manifestations rather than a bimodal distribution between active and latent TB7. TB disease is more usefully described as a spectrum of responses to Mycobacterium tuberculosis (Mtb) infection, with clear implications for preventive therapy and treatment shortening strategies1,8. Disease trajectories diverge from the point of infection. Some take an accelerated course, while others may take a more chronic or undulating course and may self-clear or self-heal with or without the development of symptoms or an adaptive immune response9–11. Active symptomatic disease itself is profoundly heterogeneous.
The drivers of this heterogeneity are poorly understood and therefore individual trajectories remain largely unpredictable12. Global trends point to geographical differences in disease progression and severity13, which may be related to health seeking behavior (delayed presentation results in increased disease extent), Mtb lineage,14–16 and may also be a consequence of host-pathogen co-evolution17,18. For instance, the occurrence of severe cavitary TB (characterized by hollow spaces filled with necrotic material, gas and Mtb bacteria surrounded by a discrete fibrotic wall) is overall higher in African than in Asian patient populations19, and lineage 1 was recently associated with higher rates of cavitary TB and mortality16. Patients with comorbidities also follow differential trajectories, with unique features and disease progression profiles. HIV infection is the strongest known risk factor for progressing from latent to active TB disease. Likewise, the relative odds of developing TB in people with diabetes compared to those without diabetes ranges from 2.4 to 8.320. Finally, infants, young children, primary school children and adolescents each present markedly different rates of TB incidence, disease presentation and patterns of disease progression21.
Considerations relating to the site of disease have recently begun to influence drug regimen design in clinical trials. TB meningitis is the most devastating and difficult-to-treat form of the disease, with poor treatment response, high mortality and morbidity, and life-long neurological sequelae (in the form of cognitive impairment, motor deficit and optic atrophy) occurring in a large fraction of survivors. Treatment recommendations call for the current pulmonary standard of care (SOC) and an extended 10-month treatment continuation phase in adults22, but with increasing awareness that these regimens require substantial overhaul to reach therapeutic concentrations in cerebral sites of infection including spinal compartments23–25. At the other end of the severity spectrum, intrathoracic lymph node TB without airway obstruction or peripheral lymphadenopathy, and TB pleural effusion without pulmonary TB are often paucibacillary and sputum culture negative, and may be cured with simpler or shorter regimens than the current SOC26.
Classification systems for TB patients and disease, such as the American Thoracic Society’s TB 0 – TB 527, have been used by public health programs. This classification distinguishes between exposure only, non-clinically active and clinically active TB but does not consider active disease severity. A recent promising development in defining the spectrum of TB disease is the conceptual mapping of eight TB states, based on systematic literature mining, to standardize scientific communication and inform clinical research and public health practice11,28. Of these eight states, the first three (infection eliminated by the innate (State 0) or acquired (State I) immune response, or infection not eliminated but controlled by the immune system (State II) cannot be distinguished with existing diagnostic tools, but their study can influence future disease control strategies. States IV to VI are relevant to disease classification for the purpose of curative therapy): State IV: bacteriologically positive without symptoms, often described as subclinical disease; State V: signs or symptoms associated with TB or active TB, and State VI: severe or disseminated TB disease, differentiated from the previous state through terms such as cavitary or extensive disease.
Subclinical or incipient TB is increasingly recognized as a disease state that must be diagnosed and treated more effectively to preclude further transmission11,13,29. Although these patients do not experience clinical symptoms, microbiologic and radiographic characteristics vary widely, from negative sputum culture and absence of radiographic or PET/CT findings to sputum positivity and cavities present in a small fraction of patients30,31. Thus, subclinical TB comes with a complex array of bacterial burden and radiographic abnormalities, which presents considerable diagnostic challenges32 and impacts disease trajectories33 and therapeutic strategies. The microbiological complexity is further compounded by the presence of viable Mtb in respiratory aerosols irrespective of sputum culture status and chemotherapy34. In addition, the time at which each individual patient will seek care along their disease trajectory, or whether they will seek care at all (regardless of clinical symptoms), is influenced by socio-cultural and other factors, further highlighting the critical role of active case finding.
Clinical phenotypes determine the required treatment duration
In middle to high resource countries, clinical practice already adjusts the treatment duration for drug susceptible TB based on disease severity, site of disease and patient profile. For example, the US Centers for Disease Control and American Thoracic Society guidelines recommend 4 months of treatment for chest X-ray negative and smear negative but symptomatic disease, 9 months for HIV co-infected persons, and a 3-month extension of the continuation phase for patients with large cavities at baseline and those who remain sputum culture positive after 2 months of treatment35. In low resource countries, WHO guidelines are more typically followed, which recommend 6 months of treatment for all new TB cases36, still calling for extended treatment for infections of the central nervous system, bone or joint, and 4 months of treatment for children/adolescents with ‘non-severe’ TB22.
The history of TB regimen development that led to the current SOC has been well-reviewed elsewhere37. Treatment duration was exhaustively explored during the development of the current four-drug combination therapy; the effect on relapse after drug discontinuation (for the collection of agents available at the time) was distinctly a steep, monotonic function of treatment duration (Figure 1). The distinct shape of the treatment duration-response curve likely reflects a range of disease severities where most patients are cured with 3 months of therapy, but a subset only respond after 5–6 months of treatment. Also noteworthy is that even extending treatment to 9 months with the same regimens did not prevent relapse in 1–2% of patients. Overall, this suggests that there are three basic strata of TB patients: ‘easy to treat' (ETT, 80%), ‘hard to treat’ (HTT, 15–18%) and ‘extremely hard to treat’ (XHTT, 1–2%)19.
Figure 1. Microbiological relapse rates as a function of treatment duration.
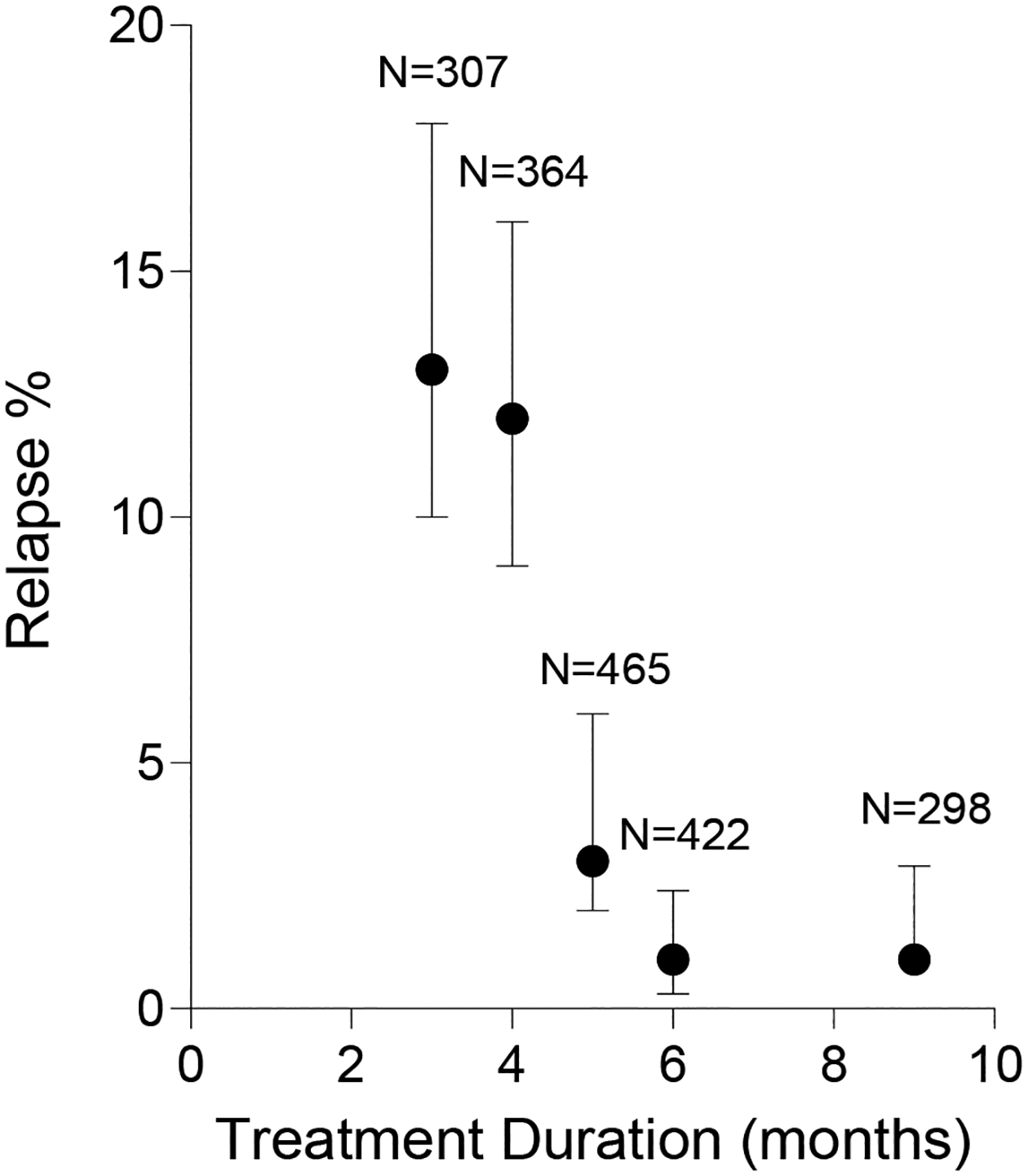
The graph shows pooled results with 95% confidence limits and the total number of participants per trial, plotted from historical data in ref108. Regimens contained various combinations of isoniazid, rifampicin, pyrazinamide, ethambutol and streptomycin. It was recognized from this analysis that more than 80% of all TB patients were cured within 3 months of combination therapy, supporting the notion that we overtreat a large fraction of patients with the current standard of care. For the remaining 20% of patient, sit wasn’t until > 5 months of therapy that relapse rates could be driven down to less than 5%, which supported 6 months as the shortest duration providing near-maximal efficacy across a heterogeneous population.
More recent clinical trials support this proposition. Three large Phase 3 trials examined the possibility that adding a fluoroquinolone to standard treatment could reduce the duration of therapy to four months38–40. All three trials failed to demonstrate non-inferiority of the shortened regimen when comparing relapse across all patients. A reanalysis of the individual patient data subsequently showed that the 4-month regimens were non-inferior in a large proportion of patients with minimal disease but were clearly inferior in patients with more extensive cavitary disease2.
HTT patients are associated with specific pathological features, especially cavitary disease, and multiple studies have shown that such patients have a higher risk of disease relapse even after six months of chemotherapy41–44. Repeated PET/CT exams in the recent PredictTB trial showed that baseline cavities, particularly those with thick walls and still containing the necrotic material known as caseum (so-called ‘dirty cavities’) are the actual site of disease relapse in patients that received 4 months of treatment19, confirming that the difference in treatment response between ETT and HTT disease centers around cavities. We posit that optimized drug regimens must be tailored to disease type: drug combinations that shorten treatment of cavitary disease may not shorten treatment of ETT disease, in which the predominant immunopathological features are inflamed airways and ‘clean’ (devoid of caseum) cavities, and vice versa (Figure 2). The notion that combining rapidly bactericidal agents selected in preclinical TB models may deliver dramatic treatment shortening for ETT but not for HTT patients is actively investigated.
Figure 2. Immunopathological features of typical easy-to-treat (ETT) and hard-to-treat (HTT) TB.

(A-B) Two patients treated for four months in the Predict TB trial, one successfully (A) and one not (B). ETT patients typically show thin-walled cavities and pulmonary nodules, visible in both the left and right lungs of the patient shown in (A). HTT patients typically show large, thick-walled cavities as shown in the left upper lobe in (B) which was also the site of disease relapse six months after this patient discontinued treatment. In the initial sputum nucleic acid amplification test (GeneXpert), patient A showed a Cycle threshold (Ct) value of 26.1 indicative of low sputum burden while patient B showed a Ct of 19.8 indicative of high sputum burden19. (C) Working model illustrating hypothetical pathological features that may create a bottleneck to cure in ETT and HTT patients and how they may overlap. Parenchymal granulomas refer to small cellular lesions. Alveolitis refers to neutrophil-driven polymorphonuclear airway disease in terminal airways and bronchitis in larger airways, the latter resulting from bronchial spread of caseous spills.
Different bacterial phenotypes may require different treatments
Tuberculosis lesions from 1500 autopsies were meticulously characterized by Georges Canetti in the 1940s. He was the first to investigate the heterogeneity of both the histopathology and the ‘histobacteriology’ of these lesions45. Canetti first described primarily cellular lesions devoid of necrosis, in which small numbers of bacilli were predominantly in macrophages. As caseation occurred (whereby necrosis results in a ‘cheese-like’ texture of the tissue or caseum) in a subset of lesions, larger numbers of bacilli were noted in the necrotic foci. As caseous lesions matured, they became paucibacillary with only 2 to 5 bacilli detected in 100 fields, but the caseum was not sterile, since injecting it to Guinea pigs resulted in disease. The remaining bacilli in these closed lesions are poised to explosively multiply upon liquefaction of the caseum. Liquefaction of caseum produces a cavity, defined by the ‘capsule’ (tissue resembling sclerosis) and an interior lining necrotic zone of variable extent, being drained with its bacillary contents into the airways. This interior necrotic zone was always associated with numbers of bacilli too numerous to count (a fact first observed by Robert Koch when he discovered the tubercle bacillus)46. Canetti also described another type of lesion, referred to as ‘polymorphonuclear alveolitis’, which was dominated by neutrophils and contained high numbers of bacilli.
Consideration of whether cavities are ‘open’ (directly connected to airways) or ‘closed’ (cut off from airways, more commonly designated as a necrotic lesion), has a poorly understood impact on the metabolic state of the bacteria. In open cavities resected from TB patients, plating the contents onto standard growth media produces colonies after the standard incubation time. However, the plated contents of many closed cavities produced colonies only after prolonged incubation for 3–10 months47. Understanding these differentially culturable bacilli in sputum is an ongoing focus of many labs (reviewed in48).
Modern imaging methods such as micro-computed tomography (CT) combined with positron-emission tomography (PET) have been used to derive 3D reconstructions of complex branched structures with often subtle connections to the airways and elucidate the dynamics of lesions over time49,50. These studies revealed that lesions can emerge from active cavities through the bronchial spread of cavity caseum, perhaps giving rise to bronchial inflammation — similar to Canetti’s ‘polymorphonuclear alveolitis’, in additional sites50. The features of these lesions imply a wide range of physiological states of the bacilli contained within51: intracellular in different immune cell types, extracellular in the vicinity of these cells and in non-vascularized caseum, actively replicating and non-replicating in a ‘persisting’ metabolically quiescent state. These populations are known to display differential susceptibilities to individual drugs, and since they are differentially abundant or present in different patients, optimizing drug regimens as function of disease phenotype is expected to deliver better cure rates and shorten treatment duration48.
Drug resistance and new TB drug regimens
The stunning success of the bedaquiline-pretomanid-linezolid combination in the NIX-TB trial52 demonstrated that > 90% cure rates can be achieved against multidrug resistant TB and that new regimens can dramatically impact treatment duration and simplicity. Yet, resistance to bedaquiline and pretomanid was detected soon after their launch. Resistance to these agents can arise during treatment, resistant organisms can be transmitted from patient to patient, and resistance is increasing in frequency in some settings53,54,55 — highlighting the need for preemptive strategies that anticipate and minimize opportunities for resistance to develop.
The evaluation of streptomycin TB monotherapy was among the first randomized controlled trials ever conducted and despite radiological improvement and a significant reduction in deaths, treatment resistance emerged in 80% of the patients37. Like most bacterial pathogens, Mtb has acquired resistance to all drugs it has been routinely exposed to56, including those only recently introduced into clinical practice (bedaquiline57, delamanid and pretomanid58) — even when used in combination with 2 or 3 other agents. The observation that mmpR5 resistance mutations predate the introduction of bedaquiline59 is helping the community draw lessons from past mistakes and develop drug susceptibility companion tests to be used in clinical trials and co-launched with future novel drugs. Deployment of such companion tests is critical because clinical resistance rates are not always consistently predicted by in vitro studies, as exemplified by isoniazid and rifampicin. In vitro, resistance acquisition rates are much higher for isoniazid than rifampicin56, but acquired monoresistance rates in patients on multidrug therapy are 0.3% for both isoniazid (95%CI 0.06 to 2.7%) and rifampicin (95%CI 0 to 0.9%) in comprehensive meta-analyses60,61. Thus, the development of a new drug or regimen should be accompanied by the development of drug susceptibility testing technologies along with the introduction of population level surveillance programs for drug susceptibility.
How can resistance emerge rapidly in the context of 3- or 4-drug therapy, as recently seen for bedaquiline, delamanid and pretomanid, seemingly faster than for the drug regimens first used in the 1960s and 1970s? First, these drugs are collectively linked to a large panel of resistance mutations: ddn, fbiABCD and fgd for delamanid/pretomanid and atpE, Rv0678, and pepQ for bedaquiline. Second, bedaquiline resistance due to drug efflux by the transporter MmpL5 (upregulated by mutations in the Rv0678 gene) was identified in geographically diverse Mtb genomes prior to its launch, attributed either to historic clofazimine use or adaptation to other environmental stressors56. Third, the frequency of resistance observed in vitro does not reliably predict acquired resistance in vivo, due to differential fitness costs (whereby resistant bacteria tend to be less ‘fit’ than their drug-sensitive counterparts). This highlights the challenges and pitfalls associated with identifying an acceptable threshold of resistance frequency in vitro to support the approval of a novel agent, be it alone or in combination. Only long-term studies assessing epidemiological and fitness costs of resistance mutations across patient populations can provide compelling answers.
In addition to these factors, de facto monotherapy at the site of infection (owing to all but one agent in the drug combination not effectively reaching the site) may drive acquired resistance. The caseous foci of necrotic lesions and cavities, which constitute treatment bottlenecks, are devoid of vascular supply and thus rely on passive equilibration of unbound drug concentrations at steady state, which can take several weeks for highly bound drugs. In addition, the very long terminal half-life of bedaquiline relative to other drugs in combination regimens creates de facto monotherapy for weeks to months following treatment completion. Combined with the profound drug tolerance of non-replicating, persisting Mtb in caseum62, these phenomena create spatial and temporal reservoirs of surviving bacteria that may fuel acquisition of resistance upon bronchogenic spread and resumption of growth. Adding a fast-acting bactericidal agent during initial treatment would increase the number of effective drugs during this period and help protect from the development of resistance. Approaches that include high-dose isoniazid, novel oral β-lactams, and second-line injectables have been proposed55.
TB is treated with multiple agents to reduce the emergence of drug resistance, but this simple idea must be tempered by the concept of pleiotropy (whereby one mutation affects two or more phenotypic traits). Acquired or intrinsic resistance to one drug may also enable cross-resistance to other drugs (within or across antibiotic classes), or could result in ‘collateral sensitivity’, whereby a mutation or pathway that confers resistance to one antibiotic increases susceptibility to another63–65. In Mtb, glutamine synthetase has been identified as a collateral metabolic vulnerability elicited by bedaquiline66. In mycobacteria, the transcriptional regulator WhiB7 coordinates an adaptive stress response to ribosome stalling, leading to multidrug intrinsic resistance and tolerance, which has important implications for the design of combinations that avoid WhiB7 induction67–69. Combination chemotherapy strategies need to incorporate these concepts early in preclinical development — not only to create more durable antibiotic pairings70, but also to reduce treatment duration by circumventing intrinsic pathways of drug tolerance. Functional genomics and chemical-genetic interactions identified by CRISPR interference71 are being exploited to identify pathways critical for fitness of resistant mutants72, suggesting rational approaches to induce sensitivity. These approaches can inform the development of mechanism-based combinations that shorten treatment and delay the emergence of resistance.
Emerging research and development strategies
As noted earlier, 80% of patients with TB likely fall into an ETT category, suggesting broad benefit from patient stratification if it can be easily incorporated into practice, considering the challenges associated with identification of those eligible for safe treatment shortening1. Patient stratification could be accomplished initially with a conceptually simple distinction between ETT and HTT disease phenotypes.
Typical PET-CT scans and sputum burden profiles of representative ETT and HTT patients are shown in Figure 2. Since increased precision comes at the cost of simplicity and feasibility, stratification and treatment algorithms may need to be adapted to clinical settings according to resource availability, or be launched at a slower pace in low resource settings. For instance, a composite signature classifies HTT disease based on cavitation by HIV status, sex, BMI and smear grade in the absence of chest X-ray2. These host factors are consistent with the guidelines from the US Centers for Disease Control and Prevention and American Thoracic Society,35 and can be collected in most clinical settings. A molecular biomarker signature captured by a regulatory-approved device is a longer-term prospect and would likely be launched gradually around the world. Table 2 describes a range of plausible stratified treatment strategies, with their associated benefits, limitations, knowledge gaps, and implementation challenges. Our hypotheses for treatment shortening echo the long-standing principles of clinical pharmacology: get the right drugs to the right patients at the right doses, and for the right duration (Figure 3). The following strategies can be envisioned to tailor drug, dose and duration to patient needs and disease phenotypes73.
Table 2.
Range of plausible treatment strategies for pulmonary TB[a]
| Treatment strategy | Strengths | Challenges | Knowledge gaps and unmet needs |
|---|---|---|---|
| One size fits all: New short regimen equally effective in rifampicin resistant and susceptible populations, including asymptomatic sputum positive patients. |
|
|
Designing regimens to protect from resistance emergence and optimizing PK/PD across all sites of infection. |
| Stratification by rifampicin resistance status (current SOC) |
|
|
Improved (faster, cheaper, easier) DST to guide therapy. |
| 1 regimen, varying durations based on patient factors |
|
Improved (faster, cheaper, easier) biomarkers to guide therapy. | |
| 1 regimen, varying doses for drug susceptible and drug resistant and patient factors |
|
|
Clinical trials to assess the efficacy of modified-dose regimens across disease severity spectrum. Routine implementation of therapeutic drug monitoring and safety monitoring. Accessible, geographically diverse DST/MICs. |
| Distinct regimens for ETT and HTT disease |
|
|
Development and validation of a globally accepted – deliverable, scalable, affordable – biomarker which allows patient stratification. Uncovering novel drug mechanisms. Systematic exploration of pharmacodynamic interactions for regimen optimization. |
| Regimen cycling (different regimens for intensive and continuation phases) |
|
|
Extensive preclinical and clinical research required to demonstrate value and optimize practice. |
| Personalized therapy (Figure 3) |
|
|
Development and validation of a globally accepted biomarker which allows patient stratification. Digital patient monitoring to assess treatment response and pharmacogenomics to enable stratified care. |
While important and deserving attention, TB meningitis and other disease manifestations require separate regimen optimization.
The new and successful 4-month regimen for drug susceptible TB (rifapentine-moxifloxacin-isoniazid-pyrazinamide36) is seldom implemented in low resource settings for that reason and due to the lack of a fixed dose combination. DST, drug susceptibility testing; DOT, directly observed therapy; NHP, non-human primate.
Figure 3. Patient stratification approach.

The figure illustrates the interplay between diagnosis, host-bacterium features, and treatment optimization for TB. As a long-term vision, algorithms that integrate disease severity and bacterial burden, comprehensive drug susceptibility profiles, pharmacogenomics, pharmacokinetics and drug-drug interactions will be developed and validated to deliver optimized, patient-tailored drug doses and regimens. Real-time monitoring of treatment response will enable adjustments of treatment duration to avoid overtreating as much as undertreating.
Develop regimens to target biological and spatial characteristics of ETT and HTT disease
Spatial and temporal within-host lesion diversity is the hallmark of Mtb infection and TB19,74,75. In these lesions, bacteria exist in various physiological and drug tolerance states, governed by intracellular and extracellular microenvironments. That new lesions appear, and cavities can form even under multidrug therapy74, reminds us that current drug regimens do not effectively suppress bacterial multiplication at all sites of infection. Retrospective analyses of recent treatment-shortening phase 3 trials indicates that regimens penetrating cavities at concentrations needed to kill drug-tolerant M. tuberculosis62 were successful across ETT and HTT populations. For instance, the first successful 4-month regimen tested in Study 3136 contains two drugs (high dose rifapentine and moxifloxacin) that achieve excellent cavity caseum coverage — meaning that they reach above the concentrations required to kill non-replicating caseum persisters for the entire dosing interval. We hypothesize that this site must be covered at adequate concentrations for resistance prevention and relapse-free cure of all patients. Retrospective analyses of large trials using simulations of drug regimen coverage in lung lesions are ongoing to confirm this hypothesis76.
To achieve relapse-free cure (no relapse within 18 months following therapy completion) while shortening treatment, a key priority is the identification of vulnerable bacterial targets in subpopulations that constitute treatment ‘bottlenecks’ in ETT and HTT disease. Preliminary findings19 point to drug-tolerant, non-replicating persisters in cavity caseum as the recalcitrant Mtb population that resists therapy and constitutes a source of relapse in HTT disease. To improve sterilization of these bacteria, we can infer vulnerable targets and pathways from successful treatment shortening trials, bactericidal assays in caseum ex vivo77 and efficacy studies in non-human primates (with longitudinal PET-CT scans as readout).
While there is clarity on the site of disease that drives outcome and is the site of relapse in HTT (‘dirty’ cavities filled with caseum19), there remains uncertainty regarding the dominant site that drives relapse in ETT. Factors associated with pathogen biology, target vulnerability and location of dominant bacterial populations in ETT versus HTT disease may require that optimized regimens for each of these features a subset of drugs with different target product profiles. Two likely scenarios must be considered. In the first scenario, treatment-relevant pathological features of ETT are all present in HTT and therefore ETT could be treated with the same regimen as HTT, either for shorter durations or with a subset of HTT drugs. In the second scenario, there exist ETT-specific pathological feature(s), and this may constitute a distinct barrier to cure — in which case drugs with ETT-specific mechanism(s) of action and site distribution profiles are required (Figure 2c). Longitudinal PET-CT imaging of patients on treatment19 suggests that neutrophil-associated airway disease (described as polymorphonuclear alveolitis by Canetti45) harbors one of the dominant bacterial populations in ETT patients. Histopathology analyses have revealed intracellular and extracellular bacilli in and around neutrophils. Together with intracellular bacilli present in pulmonary nodules – such as those within lipid laden foamy macrophages, where metabolically adapted Mtb resists treatment78–81 – neutrophil-driven pathology may constitute a cure-limiting ETT burden, while also being present in HTT disease. Further mining of data from ongoing and recently completed trials should be undertaken, followed by application of computational pipelines for testing hypotheses of regimen ranking versus outcome.
These questions and approaches to address them are an intrinsic part of a coherent strategy to enable treatment shortening in all patients by simultaneously defining the microbiology and immunopathology of ETT and HTT (and maybe even extremely HTT) and developing regimens that target the distinct biology and vulnerabilities of each.
Improve dose selection
Not all TB drugs can be safely administered at higher-than-standard doses, but key drugs with sterilizing potential such as rifampicin, rifapentine and moxifloxacin offer room, albeit modest, for dose increase. High dose rifampicin (twice the standard dose) is generally well tolerated82,83 and has been proposed for cavitary TB with high sputum burden84, after multiple studies showed that 600 mg lies in the steep part of the exposure-response curve85. High dose rifapentine is part of the first successful 4-month regimen for drug-sensitive TB36. High-dose moxifloxacin (double dose once daily or standard dose twice daily) is expected to provide superior efficacy based on site-of-disease PK-PD modeling (with a predicted safety advantage for twice daily)86 and is being tested in clinical trial NCT02409290. It is generally recognized that higher moxifloxacin doses could be safely administered if adequate monitoring is undertaken87. Given the rapid emergence of bedaquiline resistance in certain geographies, doses adjusted to local minimum inhibitory concentrations (MIC) distributions will be evaluated during the development and implementation of the next-generation diarylquinolines, TBAJ876 and TBAJ587. In clinical trials of new drug candidates and clinical studies with approved regimens, quantifying the relationship between individual drug exposure, drug susceptibility and clinical outcome should deliver an algorithm that quantifies the probability of target attainment for multiple drugs, including functions of variable MICs and composite exposure of drug regimens. Dose increase, if adequately supported by safety studies, likely carries a modest barrier to implementation.
Develop evidence-based patient stratification approaches
To reliably capture ETT and HTT disease phenotypes, a biomarker signature that integrates host and bacterial factors, and can be converted into a predictive biomarker for clinical practice, is needed. While useful, mycobacterial sputum culture comes with inherent limitations48 but with multiple prospects are emerging for better estimates of bacterial burden than achieved through sputum smear and culture status88. Indeed, the field is rapidly evolving towards a composite marker signature that overcomes current limitations89 and serves as a portable diagnostic tool. Promising avenues include immunodetection of lipoarabinomannan (a cell wall component of Mtb) in sputum and urine90, GenXpert (a nucleic acid amplification test that detects Mtb DNA in sputum and rifampin resistance in about 2 hours)91 and peripheral blood transcriptomics signatures of disease extent and TB progression, including incipient TB92. New quantitative lateral flow assays have been developed and are being evaluated to quantify host markers of disease severity and/or response to treatment93. In clinical trial settings, PET/CT can provide superior and high-resolution pathology evaluation19 to inform AI-enabled interpretation of chest Xray in programmatic settings in the future.
Small increases in morbidity/mortality associated with excessive treatment shortening or stratified treatment strategies can undermine their benefits. While the ‘stop treatment and watch’ paradigm has been successfully implemented in clinical trial settings94, observing patients after treatment completion and initiating re-treatment if needed comes with limited operational feasibility in many geographies. Therefore, the stratification algorithm should be biased towards overtreating ‘borderline’ patients. To further mitigate the risk of undertreating due to misclassification of patients with ETT disease, leading to relapse and transmission (particularly in high-burden settings)95, biomarkers capable of evaluating response and risk of relapse in real-time are critical96. Such markers could also be applied to stop treatment earlier than planned in borderline HTT patients.
We also note that a treatment algorithm that ignores asymptomatic but culture-positive patients with radiological abnormalities (e.g., subclinical TB, the ‘walking well’ or State IV11) would fall short of our objective to accelerate the decline of the epidemic. Recent active case-finding studies have revealed higher than expected fractions of participants (up to 58% in a high burden setting97) with an abnormal chest X-ray and bacteriologically confirmed TB but no other symptoms; therefore, this substantive patient population likely represents an important source of transmission.
Thus, three categories of biomarkers would enable an improved treatment algorithm: First, a composite signature to distinguish ETT and HTT at baseline and guide regimen composition; second, biomarkers of treatment response to determine when to safely stop treatment and increase confidence in ETT/HTT stratification; and third, a signature for active finding of subclinical TB cases. Clinical trials designed to assess optimized regimens in ETT and HTT patient cohorts should aim for drastic treatment shortening under the ‘stop-treatment-and-watch’ principle94. Only a dramatic treatment shortening will overcome implementation hurdles in programmatic settings. Table 3 summarizes the research needs, working hypotheses and proposed approaches to enable optimal treatment duration in patient strata.
Table 3.
Working hypotheses, biological and clinical uncertainties, and proposed approaches to better target ETT and HTT patients.
| Hypothesis | Uncertainties | Approaches to address uncertainties |
|---|---|---|
| Treatment shortening will be achieved by ensuring that ≥ 2 regimen components deliver relevant exposures to caseous lesions. |
|
|
| Treatment shortening will be achieved by ensuring regimens target MOAs associated with the predominant pathologies and Mtb phenotypes in ETT and HTT patients. |
|
|
| Treatment shortening will be achieved by ensuring all regimen components are dosed optimally. |
|
|
| Treatment shortening can be achieved by distinguishing 2 categories of patients for treatment allocation: ETT and HTT |
|
|
| Treatment shortening will be achieved through the availability of a biomarker(s) of relapse-free cure. |
|
|
Conclusion
The goal of TB research organizations and collaborations – including the TB Drug Accelerator (TBDA) and the WHO – is to discover and develop markedly shorter ‘pan-TB’ treatment regimens3. In the ideal world, drug regimen, doses and treatment duration would be optimized for each patient based on an algorithm that integrates host and pathogen factors. Recognizing the realities of TB control, we propose a series of short and long-term strategies, from least to most complex, and highlight implementation tradeoffs and challenges. We provide pointers to the research community to fill critical gaps in our fundamental knowledge of bacterial vulnerabilities in ETT and HTT disease. We also share our vision of preclinical, computational and translational approaches to build target product profiles and drug regimens with the best potential to cure each disease phenotype. As bioassay technologies improve and AI grows, the barriers to implementing an accurate stratification algorithm that minimizes overtreating while avoiding under-treatment will lessen.
Table 1.
Randomized control trials evaluating shorter-than-standard treatment duration in participants with limited disease.
| Indication | Duration | Trial acronym / Sponsor | Drug(s) | Primary endpoint / outcome[a] | Study identifier or reference |
|---|---|---|---|---|---|
| Completed | |||||
| Non-severe pediatric DS-TB (HIV+ and HIV–) | 4 mo vs 6 mo | SHINE (Phase 3) | 2HRZ (+/−E)/4HR (6 mo)2HRZ (+/−E)/2HR (4 mo) | no difference in the effect of randomized treatment duration on treatment outcomes by HIV status: 6 mo: unfavorable outcome = 2% in non-HIV (n = 540), and 11% in HIV (n = 62)4 mo: unfavorable outcome = 2% in non-HIV (n = 537), and 7% in HIV (n = 65) | 26 |
| DS-TB Non-cavitaryHIV–Smear– / culture + Culture – at 2 mo | 4 mo vs 6 mo | Tuberculosis Research Unit, Case Western Reserve University | 2HRZE/4HR (6 mo)2HRZE/2HR (4 mo) | Bacteriological or clinical relapse over 30 mo:6 mo: 1.6% (n=185)4 mo: 7% (n=185)(Study stopped early) | 98 |
| DS-TB Smear– / culture + | 4 mo vs 6 mo | Hong Kong Chest Service | 6SHRZ3 (6mth)4SHRZ3 (4mth) | Bacteriological, clinical or radiological relapse over 60 mo:6 mo: 5% (n=296)4 mo: 2% (n=293) | 99 |
| DS-TB Smear– / culture + | 4 mo vs 6 mo | Singapore Alexandra Hospital | 2HRZ/4H3R3 (6 mo)2HRZ/2HR (4 mo) | Bacteriological relapse over 60 mo: 6 mo: 2% (n=53)4 mo: 0% (n=57) | 100 |
| Ongoing | |||||
| DS-TB | 4 mo | Predict-TB (Phase 2) | HRZE | Change in radiographic characteristics from baseline to one month, and markers of residual bacterial load at the end of treatment, to identify patients who are cured | NCT02821832 |
| XDR-TB | 6 mo vs 9 mo | endTB-Q (Phase 3) | BeDeCLi vs WHO-recommended treatment | Randomized, controlled, non-inferiority open-label trial evaluating efficacy according to extent-of-disease phenotype | NCT03896685 |
| DS-TB | 4 mo | PRESCIENT (Phase 2c) | BCZD vs SOC (HRZE) | Open-label, randomized study (stratification by presence of lung cavitation and HIV status). The primary analysis will focus on time to liquid culture conversion at week 8 in the BCZD arm vs HRZE. | NCT05556746 |
outcome described for completed trials only.
DS-TB, drug sensitive TB; DR-TB, drug-resistant TB; XDR-TB, extensively resistant TB; SOC: standard of care; 2HRZ; isoniazid, rifampicin, pyrazinamide for 2 months; 4HR or 2 HR: isoniazid and rifampicin for 4 or 2 months; 6SHRZ3 or 4SHRZ3: streptomycin, isoniazid, rifampicin daily and pyrazinamide thrice weekly for 6 months or 4 months; 4H3R3: isoniazid and rifampicin thrice weekly for 4 months; BeDeCLi: bedaquiline, delamanid, clofazimine, linezolid; BCZD: bedaquiline, clofazimine, pyrazinamide, delamanid.
Acknowledgements
We are grateful to Drs Graeme Meintjes, Mark Hatherill and Digby Warner from the University of Cape Town for thoroughly reviewing the manuscript and providing constructive suggestions. We express our gratitude to all members of the TBDA (https://www.tbdrugaccelerator.org/) for their research input, thoughts and fruitful discussions that helped to define the scope and direction of this Perspective, which reflects the views of the TBDA membership. The listed authors are members who directly contributed to the manuscript.
Footnotes
Ethics Statement
The authors declare no conflicting interests
REFERENCES
- 1.Esmail H, Macpherson L, Coussens AK & Houben R Mind the gap - Managing tuberculosis across the disease spectrum. EBioMedicine 78, 103928, doi: 10.1016/j.ebiom.2022.103928 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imperial MZ et al. A patient-level pooled analysis of treatment-shortening regimens for drug-susceptible pulmonary tuberculosis. Nat Med 24, 1708–1715, doi: 10.1038/s41591-018-0224-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. Target regimen profile for tuberculosis treatment, https://www.who.int/publications/i/item/9789240081512. (Geneva, 2023). [Google Scholar]
- 4.Aldridge BB et al. The Tuberculosis Drug Accelerator at year 10: what have we learned? Nat Med, doi: 10.1038/s41591-021-01442-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger CA et al. Variation in tuberculosis treatment outcomes and treatment supervision practices in Uganda. J Clin Tuberc Other Mycobact Dis 21, 100184, doi: 10.1016/j.jctube.2020.100184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO. Global Tuberculosis Report 2023, https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023. (Geneva, 2023). [Google Scholar]
- 7.Kendall EA, Shrestha S & Dowdy DW The Epidemiological Importance of Subclinical Tuberculosis. A Critical Reappraisal. Am J Respir Crit Care Med 203, 168–174, doi: 10.1164/rccm.202006-2394PP (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barry CE 3rd et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 7, 845–855, doi: 10.1038/nrmicro2236 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drain PK et al. Incipient and Subclinical Tuberculosis: a Clinical Review of Early Stages and Progression of Infection. Clin Microbiol Rev 31, doi: 10.1128/CMR.00021-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emery JC et al. Self-clearance of Mycobacterium tuberculosis infection: implications for lifetime risk and population at-risk of tuberculosis disease. Proc Biol Sci 288, 20201635, doi: 10.1098/rspb.2020.1635 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaidi SMA et al. Beyond latent and active tuberculosis: a scoping review of conceptual frameworks. EClinicalMedicine 66, 102332, doi: 10.1016/j.eclinm.2023.102332 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards AS et al. Quantifying progression and regression across the spectrum of pulmonary tuberculosis: a data synthesis study. Lancet Glob Health 11, e684–e692, doi: 10.1016/S2214-109X(23)00082-7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frascella B et al. Subclinical Tuberculosis Disease-A Review and Analysis of Prevalence Surveys to Inform Definitions, Burden, Associations, and Screening Methodology. Clin Infect Dis 73, e830–e841, doi: 10.1093/cid/ciaa1402 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long R et al. The association between phylogenetic lineage and the subclinical phenotype of pulmonary tuberculosis: A retrospective 2-cohort study. J Infect 88, 123–131, doi: 10.1016/j.jinf.2023.12.006 (2024). [DOI] [PubMed] [Google Scholar]
- 15.Reed MB et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 431, 84–87. (2004). [DOI] [PubMed] [Google Scholar]
- 16.Stanley S et al. Identification of bacterial determinants of tuberculosis infection and treatment outcomes: a phenogenomic analysis of clinical strains. Lancet Microbe, doi: 10.1016/S2666-5247(24)00022-3 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gagneux S Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci 367, 850–859, doi: 10.1098/rstb.2011.0316 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stucki D et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet 48, 1535–1543, doi: 10.1038/ng.3704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malherbe ST et al. PET/CT guided tuberculosis treatment shortening: a randomized trial. medRxiv, 2024.2010.2003.24314723, doi: 10.1101/2024.10.03.24314723 (2024). [DOI] [Google Scholar]
- 20.Zhao L, Gao F, Zheng C & Sun X The Impact of Optimal Glycemic Control on Tuberculosis Treatment Outcomes in Patients With Diabetes Mellitus: Systematic Review and Meta-Analysis. JMIR Public Health Surveill 10, e53948, doi: 10.2196/53948 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seddon JA, Chiang SS, Esmail H & Coussens AK The Wonder Years: What Can Primary School Children Teach Us About Immunity to Mycobacterium tuberculosis? Front Immunol 9, 2946, doi: 10.3389/fimmu.2018.02946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.WHO. WHO consolidated guidelines on tuberculosis, Module 4: Treatment, Drug-susceptible tuberculosis treatment, https://www.who.int/publications/i/item/9789240048126. (Geneva, 2022). [PubMed] [Google Scholar]
- 23.Jain SK et al. Tuberculous meningitis: a roadmap for advancing basic and translational research. Nat Immunol 19, 521–525, doi: 10.1038/s41590-018-0119-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wasserman S & Harrison TS Tuberculous Meningitis - New Approaches Needed. N Engl J Med 389, 1425–1426, doi: 10.1056/NEJMe2310262 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Wasserman S et al. Advancing the chemotherapy of tuberculous meningitis: a consensus view. Lancet Infect Dis, doi: 10.1016/S1473-3099(24)00512-7 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chabala C et al. Clinical outcomes in children living with HIV treated for non-severe tuberculosis in the SHINE Trial. Clin Infect Dis, doi: 10.1093/cid/ciae193 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MOREY SS ATS Adopts Diagnostic Standards for Tuberculosis. Am Fam Physician. 63, 979–980 (2001). [PubMed] [Google Scholar]
- 28.Coussens AK et al. Classification of early tuberculosis states to guide research for improved care and prevention: an international Delphi consensus exercise. Lancet Respir Med 12, 484–498, doi: 10.1016/S2213-2600(24)00028-6 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sossen B et al. The natural history of untreated pulmonary tuberculosis in adults: a systematic review and meta-analysis. Lancet Respir Med 11, 367–379, doi: 10.1016/S2213-2600(23)00097-8 (2023). [DOI] [PubMed] [Google Scholar]
- 30.Lau A et al. The Radiographic and Mycobacteriologic Correlates of Subclinical Pulmonary TB in Canada: A Retrospective Cohort Study. Chest 162, 309–320, doi: 10.1016/j.chest.2022.01.047 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Esmail H et al. High resolution imaging and five-year tuberculosis contact outcomes. medRxiv, doi: 10.1101/2023.07.03.23292111 (2023). [DOI] [Google Scholar]
- 32.Nathavitharana RR, Garcia-Basteiro AL, Ruhwald M, Cobelens F & Theron G Reimagining the status quo: How close are we to rapid sputum-free tuberculosis diagnostics for all? EBioMedicine 78, 103939, doi: 10.1016/j.ebiom.2022.103939 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryckman TS, Dowdy DW & Kendall EA Infectious and clinical tuberculosis trajectories: Bayesian modeling with case finding implications. Proc Natl Acad Sci U S A 119, e2211045119, doi: 10.1073/pnas.2211045119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patterson B et al. Aerosolization of viable Mycobacterium tuberculosis bacilli by tuberculosis clinic attendees independent of sputum-Xpert Ultra status. Proc Natl Acad Sci U S A 121, e2314813121, doi: 10.1073/pnas.2314813121 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nahid P et al. Executive Summary: Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin Infect Dis 63, 853–867, doi: 10.1093/cid/ciw566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dorman SE et al. Four-Month Rifapentine Regimens with or without Moxifloxacin for Tuberculosis. N Engl J Med 384, 1705–1718, doi: 10.1056/NEJMoa2033400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox W, Ellard GA & Mitchison DA Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946–1986, with relevant subsequent publications. Int J Tuberc Lung Dis 3, S231–279 (1999). [PubMed] [Google Scholar]
- 38.A four-month gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med 372, 1677, doi: 10.1056/NEJMx150015 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Gillespie SH et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med 371, 1577–1587, doi: 10.1056/NEJMoa1407426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jindani A et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med 371, 1599–1608, doi: 10.1056/NEJMoa1314210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gopalan N et al. Predictors of unfavorable responses to therapy in rifampicin-sensitive pulmonary tuberculosis using an integrated approach of radiological presentation and sputum mycobacterial burden. PLoS One 16, e0257647, doi: 10.1371/journal.pone.0257647 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romanowski K et al. Predicting tuberculosis relapse in patients treated with the standard 6-month regimen: an individual patient data meta-analysis. Thorax 74, 291–297, doi: 10.1136/thoraxjnl-2017-211120 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Campbell JR et al. Association of indicators of extensive disease and rifampin-resistant tuberculosis treatment outcomes: an individual participant data meta-analysis. Thorax 79, 169–178, doi: 10.1136/thorax-2023-220249 (2024). [DOI] [PubMed] [Google Scholar]
- 44.Chang KC, Leung CC, Yew WW, Ho SC & Tam CM A nested case-control study on treatment-related risk factors for early relapse of tuberculosis. Am J Respir Crit Care Med 170, 1124–1130 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Canetti G The Tubercle Bacillus. (Springer Publishing COmpany, 1955). [Google Scholar]
- 46.Koch R in Berliner klinische Wochenschrift, (ed Ewald CA) Ch. 15, 221–230 (1882). [Google Scholar]
- 47.Loring WW, Melvin I, Vandiviere HM & Willis HS The death and resurrection of the tubercle bacillus. Trans Am Clin Climatol Assoc 67, 132–138 (1955). [PMC free article] [PubMed] [Google Scholar]
- 48.Mishra S & Saito K Clinically encountered growth phenotypes of tuberculosis-causing bacilli and their in vitro study: A review. Front Cell Infect Microbiol 12, 1029111, doi: 10.3389/fcimb.2022.1029111 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wells G et al. μCT analysis of the human tuberculous lung reveals remarkable heterogeneity in 3D granuloma morphology. American Journal of Respiratory and Critical Care Medicine (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen RY et al. Radiological and functional evidence of the bronchial spread of tuberculosis: an observational analysis. The Lancet Microbe, doi: 10.1016/S2666-5247(21)00058-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prosser G et al. The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect 19, 177–192, doi: 10.1016/j.micinf.2016.10.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conradie F et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N Engl J Med 382, 893–902, doi: 10.1056/NEJMoa1901814 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goig GA et al. Transmission as a Key Driver of Resistance to the New Tuberculosis Drugs. N Engl J Med 392, 97–99, doi: 10.1056/NEJMc2404644 (2025). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goig GA et al. Transmission is a key driver of extensively drug-resistant tuberculosis. medRxiv, 2024.2006.2028.24309543, doi: 10.1101/2024.06.28.24309543 (2024). [DOI] [Google Scholar]
- 55.Shaw ES et al. Bedaquiline: what might the future hold? Lancet Microbe, 100909, doi: 10.1016/S2666-5247(24)00149-6 (2024). [DOI] [PubMed] [Google Scholar]
- 56.Nimmo C et al. Evolution of Mycobacterium tuberculosis drug resistance in the genomic era. Front Cell Infect Microbiol 12, 954074, doi: 10.3389/fcimb.2022.954074 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perumal R et al. Baseline and treatment-emergent bedaquiline resistance in drug-resistant tuberculosis: a systematic review and meta-analysis. Eur Respir J 62, doi: 10.1183/13993003.00639-2023 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao B et al. Prevalence and genetic basis of Mycobacterium tuberculosis resistance to pretomanid in China. Ann Clin Microbiol Antimicrob 23, 40, doi: 10.1186/s12941-024-00697-0 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miotto P, Cirillo DM, Schon T & Koser CU The exceptions that prove the rule-a historical view of bedaquiline susceptibility. Genome Med 16, 39, doi: 10.1186/s13073-024-01311-w (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rockwood N, Abdullahi LH, Wilkinson RJ & Meintjes G Risk Factors for Acquired Rifamycin and Isoniazid Resistance: A Systematic Review and Meta-Analysis. PLoS One 10, e0139017, doi: 10.1371/journal.pone.0139017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barilar I, et al. , CRyPTIC Consortium. Quantitative measurement of antibiotic resistance in Mycobacterium tuberculosis reveals genetic determinants of resistance and susceptibility in a target gene approach. Nat Commun 15, 488, doi: 10.1038/s41467-023-44325-5 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarathy JP et al. Extreme Drug Tolerance of Mycobacterium tuberculosis in Caseum. Antimicrob Agents Chemother 62, e02266–02217, doi: 10.1128/AAC.02266-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waller NJE, Cheung CY, Cook GM & McNeil MB The evolution of antibiotic resistance is associated with collateral drug phenotypes in Mycobacterium tuberculosis. Nat Commun 14, 1517, doi: 10.1038/s41467-023-37184-7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roemhild R, Bollenbach T & Andersson DI The physiology and genetics of bacterial responses to antibiotic combinations. Nat Rev Microbiol, doi: 10.1038/s41579-022-00700-5 (2022). [DOI] [PubMed] [Google Scholar]
- 65.Sullivan GJ, Delgado NN, Maharjan R & Cain AK How antibiotics work together: molecular mechanisms behind combination therapy. Curr Opin Microbiol 57, 31–40, doi: 10.1016/j.mib.2020.05.012 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Wang Z et al. Mode-of-action profiling reveals glutamine synthetase as a collateral metabolic vulnerability of M. tuberculosis to bedaquiline. Proc Natl Acad Sci U S A 116, 19646–19651, doi: 10.1073/pnas.1907946116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poulton NC et al. Beyond antibiotic resistance: the whiB7 transcription factor coordinates an adaptive response to alanine starvation in mycobacteria. bioRxiv, doi: 10.1101/2023.06.02.543512 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morris RP et al. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 102, 12200–12205, doi: 10.1073/pnas.0505446102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schrader SM et al. Multiform antimicrobial resistance from a metabolic mutation. Sci Adv 7, eabh2037, doi: 10.1126/sciadv.abh2037 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dheda K et al. Multidrug-resistant tuberculosis. Nat Rev Dis Primers 10, 22, doi: 10.1038/s41572-024-00504-2 (2024). [DOI] [PubMed] [Google Scholar]
- 71.Li S et al. CRISPRi chemical genetics and comparative genomics identify genes mediating drug potency in Mycobacterium tuberculosis. Nat Microbiol 7, 766–779, doi: 10.1038/s41564-022-01130-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eckartt KA et al. Compensatory evolution in NusG improves fitness of drug-resistant M. tuberculosis. Nature 628, 186–194, doi: 10.1038/s41586-024-07206-5 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dooley KE, Hanna D, Mave V, Eisenach K & Savic RM Advancing the development of new tuberculosis treatment regimens: The essential role of translational and clinical pharmacology and microbiology. PLoS Med 16, e1002842, doi: 10.1371/journal.pmed.1002842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xie YL et al. Fourteen-day PET/CT imaging to monitor drug combination activity in treated individuals with tuberculosis. Sci Transl Med 13, doi: 10.1126/scitranslmed.abd7618 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin PL et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 20, 75–79, doi: 10.1038/nm.3412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ernest JP et al. Development of New Tuberculosis Drugs: Translation to Regimen Composition for Drug-Sensitive and Multidrug-Resistant Tuberculosis. Annu Rev Pharmacol Toxicol 61, 495–516, doi: 10.1146/annurev-pharmtox-030920-011143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sarathy JP et al. A Novel Tool to Identify Bactericidal Compounds against Vulnerable Targets in Drug-Tolerant M. tuberculosis found in Caseum. mBio 14, e0059823, doi: 10.1128/mbio.00598-23 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lovewell RR, Sassetti CM & VanderVen BC Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Curr Opin Microbiol 29, 30–36, doi: 10.1016/j.mib.2015.10.002 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Gold B & Nathan C Targeting Phenotypically Tolerant Mycobacterium tuberculosis. Microbiol Spectr 5, doi: 10.1128/microbiolspec.TBTB2-0031-2016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lanni F et al. Adaptation to the intracellular environment of primary human macrophages influences drug susceptibility of Mycobacterium tuberculosis. Tuberculosis (Edinb) 139, 102318, doi: 10.1016/j.tube.2023.102318 (2023). [DOI] [PubMed] [Google Scholar]
- 81.Liu Y et al. Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J Exp Med 213, 809–825, doi: 10.1084/jem.20151248 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perumal Kannabiran B et al. Safety and Efficacy of 25 mg/kg and 35 mg/kg vs 10 mg/kg Rifampicin in Pulmonary TB: A Phase IIb Randomized Controlled Trial. Open Forum Infect Dis 11, ofae034, doi: 10.1093/ofid/ofae034 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Onorato L et al. Standard versus high dose of rifampicin in the treatment of pulmonary tuberculosis: a systematic review and meta-analysis. Clin Microbiol Infect 27, 830–837, doi: 10.1016/j.cmi.2021.03.031 (2021). [DOI] [PubMed] [Google Scholar]
- 84.Espinosa-Pereiro J et al. Safety of Rifampicin at High Dose for Difficult-to-Treat Tuberculosis: Protocol for RIAlta Phase 2b/c Trial. Pharmaceutics 15, doi: 10.3390/pharmaceutics15010009 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diacon AH et al. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob Agents Chemother 51, 2994–2996 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yun HY et al. Model-Based Efficacy and Toxicity Comparisons of Moxifloxacin for Multidrug-Resistant Tuberculosis. Open Forum Infect Dis 9, ofab660, doi: 10.1093/ofid/ofab660 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kusmiati T et al. Moxifloxacin concentration correlate with QTc interval in rifampicin-resistant tuberculosis patients on shorter treatment regimens. J Clin Tuberc Other Mycobact Dis 28, 100320, doi: 10.1016/j.jctube.2022.100320 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chang A et al. Circulating Cell-Free RNA in Blood as a Host Response Biomarker for the Detection of Tuberculosis. medRxiv, doi: 10.1101/2023.01.11.23284433 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang F, Zhang F, Dong Y, Li L & Pang Y New Insights into Biomarkers for Evaluating Therapy Efficacy in Pulmonary Tuberculosis: A Narrative Review. Infect Dis Ther 12, 2665–2689, doi: 10.1007/s40121-023-00887-x (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Corrigan DT, Ishida E, Chatterjee D, Lowary TL & Achkar JM Monoclonal antibodies to lipoarabinomannan/arabinomannan - characteristics and implications for tuberculosis research and diagnostics. Trends Microbiol 31, 22–35, doi: 10.1016/j.tim.2022.07.001 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gong X, He Y, Zhou K, Hua Y & Li Y Efficacy of Xpert in tuberculosis diagnosis based on various specimens: a systematic review and meta-analysis. Front Cell Infect Microbiol 13, 1149741, doi: 10.3389/fcimb.2023.1149741 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tabone O et al. Blood transcriptomics reveal the evolution and resolution of the immune response in tuberculosis. J Exp Med 218, doi: 10.1084/jem.20210915 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pierneef L et al. Host biomarker-based quantitative rapid tests for detection and treatment monitoring of tuberculosis and COVID-19. iScience 26, 105873, doi: 10.1016/j.isci.2022.105873 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Paton NI et al. Treatment Strategy for Rifampin-Susceptible Tuberculosis. N Engl J Med 388, 873–887, doi: 10.1056/NEJMoa2212537 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hermans S, Horsburgh CR Jr. & Wood R A Century of Tuberculosis Epidemiology in the Northern and Southern Hemisphere: The Differential Impact of Control Interventions. PLoS One 10, e0135179, doi: 10.1371/journal.pone.0135179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gillespie SH et al. Developing biomarker assays to accelerate tuberculosis drug development: defining target product profiles. Lancet Microbe, doi: 10.1016/S2666-5247(24)00085-5 (2024). [DOI] [PubMed] [Google Scholar]
- 97.Moyo S et al. Prevalence of bacteriologically confirmed pulmonary tuberculosis in South Africa, 2017–19: a multistage, cluster-based, cross-sectional survey. Lancet Infect Dis 22, 1172–1180, doi: 10.1016/S1473-3099(22)00149-9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnson JL et al. Shortening treatment in adults with noncavitary tuberculosis and 2-month culture conversion. Am J Respir Crit Care Med 180, 558–563, doi: 10.1164/rccm.200904-0536OC (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.A controlled trial of 3-month, 4-month, and 6-month regimens of chemotherapy for sputum-smear-negative pulmonary tuberculosis. Results at 5 years. Hong Kong Chest Service/Tuberculosis Research Centre, Madras/British Medical Research Council. Am Rev Respir Dis 139, 871–876, doi: 10.1164/ajrccm/139.4.871 (1989). [DOI] [PubMed] [Google Scholar]
- 100.Teo SK, Tan KK & Khoo TK Four-month chemotherapy in the treatment of smear-negative pulmonary tuberculosis: results at 30 to 60 months. Ann Acad Med Singap 31, 175–181 (2002). [PubMed] [Google Scholar]
- 101.James LP et al. Impact and cost-effectiveness of the 6-month BPaLM regimen for rifampicin-resistant tuberculosis in Moldova: A mathematical modeling analysis. PLoS Med 21, e1004401, doi: 10.1371/journal.pmed.1004401 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Knight GM et al. The Impact and Cost-Effectiveness of a Four-Month Regimen for First-Line Treatment of Active Tuberculosis in South Africa. PLoS One 10, e0145796, doi: 10.1371/journal.pone.0145796 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goh JJN et al. Prospectively predicting BPaMZ phase IIb/III trial outcomes using a translational mouse-to-human platform. Antimicrob Agents Chemother 68, e0061524, doi: 10.1128/aac.00615-24 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walter ND et al. Mycobacterium tuberculosis precursor rRNA as a measure of treatment-shortening activity of drugs and regimens. Nat Commun 12, 2899, doi: 10.1038/s41467-021-22833-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zainabadi K et al. Transcriptional Biomarkers of Differentially Detectable Mycobacterium tuberculosis in Patient Sputum. mBio 13, e0270122, doi: 10.1128/mbio.02701-22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zainabadi K et al. Characterization of Differentially Detectable Mycobacterium tuberculosis in the Sputum of Subjects with Drug-Sensitive or Drug-Resistant Tuberculosis before and after Two Months of Therapy. Antimicrob Agents Chemother 65, e0060821, doi: 10.1128/AAC.00608-21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peters JS et al. Differentially culturable tubercle bacteria as a measure of tuberculosis treatment response. Front Cell Infect Microbiol 12, 1064148, doi: 10.3389/fcimb.2022.1064148 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fox W Whither short-course chemotherapy? Br J Dis Chest 75, 331–357, doi: 10.1016/0007-0971(81)90022-x (1981). [DOI] [PubMed] [Google Scholar]