

Abstract
We have shown that the small GTPase Rap1b, a protein known to antagonize the mitogenic and transforming activity of Ras, is endowed with both mitogenic and tumorigenic properties. Rap1b can be activated by cAMP, an intracellular message known to either stimulate or inhibit cell proliferation. The oncogenic property of Rap1b was revealed in a model system in which cAMP stimulates cell proliferation and was linked to Rap's ability to promote S phase entry. We have now tested the significance of the mitogenic action of Rap1b in a physiologically relevant model, the differentiated thyroid follicular cells, a system that requires thyroid-stimulating hormone (TSH), acting via cAMP, to mediate a full mitogenic response. Here we report that cAMP-dependent hormonal stimulation of DNA synthesis requires Rap1b in a manner dependent on its phosphorylation by protein kinase A.
The GTPase Rap1b is a GTP-binding protein that can be activated by agonists that increase the intracellular levels of cAMP, calcium, and diacylglycerol, as well as growth factors, cytokines, and cell adhesion molecules (1). G proteins are biochemical transducers that operate as allosteric regulatory elements, switching between an inactive GDP-bound and an active GTP-bound conformation (2). The rate-limiting step for G protein activation is the dissociation of GDP, catalyzed by several guanine nucleotide exchange factors (GEFs) (3). The variety of Rap1-GEFs (1) might explain how numerous stimuli activate Rap1b, and points to a complex role for Rap1b in coordinating different incoming signals.
Rap1b was originally isolated as a Ras revertant clone (4). Subsequent studies involving sequence comparisons, mutagenesis, and structural analysis revealed that, although Rap1b shares 50% overall identity with Ras, their effector domains are virtually identical (5). These and other studies demonstrated that Rap1b interacted with proteins that are typical modulators or effectors of Ras, without leading to their activation (6–12). Thus, a model emerged where Rap1b forms nonproductive complexes with Ras effector molecules, which has led to the current concept that Rap1b is an antimitogenic protein that functions by antagonizing Ras action (13, 14).
Because agonists that increase intracellular cAMP rapidly phosphorylate (15) and activate Rap1b (16), we hypothesized that Rap1 could mediate some of cAMP's biological action (17). It has long been known that cAMP can either stimulate or inhibit cell proliferation (18, 19). Despite the recent emphasis given to the inhibitory action of cAMP on cell proliferation (20), cAMP enhances mitogenic activity in a large number of cells, particularly in primary cultures and cell lines that retain some degree of cellular differentiation (21). Interestingly, the reported negative effects of Rap1b on mitogenesis are seen in systems where cAMP inhibits cell proliferation (22). We asked whether cAMP stimulation of mitogenesis could also be associated with Rap1b activity, and hypothesized that Rap's mitogenic action might depend on cell-specific signal transduction programs, such that Rap1b, like cAMP, can either stimulate or inhibit cell proliferation (17).
This apparent parallelism between the effects of Rap1b and cAMP was first tested in the Swiss 3T3 fibroblast model, a system in which cAMP is a positive regulator of cell proliferation (23). Exogenous expression of Rap1b in Swiss 3T3 fibroblasts was sufficient to endow these cells with both mitogenic and tumorigenic properties (17). In that report, we emphasized that the mitogenic effects of Rap1b were linked to its ability to promote S phase entry and introduced the hypothesis that Rap1b can be viewed as a conditional oncogene.
These findings raised several questions. First, can the mitogenic effect of Rap1 be recapitulated in a more differentiated and physiologically relevant model, in which its mitogenic response strictly depends on a trophic hormone that typically acts via cAMP? Second, does the mitogenic response to such a hormone require Rap1b? Third, is the PKA phosphorylation site on Rap1b essential for its mitogenic action? In this report, we extend the notion that Rap1b has mitogenic properties to the thyroid follicular cell, a prototypical cAMP responsive system that requires thyroid-stimulating hormone (TSH) for a full mitogenic response (24). We provide evidence that in this system, cAMP stimulation of DNA synthesis requires the action of Rap1b in a manner dependent on its phosphorylation by PKA.
Methods
Plasmids Construction, PCCL3 Cells, and Derived Cell Lines.
All mutations were introduced by PCR as described (15). PCCL3 cells were routinely grown in Coon's modified F-12 medium (Irvine Scientific), supplemented with 5% FBS and the combination of four hormones: TSH (1 mIU/ml; IU = international units), insulin (1 μg/ml), transferrin (5 μg/ml), and hydrocortisone (1 nM). Cells were kept at 37°C in a 5% CO2/95% humidified air environment. Cells were transfected in 6-well dishes (≈4 × 105 cells per well, plated the day before) with the Tfx-20 reagent (Promega) according to manufacturer's instructions. For stable expression, Rap1b sequences were subcloned in frame into the Emerald green fluorescent protein (Emd-GFP) vector (BioSignal, Packard), containing a G418-resistance cassette. After transfection, pools of G418-resistant cells were sorted, and protein expression was analyzed by Western blotting and fluorescence microscopy. For transient transfections, hemagglutinin (HA)-tagged constructs were transfected as described and then analyzed by immunofluorescence. In both transient and stable expression, control cells (CL3) received empty vector.
Thymidine Incorporation and BrdUrd-Labeling Assays.
Cells were plated in 96-well dishes (≈2.5 × 104 cells per well) and grown for 2 days until confluency. After a 20-h starvation in Coon's medium/0.2% BSA, cells were agonist-stimulated for 16 h and labeled with thymidine (methyl-[3H]thymidine, Amersham Pharmacia, 1 μM − 1 μCi/ml; 1 Ci = 37 GBq). Twenty-four hours later, cells were collected (cell harvester), and filters were counted by scintillation. G1/S entry was also evaluated by BrdUrd incorporation as described (17). Briefly, for the transient assays, HA-Rap1b or HA-Rap-GAP plasmids were transfected into PCCL3 cells as described. The day after transfection, cells were starved in Coon's medium/0.2% BSA for 16–18 h. Cells were agonist-stimulated for 24 h, and 100 μM BrdUrd was added for the last 12 h of stimulation. After fixing, cells were double stained for BrdUrd and HA by immunofluorescence as described below. All statistical analysis was done with graphpad prism. Error bars represent SD, n = 3.
Rap1b Activation Assay.
The Rap1b-binding domain (RBD) of RalGDS fused to glutathione S-transferase (GST) was used to isolate active Rap1b as described (25, 26). Cells were transfected with HA-WT-Rap1b (WT, wild type). On the next day, cells were serum starved (20 h) and then stimulated with TSH (1 mIU/ml) at 37°C for the indicated time. Cells were lysed in an Nonidet P-40-based buffer in the presence of purified GST-RalGDS-RBD, and the Rap1-GTP/GST-RBD complex was purified with glutathione-Sepharose beads. The beads were rinsed four times with lysis buffer, and eluted proteins were subjected to SDS/PAGE and Western blotting with HA antibodies (HA11, BabCo, Richmond, CA).
Immunofluorescence.
Cells grown on coverslips were washed at the end of the experiment in PBS and fixed for 10 min in 4% paraformaldehyde/PBS. Permeabilization was performed by incubation with 0.5% Triton X-100/PBS for 5 min, followed by extensive washes in 0.05% Tween 20/PBS and PBS. All incubations were performed at 25°C. Primary and secondary antibody incubations were performed for 30 min at 37°C in PBS/2% BSA as diluent. HA antibody was diluted 1:250. Anti-BrdUrd antibody (BioDesign International, Kennebunkport, ME) was used at 1:100 dilution in solution containing RQ-1 RNase-free DNase (10 IU/ml, Promega). Anti-thyroglobulin serum (Dako) was used at 1:200 dilution. All secondary antibodies were Alexa-conjugated antibodies (Molecular Probes) and were used at 1:2,000 to 1:4,000 dilution. DAPI (4′,6-diamidino-2-phenylindole) was included for nuclear staining (0.1 μg/ml). After extensive washes, cells were mounted in PermaFluor (Thomas Scientific) and analyzed by epifluorescence.
Results and Discussion
A Physiological Model to Test the Notion That Rap1b Mediates the Mitogenic Action of cAMP.
TSH, acting via cAMP, induces proliferation of thyroid follicular cells while maintaining their differentiated state (27). The mitogenic responses induced by TSH and cAMP require protein kinase A (PKA) activity (28). However, microinjection of the PKA catalytic subunit is not sufficient to stimulate mitogenesis in thyroid cells, suggesting that cAMP engages events in addition to stimulation of PKA for its mitogenic action (29, 30). A recently identified family of Ras and Rap guanine nucleotide exchange factors can be activated by direct binding of cAMP, independent of PKA action (31, 32). In fact, Ras can be activated by cAMP in a PKA-independent manner and it seems to be required for TSH action (28, 30, 33). However, expression of a constitutively active Ras mutant does not fully mimic the mitogenic action of TSH, but instead induces dedifferentiation (34–37). This observation suggests that some other cAMP-dependent processes must be active which, in collaboration with Ras action, should amplify proliferation while maintaining the differentiated phenotype. In this way, Rap1b could represent a physiological effector in the cAMP mitogenic response.
To explore this possibility, several Rap1b expression vectors were constructed as GFP-fusion and HA-epitope-tagged proteins, and were expressed into PCCL3 thyroid cells. This cell line retains many of the differentiated and physiological properties of the thyrocyte (27, 37), including a strict dependency on TSH and cAMP for growth (Fig. 1 A and B). Moreover, stimulation of these cells with TSH results in a rapid and transient activation of Rap1b, as measured by the RalGDS-RBD binding assay. Maximal Rap1b activation occurs within 1 min, returning to basal values 5 min after stimulation (Fig. 1C). Interestingly, this kinetic profile contrasts with our previous observations in NIH 3T3 cells (16), where cAMP and Rap1b negatively modulate cell growth (38–40). In these cells, cAMP stimulation of Rap1b shows an initial peak similar to the one described here for thyroid cells, followed by a second wave of sustained activation (16, 39). Whether this difference in the kinetics of Rap1b activation (transient vs. sustained) accounts for why cAMP stimulates or inhibits mitogenesis in cells requires further investigation. We have established PCCL3 cell lines expressing GFP-Rap1b fusion proteins with the expected molecular mass and at similar expression levels (Fig. 2 Top). There were no significant morphological differences associated with the different pools of established cells. GFP-Rap1b immunofluorescence revealed the typical perinuclear distribution as described for HA-Rap1 (15). No significant differences were observed in the subcellular localization pattern among cells expressing WT-Rap1b, G12V-Rap1b, or the phosphorylation mutant G12V/S179A-Rap1b constructs. Moreover, no significant changes were observed on stimulation of Rap1b-G12V expressing cells with forskolin (20 min at 37°C) (Fig. 2 Middle). The data show that, at this level of resolution, expression of Rap1b mutants affecting either the nucleotide content (WT vs. G12V) or its phosphorylation state (G12V vs. G12V/S179A), do not significantly alter their subcellular localization. Although the stoichiometry of Rap1b/S179 phosphorylation is not known in these experiments, the results may suggest that cAMP-mediated activation and/or phosphorylation of endogenous Rap1b does not significantly modify its subcellular localization.
Figure 1.

PCCL3 cells as a model to test the notion that Rap1b mediates the cAMP mitogenic response. (A) TSH-dependent G1/S progression. TSH dose responses in the presence of insulin (1 μg/ml) or insulin plus FBS (Insulin + FBS = 1 μg/ml insulin/5% FBS) were used to assess TSH-dependent G1/S entry by [3H]thymidine incorporation. Results are expressed as mean ± SD (n = 3). (B) TSH-dependent cell growth. Cell growth was assessed in complete medium (+TSH) or same medium without TSH (−TSH) by cell counting. Fresh medium was replaced daily. (C) TSH-dependent transient activation of Rap1b. PCCL3 cells were transiently transfected with HA-WT-Rap1b. Cells were starved for 16 h, and upon TSH stimulation, HA-Rap1-GTP was assessed with the RalGDS-RBD pull down assay as described in Methods. (Inset) The actual HA blot on the RBD pull down step.
Figure 2.

Expression of Rap1b in thyroid cells is not accompanied by loss of differentiation. (Top) Western blot of lysates from GFP-Rap1b-expressing cells after G418 selection visualized with an anti-GFP antibody. An Emd-WT sample was enriched by a cycle of cell sorting (FITC channel). Arrow indicates the position of the specific fusion protein (GFP-Rap1b). Numbers on the right represent molecular masses (kDa). A nonspecific protein was observed in all lanes (NS). (Middle) The typical perinuclear subcellular localization was observed in all of the established cells by immunofluorescence of GFP or HA staining. Representative images are shown. Forskolin treatment was for 20 min at 37°C. (Bottom) Thyroglobulin expression in parental or G12V-Rap1b-expressing cells grown in TSH-containing medium was analyzed by indirect immunofluorescence using a polyclonal anti-thyroglobulin serum, as described in Methods.
Rap1b Enhances the Mitogenic Response to cAMP While Maintaining Differentiation.
Expression of TSH receptor and thyroglobulin are routinely used as positive differentiation markers. The Rap1b cells established here express the TSH receptor, as they are fully responsive to TSH (see below). Thyroglobulin expression in parental and G12V-Rap1b-expressing cells, grown in TSH-containing medium, was assessed by indirect immunofluorescence. These cells expressed similar levels of thyroglobulin and displayed an undistinguishable subcellular pattern (Fig. 2 Bottom). Northern analysis for thyroglobulin and thyroperoxidase mRNA also failed to show differences between control and Rap1b expressing cells (data not shown). Thus, unlike the expression of the active G12V-Ras mutant (36), G12V-Rap1b expression is not accompanied by a loss of a differentiated phenotype.
TSH, insulin/insulin-like growth factor 1, or FBS show very low mitogenic activity when tested as the sole stimulant of mitogenesis, whereas TSH exerts a strong synergistic effect when tested in combination with insulin and/or FBS (Fig. 3). If Rap1b is able to transduce the cAMP mitogenic signal, an increase in its cellular concentration ought to enhance the synergistic component of the mitogenic response. Expression of Rap1b strongly increased the maximal response whether the mitogenic stimuli were insulin, FBS, or both and, irrespective if the cAMP-elevating agent used was TSH or forskolin (Fig. 3A). This response appears to require PKA activity because it is blocked by treatment with a specific PKA inhibitor (Fig. 3B). Interestingly, both the mitogenic (Fig. 3B) and the antimitogenic properties of Rap1b require PKA (39). In contrast to our results, a recent report found that microinjection of GST-Rap1b in primary dog thyroid cells, alone or in combination with PKA, failed to induce an effect on either proliferation or differentiation (41). This difference may be a consequence of different model systems used (primary dog vs. established rat cells) or improper targeting of GST-Rap1b and/or PKA on microinjection. Other reports have suggested that Rap1a expression in Wistar Rat thyroid (WRT) cells promotes differentiation without affecting cell proliferation (42). Nevertheless, the present results are consistent with our previous observation using Swiss 3T3 cells (17) and extend the mitogenic effect of Rap1b to a differentiated system, the thyroid cell, where cAMP physiologically acts as a bona fide comitogen. In this model, Rap1b enhances the mitogenic response while maintaining differentiation.
Figure 3.

G12V-Rap1b increases G1/S progression in thyroid cells. (A) Stable cells expressing GFP-G12V-Rap1b (G12V) were assayed for agonist-mediated G1/S entry by [3H]thymidine incorporation, as described in Methods. Agonists used were as follows: insulin (INS), 1 μg/ml; FBS, 5%; TSH , 1 mIU/ml; forskolin (FK), 10 μM. (B) PKA is required for the TSH mitogenic response. PCCL3 and stable cells expressing GFP-G12V-Rap1b (G12V) were assayed for agonist-mediated (TSH+FBS) G1/S entry by [3H]thymidine incorporation, as described in Methods. The PKA inhibitor H89 (25 μM) was included with the agonists (1 mIU/ml TSH/5% FBS).
Rap1b Is a Physiological Effector for the Mitogenic Action of cAMP.
The key feature of cAMP in mitogenesis is to interact synergistically with other mitogens (43, 44). If Rap1b is able to transduce the mitogenic signal of cAMP, a synergisitc interaction should be evident between Rap1b and cAMP generated by either forskolin or TSH. For this, forskolin dose-response curves were performed at a saturating concentration of a comitogen such as FBS. Basal thymidine incorporation in cells expressing the active mutant G12V-Rap1b in the presence of 5% FBS was increased by about 2-fold over control cells. Addition of varying concentrations of forskolin markedly increased the effect of G12V-Rap, which peaked at 10-fold over control between 0.2 and 0.4 μM forskolin, and then gradually decreased to values similar to those seen without forskolin (Fig. 4). The TSH dose-response curves were similar to those obtained for forskolin with the maximal effects of G12V observed at low TSH concentrations (Fig. 5A, ratios). These responses were blunted in the presence of the PKA inhibitor H-89 (data not shown). These results indicate that Rap1b is able to transduce the cAMP mitogenic signal. We propose that Rap1b is a genuine physiological effector for cAMP and PKA actions on mitogenesis.
Figure 4.

The active Rap1b-G12V mutant increases the maximal cAMP mitogenic response. The effect of varying concentrations of forskolin on [3H]thymidine incorporation in the presence of 5% FBS (Right) was assessed in TSH-responsive cells (Left) to test for a synergistic role of cAMP in the action of Rap1b to promote G1/S entry. Results are means ± SD of three independent experiments, each assayed in triplicate. Note the presence of marked synergy (ratio) between cAMP (forskolin) and G12V (●).
Figure 5.

Phosphorylation-dependent mitogenic effects of G12V-Rap1b in thyroid cells. (A) TSH dose-response (constant 5% FBS) was used to analyze the role of Rap1b phosphorylation in the TSH-FBS synergistic behavior on G1/S entry, as assayed by [3H]thymidine incorporation. The observed effect of G12V cells is lost in the phosphorylation-deficient mutant Rap1b-G12V-S179A. Note the presence of Rap1b-G12V markedly enhanced the TSH-FBS synergy as indicated by the ratio G12V/CL3 (●). (B) Single-cell G1/S entry assessment by transient transfection of HA-constructs and BrdUrd labeling. Results are expressed as %BrdUrd/HA on double immunofluorescence staining as described in Methods. A representative experiment is shown.
Phosphorylation of Rap1b Is Required for Its Mitogenic Action.
Rap1b can be phosphorylated by PKA both in vivo an in vitro on residue S179 (15). To determine whether Rap1b phosphorylation is required for its mitogenic effect, a G12V-S179A mutation was made and stable cells were established for mitogenic assays. The phosphorylation-deficient Rap1b mutant, expressed at levels that were similar to those of G12V (Fig. 2 Top), was unable to enhance the mitogenic response, regardless whether the comitogen used was TSH (Fig. 5A) or forskolin (data not shown). The ability of G12V to enhance G1/S transition was confirmed by single cell BrdUrd staining experiments, excluding any potential differences in specific activity of [3H]thymidine and/or cell number. In these experiments, the effect of a phosphomimetic mutant G12V-S179E was also investigated, showing similar results as the phosphodeficient G12V-S179A construct (Fig. 5B). Thus, at least with respect to G1/S entry, both phosphodeficient and phosphomimetic constructs behave as loss-of-function mutants. These results suggest that reversible phosphorylation of Rap1b is required for its ability to enhance the mitogenic response to agents that act via cAMP.
These results demonstrate that a phosphorylation-dependent step is required for Rap1b signaling events leading to a TSH-FBS mediated G1/S entry. Phosphorylation might exert its effects on agonist-dependent Rap1b activation, Rap1b targeting/localization, and/or effector coupling/activation. Rap1 activation is mediated by a number of exchange factors responding to specific incoming signals (1). Of these, the effects of Rap1 phosphorylation were reported only for the cAMP-dependent Epac, an exchange factor highly enriched in thyroid (45). Results from that study showed no role of Rap1 phosphorylation on Epac-dependent Rap1 GTP binding activity (31). Moreover, experiments performed with the mutant S17N/S179A-Rap1b showed the same inhibitory effects as S17N on TSH-dependent G1/S entry (see below). This result suggests that, regardless of the exchange factor involved, Rap1b phosphorylation does not play a major role in the activation step. GFP-Rap immunofluorescence revealed that intracellular localization of the phosphorylation mutant is similar to WT and constitutively active Rap1b forms (Fig. 2 Middle). Furthermore, the subcellular localization of G12V-Rap1b, which display significant GTP-binding activity on cAMP stimulation (16, not shown) was unchanged by treatment of cells with forskolin. Another possibility is that Rap1b phosphorylation might be involved at the effector-coupling step. The simplest likely scenario is that phosphorylation of Rap1b might influence the affinity of binding to effector(s) involved in the mitogenic response. The protein kinase c-Raf is a known Rap target (46, 47) and, phosphorylation of Rap1a by PKA reduces its affinity for c-Raf and blocks its ability to inhibit Ras-dependent Raf activation (48). On the other hand, a recent report suggests that phosphorylation of Rap1b is necessary for activation of another known Rap target, B-Raf (49). These proteins are involved in the activation of mitogen-activated protein kinases (MAPKs). However, in thyroid, the effects of TSH and cAMP on mitogenesis appear to be unrelated to MAPK activation (27, 50, 51). Recently, a role for Rap1a phosphorylation in negative feedback regulation of its GTP binding activity was also suggested (42). Even though the effectors involved in the phosphorylation-dependent responses described in this study are unknown, it is tempting to propose the hypothesis that Rap1b phosphorylation might represent a molecular switch mediating the dynamics of specific effector-coupling. This hypothesis is consistent with the lack of effect of the phosphomimetic mutant in G1/S entry (Fig. 5B), despite its ability to affect the interaction between Rap1a and Raf in vitro (48). This scenario raises a new challenge to identify effector molecule/s that mediate the phosphorylation-dependent Rap1b effects in mitogenesis.
Rap1b Is Required for the Mitogenic Effects of TSH and cAMP.
We examined whether Rap1b is required for the mitogenic actions of TSH and cAMP. A transient approach was used because establishment of stable cell lines expressing a dominant negative Rap1b construct (S17N-Rap1b) was unsuccessful. HA-epitope tagged S17N-Rap1b was transiently transfected into cells, and its effect on the mitogenic activity of TSH monitored by BrdUrd staining, and compared with HA-WT-Rap1b, that did not show any mitogenic effect and was therefore used as control. Expression of S17N-Rap1b considerably inhibited TSH activity at all doses tested, approximately 75% at maximal TSH concentration (Fig. 6). Essentially identical results to those seen with S17N-Rap1b were obtained on expression of a dominant negative and phosphodeficient double mutant S17N/S179A-Rap1b (data not shown). That S17N-Rap1b did not completely block the TSH effect may be explained by the complexity of the recently identified family of Rap1-specific exchange factors, some of them resistant to the inhibitory effect of the S17N mutation (52, 53). Likewise, Rap1b-mediated G1/S entry activity was also reduced (50% of control) by expression of Rap1-GAP (Fig. 6). Similar results to those with TSH were obtained when forskolin was used as a cAMP-elevating agent (data not shown). Therefore, because inhibition of an upstream activator and expression of a negative regulator, both inhibited cAMP-dependent G1/S entry, we conclude that Rap1b activity is required for the mitogenic response induced by TSH and cAMP in thyroid cells.
Figure 6.
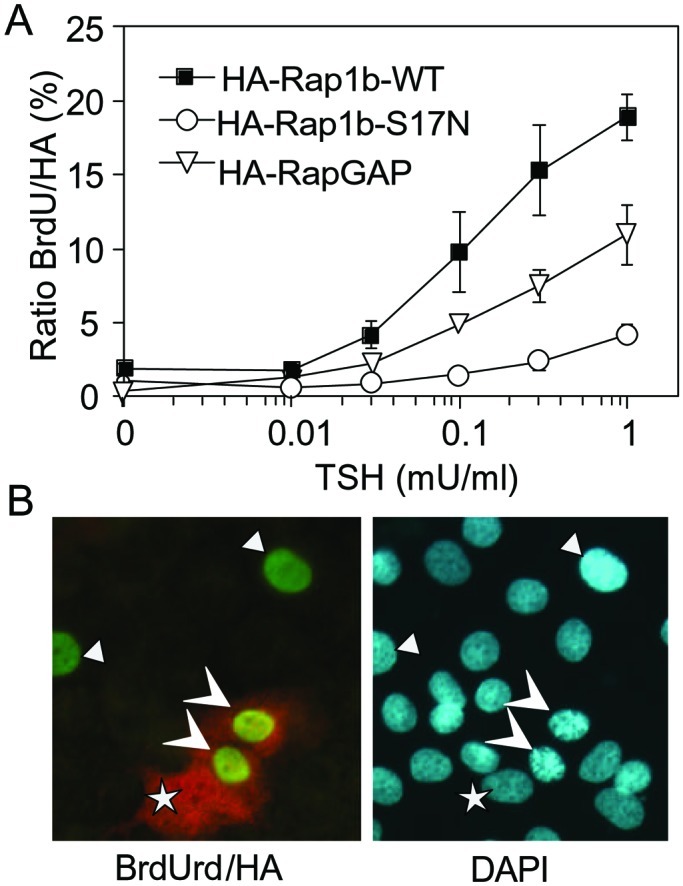
Rap1b is required for the TSH mitogenic effect. (A) PCCL3 cells were transiently transfected with HA-Rap1b WT (control), S17N (dominant negative), with HA-Rap1-GAP. TSH dose-responses were evaluated by BrdUrd labeling and double-immunofluorescent staining (BrdUrd and HA) as described in Methods. Results are means of %BrdUrd/HA ± SD of three independent experiments in each of which 500 cells were scored per point. (B) A representative field of cells with negative, single positive (BrdUrd), and double positive cells (BrdUrd+HA). DAPI staining was used to reveal all of the nuclei in the same field. Arrowheads denote BrdUrd+, HA+ cells; triangles denote BrdUrd+, HA− cells; and the stars represent BrdUrd−, HA+ cells.
The results from the present work demonstrate that Rap1b enhances the maximal mitogenic response to cAMP-elevating agents and that the cAMP synergistic effect requires Rap1b and its PKA phosphorylation site. It follows from these findings that activation of Rap1b, as measured solely by GTP loading, is not sufficient for the transduction of the cAMP signal, because Rap1b is required on its active and phosphorylated forms. The characteristic action of cAMP in mitogenesis is to act in synergy with other comitogens that do not alter the cellular levels of cAMP (43). It is this fundamental aspect of the cAMP mitogenic action that is affected by Rap1b, because its expression markedly increased the maximal mitogenic response without significantly altering the apparent EC50 for cAMP-elevating agents (Figs. 4 and 5). Because the mitogenic action of the comitogens used in this work depends on Ras, the results suggest that Rap1b synergizes with Ras signals for a full mitogenic response. Although it is widely believed that Rap1b is an antimitogenic and antitransforming protein that antagonizes the actions of Ras (14), we have provided a rationale and experimental evidence suggesting that such a view is too restrictive. We had previously proposed that Rap1b effects on mitogenesis might be conditional to the role that cAMP has in cell proliferation and, thus, Rap1b would act in synergy with or as an antagonist to the action of other mitogens (17). Half of this equation was recently confirmed, as Rap1b mediates the cAMP-dependent inhibition of cell proliferation in NIH 3T3 cells (39). Our studies with Swiss 3T3 fibroblasts first, and now with thyroid cells, extend and complement the notion that Rap1b also mediates the mitogenic effects of cAMP, thus lending a strong support to the theory that Rap1b is a conditional mitogen (17). Fig. 7 summarizes our hypothesis on regulation of mitogenesis by cAMP via Rap1b. It highlights the dual role of cAMP in Rap1b action (via Epac and PKA), and its cooperation with comitogens acting via tyrosine kinase receptors (RTKs) and via the Ras GTPase instead of Rap1b.
Figure 7.

Model of dual action of cAMP on Rap1b's ability to transduce its comitogenic activity (see text).
We propose that the mitogenic action of Rap1b might not be restricted only to Swiss 3T3 (17) or thyroid cells, as demonstrated in the present work. Indeed, it may occur in all cells in which cAMP acts as comitogen. An important consequence of this reasoning is to emphasize, as presented in Fig. 7, that some effects of cAMP may be critically dependent on the cooperative action of Rap1 and Ras.
Acknowledgments
This work is dedicated to the memory of Dr. Martin Rodbell. We thank Dr. J. L. Bos (The Netherlands) for providing protocols and reagents for the Rap1 activation assay, and Dr. J. Fagin for the PCCL3 cells. This work was supported by National Institutes of Health Grant R29 CA71649 and American Cancer Society Grant RPG00-291-01-TBE (to D.L.A.), and by the Laboratory of Signal Transduction, National Institute of Environmental Health Sciences, National Institutes of Health.
Abbreviations
- GFP
green fluorescent protein
- HA
hemagglutinin
- GST
glutathione S-transferase
- RBD
Rap1b-binding domain
- WT
wild type
- IU
international units
References
- 1.Bos J L, de Rooij J, Reedquist K A. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 2.Bourne H R, Sanders D A, McCormick F. Nature (London) 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 3.Feig L A. Curr Opin Cell Biol. 1994;6:204–211. doi: 10.1016/0955-0674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 4.Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. Cell. 1989;56:77–84. doi: 10.1016/0092-8674(89)90985-9. [DOI] [PubMed] [Google Scholar]
- 5.Valencia A, Chardin P, Wittinghofer A, Sander C. Biochemistry. 1991;30:4637–4648. doi: 10.1021/bi00233a001. [DOI] [PubMed] [Google Scholar]
- 6.Frech M, John J, Pizon V, Chardin P, Tavitian A, Clark R, McCormick F, Wittinghofer A. Science. 1990;249:169–171. doi: 10.1126/science.2164710. [DOI] [PubMed] [Google Scholar]
- 7.Hata Y, Kikuchi A, Sasaki T, Schaber M D, Gibbs J B, Takai Y. J Biol Chem. 1990;265:7104–7107. [PubMed] [Google Scholar]
- 8.Ikeda M, Koyama S, Okazaki M, Dohi K, Kikuchi A. FEBS Lett. 1995;375:37–40. doi: 10.1016/0014-5793(95)01169-f. [DOI] [PubMed] [Google Scholar]
- 9.Spaargaren M, Bischoff J R. Proc Natl Acad Sci USA. 1994;91:12609–12613. doi: 10.1073/pnas.91.26.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urano T, Emkey R, Feig L A. EMBO J. 1996;15:810–816. [PMC free article] [PubMed] [Google Scholar]
- 11.Wolthuis R M, Bauer B, van't Veer L J, de Vries-Smits A M, Cool R H, Spaargaren M, Wittinghofer A, Burgering B M, Bos J L. Oncogene. 1996;13:353–362. [PubMed] [Google Scholar]
- 12.Zwartkruis F J, Wolthuis R M, Nabben N M, Franke B, Bos J L. EMBO J. 1998;17:5905–5912. doi: 10.1093/emboj/17.20.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noda M. Biochem Biophys Acta. 1993;1155:97–109. doi: 10.1016/0304-419x(93)90024-7. [DOI] [PubMed] [Google Scholar]
- 14.Zhang K, Noda M, Vass W C, Papageorge A G, Lowy D R. Science. 1990;249:162–165. doi: 10.1126/science.2115210. [DOI] [PubMed] [Google Scholar]
- 15.Altschuler D, Lapetina E G. J Biol Chem. 1993;268:7527–7531. [PubMed] [Google Scholar]
- 16.Altschuler D L, Peterson S N, Ostrowski M C, Lapetina E G. J Biol Chem. 1995;270:10373–10376. doi: 10.1074/jbc.270.18.10373. [DOI] [PubMed] [Google Scholar]
- 17.Altschuler D L, Ribeiro-Neto F. Proc Natl Acad Sci USA. 1998;95:7475–7479. doi: 10.1073/pnas.95.13.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pastan I H, Johnson G S, Anderson W B. Annu Rev Biochem. 1975;44:491–522. doi: 10.1146/annurev.bi.44.070175.002423. [DOI] [PubMed] [Google Scholar]
- 19.Boynton A L, Whitfield J F. Adv Cyclic Nucleotide Res. 1983;15:193–294. [Google Scholar]
- 20.Graves L, Lawrence J., Jr Trends Endocrinol Metab. 1996;7:43–50. doi: 10.1016/1043-2760(95)00204-9. [DOI] [PubMed] [Google Scholar]
- 21.Dumont J E, Jauniaux J C, Roger P P. Trends Biochem Sci. 1989;14:67–71. doi: 10.1016/0968-0004(89)90046-7. [DOI] [PubMed] [Google Scholar]
- 22.Cook S J, Rubinfeld B, Albert I, McCormick F. EMBO J. 1993;12:3475–3485. doi: 10.1002/j.1460-2075.1993.tb06022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rozengurt E, Legg A, Strang G, Courtenay-Luck N. Proc Natl Acad Sci USA. 1981;78:4392–4396. doi: 10.1073/pnas.78.7.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumont J E, Lamy F, Roger P, Maenhaut C. Physiol Rev. 1992;72:667–697. doi: 10.1152/physrev.1992.72.3.667. [DOI] [PubMed] [Google Scholar]
- 25.van Triest M, de Rooij J, Bos J L. Methods Enzymol. 2001;333:343–348. doi: 10.1016/s0076-6879(01)33068-9. [DOI] [PubMed] [Google Scholar]
- 26.Franke B, Akkerman J W, Bos J L. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medina D L, Santisteban P. Eur J Endocrinol. 2000;143:161–178. doi: 10.1530/eje.0.1430161. [DOI] [PubMed] [Google Scholar]
- 28.Kupperman E, Wen W, Meinkoth J L. Mol Cell Biol. 1993;13:4477–4484. doi: 10.1128/mcb.13.8.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dremier S, Pohl V, Poteet-Smith C, Roger P P, Corbin J, Doskeland S O, Dumont J E, Maenhaut C. Mol Cell Biol. 1997;17:6717–6726. doi: 10.1128/mcb.17.11.6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsygankova O M, Kupperman E, Wen W, Meinkoth J L. Oncogene. 2000;19:3609–3615. doi: 10.1038/sj.onc.1203680. [DOI] [PubMed] [Google Scholar]
- 31.de Rooij J, Zwartkruis F J, Verheijen M H, Cool R H, Nijman S M, Wittinghofer A, Bos J L. Nature (London) 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki H, Springett G M, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman D E, Graybiel A M. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 33.al-Alawi N, Rose D W, Buckmaster C, Ahn N, Rapp U, Meinkoth J, Feramisco J R. Mol Cell Biol. 1995;15:1162–1168. doi: 10.1128/mcb.15.3.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Missero C, Pirro M T, Di Lauro R. Mol Cell Biol. 2000;20:2783–2793. doi: 10.1128/mcb.20.8.2783-2793.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Missero C, Cobellis G, De Felice M, Di Lauro R. Mol Cell Endocrinol. 1998;140:37–43. doi: 10.1016/s0303-7207(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 36.Kupperman E, Wofford D, Wen W, Meinkoth J L. Endocrinology. 1996;137:96–104. doi: 10.1210/endo.137.1.8536648. [DOI] [PubMed] [Google Scholar]
- 37.Fusco A, Berlingieri M T, Di Fiore P P, Portella G, Grieco M, Vecchio G. Mol Cell Biol. 1987;7:3365–3370. doi: 10.1128/mcb.7.9.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Iyengar R. Science. 1994;263:1278–1281. doi: 10.1126/science.8122111. [DOI] [PubMed] [Google Scholar]
- 39.Schmitt J M, Stork P J. Mol Cell Biol. 2001;21:3671–3683. doi: 10.1128/MCB.21.11.3671-3683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J, Dent P, Jelinek T, Wolfman A, Weber M J, Sturgill T W. Science. 1993;262:1065–1069. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- 41.Dremier S, Vandeput F, Zwartkruis F J, Bos J L, Dumont J E, Maenhaut C. Biochem Biophys Res Commun. 2000;267:7–11. doi: 10.1006/bbrc.1999.1919. [DOI] [PubMed] [Google Scholar]
- 42.Tsygankova O M, Saavedra A, Rebhun J F, Quilliam L A, Meinkoth J L. Mol Cell Biol. 2001;21:1921–1929. doi: 10.1128/MCB.21.6.1921-1929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rozengurt E. Science. 1986;234:161–166. doi: 10.1126/science.3018928. [DOI] [PubMed] [Google Scholar]
- 44.Kimura T, Dumont J E, Fusco A, Golstein J. Eur J Endocrinol. 1999;140:94–103. doi: 10.1530/eje.0.1400094. [DOI] [PubMed] [Google Scholar]
- 45.Kawasaki H, Springett G M, Toki S, Canales J J, Harlan P, Blumenstiel J P, Chen E J, Bany I A, Mochizuki N, Ashbacher A, et al. Proc Natl Acad Sci USA. 1998;95:13278–13283. doi: 10.1073/pnas.95.22.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okada T, Hu C D, Jin T G, Kariya K, Yamawaki-Kataoka Y, Kataoka T. Mol Cell Biol. 1999;19:6057–6064. doi: 10.1128/mcb.19.9.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. Nature (London) 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 48.Hu C D, Kariya K, Okada T, Qi X, Song C, Kataoka T. J Biol Chem. 1999;274:48–51. doi: 10.1074/jbc.274.1.48. [DOI] [PubMed] [Google Scholar]
- 49.Vossler M R, Yao H, York R D, Pan M G, Rim C S, Stork P J. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 50.Lamy F, Wilkin F, Baptist M, Posada J, Roger P P, Dumont J E. J Biol Chem. 1993;268:8398–8401. [PubMed] [Google Scholar]
- 51.Miller M J, Rioux L, Prendergast G V, Cannon S, White M A, Meinkoth J L. Mol Cell Biol. 1998;18:3718–3726. doi: 10.1128/mcb.18.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seidel M G, Klinger M, Freissmuth M, Holler C. J Biol Chem. 1999;274:25833–25841. doi: 10.1074/jbc.274.36.25833. [DOI] [PubMed] [Google Scholar]
- 53.van den Berghe N, Cool R H, Horn G, Wittinghofer A. Oncogene. 1997;15:845–850. doi: 10.1038/sj.onc.1201407. [DOI] [PubMed] [Google Scholar]