

Abstract
TSG101 was discovered in a screen for tumor susceptibility genes and has since been shown to have a multiplicity of biological effects. However, the basis for TSG101's ability to regulate cell growth has not been elucidated. We report here that the TSG101 protein binds to the cyclin/cyclin-dependent kinase (CDK) inhibitor (CKI) p21Cip1/WAF1 and increases stability of the p21 protein in HEK293F cells and differentiating primary keratinocytes, suppressing differentiation in a p21-dependent manner. In proliferating keratinocytes where the p21 protein is relatively stable, TSG101 does not affect the stability or expression of p21 but shows p21-dependent recruitment to cyclin/CDK complexes, inhibits cyclin/CDK activity, and causes strong growth suppression to a much greater extent in p21+/+ than in p21−/− cells. Conversely, suppression of endogenous TSG101 expression by an antisense TSG101 cDNA causes doubling of the fraction of keratinocytes in the S phase of the cell cycle as occurs during p21 deficiency. Our results indicate that TSG101 has a direct role in the control of growth and differentiation in primary epithelial cells, and that p21 is an important mediator of these TSG101 functions.
The first cyclin/cyclin-dependent kinase (CDK) inhibitor (CKI) to be identified was p21Cip1/WAF1, which is a well characterized mediator of p53-induced growth arrest (1). The amino-terminal domain of p21, similar to the corresponding domains of p27 and p57, is both necessary and sufficient to bind and inhibit cyclin/CDK complexes. The unique carboxyl-terminal domain of p21 associates with the proliferating cell nuclear antigen (PCNA), a subunit of DNA polymerase, and can inhibit DNA replication directly (1). In addition, p21 participates in a number of other specific protein–protein interactions that relate to cell-cycle control, apoptosis, and differentiation (2). This complexity of functions is well illustrated by the role of p21 in keratinocyte self-renewal and differentiation; induction of p21 expression is one of the earliest cell-cycle regulatory events that contributes to differentiation-associated growth arrest (3, 4), and analysis of p21 null keratinocytes indicates that p21 is required for the intrinsic commitment of keratinocytes to differentiate (5). However, at later stages of differentiation, the p21 protein is down-modulated by proteasome-mediated degradation, and this down-modulation is required for differentiation (6). Sustained p21 expression under these conditions blocks terminal differentiation marker expression at the level of gene transcription in a manner that is unlinked to its effects on the cell cycle (6).
To gain further insights into how p21 may function in keratinocytes, we used a yeast two-hybrid screen to identify new interacting proteins with p21. One of the new interactors corresponds to TSG101, a multifunctional domain protein discovered in a screen for tumor-susceptibility genes in NIH 3T3 fibroblasts (7–10). The TSG101 protein plays an essential role during development, as demonstrated by the early lethality observed in null mutant mouse embryos (11). TSG101 protein levels normally are controlled posttranscriptionally within a narrow range (12), and both increased and suppressed expression of TSG101 in NIH 3T3 cells lead to abnormal cell growth (7). TSG101 deficiency by antisense cDNA expression causes a variety of nuclear, microtubule, and mitotic spindle abnormalities (13). Sequence analysis indicates that the TSG101 protein contains an amino-terminal region of homology to ubiquitin-conjugating enzymes (UBC E2), a proline-rich sequence with the features of a transcription transactivation domain, a leucine zipper, and a central coiled-coil region (7, 14). Recent experiments have shown that TSG101 has an important role in ubiquitin-mediated endosomal sorting pathways (15–17). There also is evidence implicating the TSG101 UBC domain on regulation of the p53/MDM2 feedback control loop in human tumor cells (18). However, the mechanism(s) by which TSG101 acts to affect proliferation of normal cells has not been elucidated. We report here that in primary epithelial cells (keratinocytes) TSG101 can function as a direct negative regulator of both cell growth and differentiation, and that p21 is an important mediator of these effects.
Materials and Methods
Yeast Two-Hybrid Screening, Plasmids, Recombinant Proteins, and Adenovirus (Ad) Construction.
Yeast two-hybrid analysis was carried out by using a primary mouse keratinocyte cDNA library inserted into the pPC86 plasmid (19). Vectors for truncated p21 mutants were obtained by PCR amplification from the human full-length p21 cDNA and inserting the resulting fragments into the pcDNA3.1/V5/His-Topo plasmid (Invitrogen). The cytomegalovirus (CMV) expression vector for mouse p27 was a gift from Dr. Yin Sun (Whitehead Institute, Massachusetts Institute of Technology, Cambridge, MA). The recombinant Ad-expressing green fluorescent protein (GFP)-TSG101 was obtained by inserting the corresponding full-length cDNA (13) into the pCMV-shuttle vector, followed by homologous recombination with the adenoviral backbone plasmid pAdEasy-1 (20).
Cell Culture and Transfection.
The conditions for cultivation of cells, Ad infection, suspension-induced differentiation, and proteasome inhibitor treatment were as described previously (6). For transient transfection of plasmid DNA into keratinocytes, the lipofection technique (GIBCO/BRL) was used. At 72 h after transfection, keratinocytes were trypsinized, and transfected red fluorescent protein (RFP)-positive cells were separated from untransfected cells by using a MoFlo cell sorter (Cytomation, Fort Collins, CO).
Kinase Assays, Immunoprecipitation, and Antibodies.
CDK2 kinase assays and coimmunoprecipitations were performed as described (3). Antibodies against mouse p21, human p21, human p27, TSG101, cyclin E, CDK2, PCNA, RasGTPase-activating protein (RasGAP), and nonimmune mouse and rabbit IgG were from Santa Cruz Biotechnology. Antibodies against Phospho-Rb and control Rb were from Cell Signaling Technology (Beverly, MA). The anti-BrdUrd antibody was from Becton Dickinson.
Results
The TSG101 Protein Associates with p21 and Increases Its Stability.
Primary mouse keratinocytes provide a well characterized model system for growth/differentiation control of normal epithelial cells (21). To gain further insights into the function of p21 in this system, we screened a mouse keratinocyte cDNA library with a full-length p21 bait. The screening yielded ≈200 independent cDNA clones that were confirmed to code for p21-interacting proteins when reintroduced into yeast. Fifty of these cDNAs were sequenced and of these, six (≈12%) were found to code for mouse TSG101 (7). To assess the interaction between these two proteins in mammalian cells, expression vectors for full-length p21 and a TSG101-GFP fusion protein were cotransfected into HEK293F cells followed by immunoprecipitation of the p21 protein and immunoblotting for TSG101. To determine whether the endogenous TSG101 and p21 proteins can also associate, extracts from larger amounts of untransfected HEK293F cells or from mouse primary keratinocytes were analyzed similarly. As shown in Fig. 1 A and B, both exogenously expressed and endogenous TSG101 proteins were found in the p21 immunoprecipitates. Similar cotransfection experiments with HEK293F cells using vectors that expressed progressively truncated forms of p21 showed that the carboxyl terminus of p21 is dispensable for TSG101 binding, and that the first 86-aa region of p21 binds to TSG101 as efficiently as does the full-length protein (Fig. 1A). Consistent with the significant homology between the amino-terminal domain of p21 and the related CKI p27 (1), we found that p27 also associates with TSG101 when both proteins are coexpressed in HEK293F cells (Fig. 1A). HEK293F cells were transfected additionally with increasing amounts of the p21 expression vector in the presence or absence of a fixed amount of the vector expressing TSG101. Immunoprecipitation with antibodies against p21 followed by immunoblotting with the same antibodies revealed that p21 protein levels were significantly higher in cells that concomitantly overexpressed TSG101 (Fig. 1C). Unlike p21, p27 expression was not increased by the presence of TSG101 (Fig. 1C). Immunoblotting of the p21 immunoprecipitates with antibodies against CDK2, cyclin E, and PCNA indicated that the association of p21 with these molecules was not disrupted by increased TSG101 expression (Fig. 1D).
Figure 1.

TSG101 associates with p21 and increases its stability in HEK293F cells. (A) Association of TSG101 with p21 in HEK293F cells. HEK293F cells were transiently transfected by the calcium phosphate DNA precipitation method with expression vectors for p21 and p27 proteins and for p21 mutants with progressive deletion of the carboxyl-terminal domain to the indicated amino acid positions with or without an expression plasmid for a GFP-TSG101 fusion protein. Total cell extracts (2 mg) were immunoprecipitated with anti-p21 antibodies and immunoblotted in parallel with nonprecipitated cell extracts transfected with a TSG101-expressing plasmid (TSG extr.) with anti-TSG101 antibodies. (B) Association of endogenous TSG101 and p21 proteins. Total cell extracts from HEK293F cells (40 mg) and primary keratinocytes (10 mg) were immunoprecipitated (IP) with nonimmune IgGs (NI IgG) or anti-p21 antibodies and immunoblotted (IB) in parallel with nonprecipitated cell extracts (Extr.) with antibodies against TSG101 (Upper) or p21 (Lower). (C) Stabilization of the p21 protein in the presence of TSG101. HEK293F cells were transiently transfected with increasing amounts of expression vectors for full-length p21 or p27 with or without a fixed amount (10 μg) of vector DNA for GFP-TSG101. Extracts (2 mg) were immunoprecipitated with anti-p21 or anti-p27 antibodies and immunoblotted with the same antibodies. (D) Association of p21 with cyclin E, CDK2, and PCNA. The same p21 immunoprecipitates used in C were immunoblotted sequentially with anti-cyclin E, anti-CDK2, and anti-PCNA antibodies. (E) Stability of the p21 protein in the absence or presence of TSG101. HEK293F cells were transfected with the expression vector for full-length p21 (15 μg) with or without the vector for GFP-TSG101 (15 μg). At 24 h after transfection, cells were pulse-labeled with [35S]methionine [100 μCi/ml (1 Ci = 37 GBq)] in methionine-free medium for 2 h. Cells were chased in fully supplemented medium for various amounts of time before immunoprecipitation with anti-p21 antibodies, 12% SDS/PAGE, and autoradiography. The results were quantified by densitometric scanning of the autoradiographs and expressed as arbitrary unit amounts (Right). For all transient transfection of HEK293F cells, empty vector DNA was added such that all cells received the same amount of total DNA.
The increased p21 protein level observed in HEK293F cells that concomitantly overexpressed TSG101 may reflect posttranscriptional stabilization. To test this possibility, HEK293F cells were transfected with the p21 expression vector with or without the TSG101 expression vector, labeled with [35S]methionine, and pulse–chased for various times. The half life of p21 was more than 40 min in cells overexpressing TSG101 compared with 20 min in controls, indicating that TSG101 overproduction substantially increases p21 protein stability (Fig. 1E).
Increased TSG101 Expression Counteracts the Destabilization of the p21 Protein in Terminally Differentiating Keratinocytes.
The p21 protein level is strongly down-regulated through proteasome-dependent degradation when keratinocytes lose contact with the substrate and consequently are induced to terminally differentiate (6). Consistent with the results from the HEK293F cells, overexpression of TSG101 by recombinant Ad infection reversed down-regulation of the p21 protein in differentiating keratinocytes, whereas the level of p27 was unaffected (Fig. 2A Left). A similar increase in p21 protein was associated with TSG101 expression in differentiating keratinocytes derived from p53−/− mice (Fig. 2A Middle). Moreover, overexpression of TSG101 in either attached or detached keratinocytes had no effects on p21 gene transcription (Fig. 2B), indicating that the observed effects of TSG101 on p21 expression are posttranscriptional and independent of p53 regulation. Down-modulation of the p21 protein level in differentiating keratinocytes is caused by proteasome-dependent degradation (6). Treatment with specific proteasome inhibitors, N-acetyl-leucinyl-leucinyl-norleucinal, and lactacystin, but not with the unrelated cysteine proteinase inhibitor L-trans-epoxysuccinic acid allows detection of higher molecular weight forms of the p21 protein that correspond to undegraded ubiquitin-conjugated products (6, 22, 23). In cells overexpressing the TSG101 protein and treated with the proteasome inhibitors, the high molecular weight forms of ubiquitinated p21 still were observed, although in lesser amounts than in control cells (Fig. 2C), indicating that the stabilization of the p21 protein by TSG101 is accompanied by only limited suppression of ubiquitination.
Figure 2.

TSG101 overexpression leads to increased p21 protein levels in differentiating keratinocytes but not in proliferating cells. (A) p21 protein levels in attached versus detached keratinocytes after TSG101 overexpression. (Left and Middle) Primary keratinocytes from either wild-type or p53−/− mice were infected with Ad-GFP and Ad-TSG101 at the indicated multiplicity of infection (MOI). Spontaneously detached differentiating keratinocytes at 24 h after infection were analyzed by immunoblotting with either anti-p21 or anti-p27 antibodies. (Right) Attached keratinocytes at 24 h after infection with Ad-GFP and Ad-TSG101 at the indicated MOI were analyzed by immunoblotting with anti-p21, anti-p27, or anti-p53 antibodies. (B) p21 mRNA expression levels. Primary mouse keratinocytes were infected with recombinant Ads expressing either GFP or TSG101 at 50 MOI. Total (30 μg) RNA samples isolated from attached and spontaneously detached keratinocyte populations were analyzed by Northern blotting with mouse p21 (Upper) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA probes (Lower). (C) Ubiquitination of the p21 protein. Primary keratinocytes were infected for 1 h with Ad-GFP or Ad-TSG101 at 50 MOI followed by the addition of medium with dimethyl sulfoxide solvent alone (−) or 5 μM each of L-trans-epoxysuccinic acid (E64), lactacystin (Lact.), or N-acetyl-leucinyl-leucinyl-norleucinal (LLnL). Spontaneously detached keratinocytes were harvested 24 h after treatment and analyzed by immunoblotting with anti-p21 antibodies. Higher molecular weight bands recognized by anti-p21 antibodies correspond to ubiquitinated forms of this protein.
TSG101 Functions as a p21-Dependent Suppressor of Differentiation.
The cellular level of endogenous TSG101 protein was found to decrease in parallel with the p21 level in keratinocytes induced to differentiate by detachment from the dish. This event was accompanied by induction of the terminal differentiation marker filaggrin, whereas the level of keratin 5, a marker for the proliferative layer of the epidermis that is constitutively expressed in cultured keratinocytes, remained constant (Fig. 3A). Earlier work has shown that expression of the terminal differentiation markers filaggrin and keratin 1 in keratinocytes is associated with down-modulation of the p21 protein and is suppressed by overexpression of the full-length p21 protein independently of the effects of p21 on the cell cycle (6). Similarly, overexpression of TSG101 in differentiating keratinocytes inhibited expression of these markers (Fig. 3B). In p21 null mutant cells, differentiation markers such as keratin 1 are expressed at a lower level than in wild-type keratinocytes, whereas other markers such as filaggrin are expressed in similar amounts as in wild-type cells (4). Overexpression of TSG101 in p21−/− cells did not inhibit filaggrin expression and caused no further reduction in keratin 1 (Fig. 3C), suggesting that TSG101 inhibition of keratinocyte differentiation markers requires p21.
Figure 3.
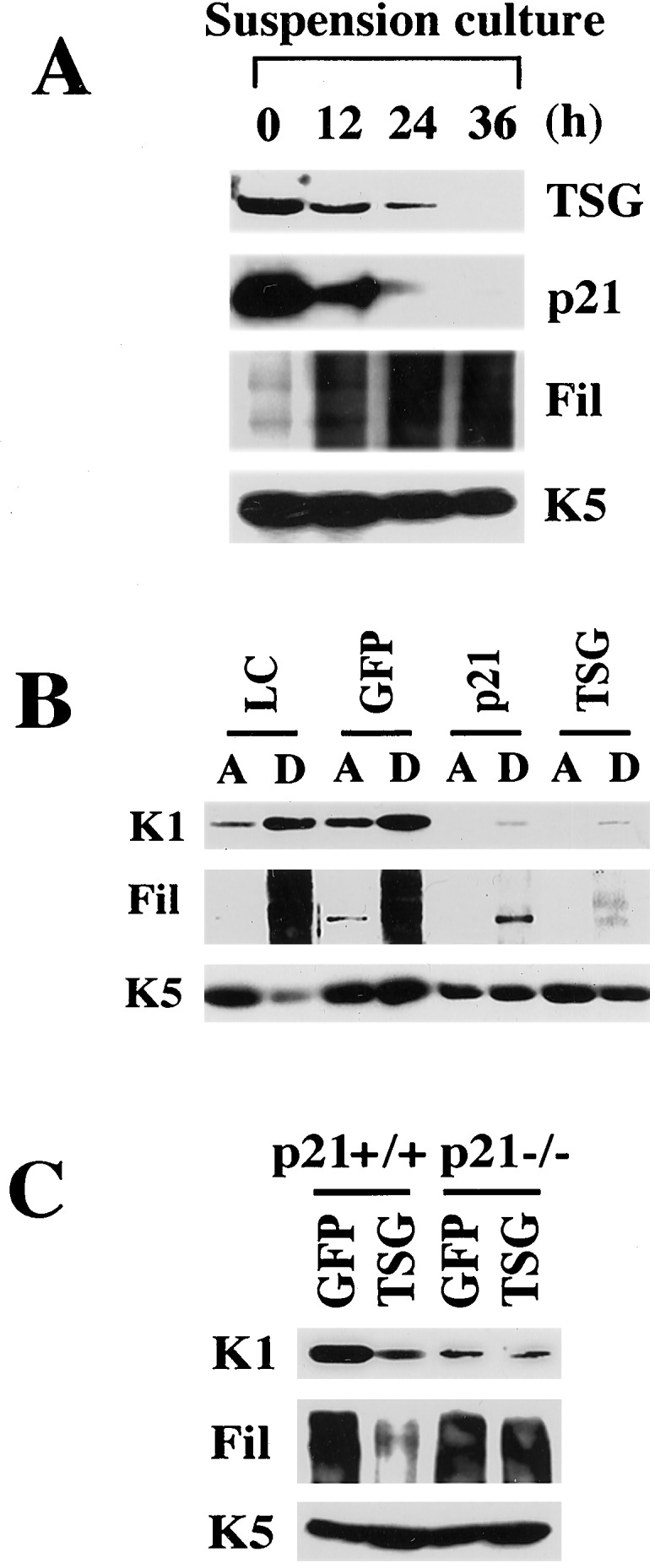
Suppression of keratinocyte differentiation marker expression by p21 and TSG101. (A) Down-modulation of endogenous p21 and TSG101 proteins in keratinocytes in suspension. Primary keratinocytes were trypsinized and cultured in suspension for the indicated times (h). Total cell extracts were analyzed by immunoblotting with antibodies against TSG101, p21, filaggrin (Fil), and keratin 5 (K5). Filaggrin is synthesized as a high molecular weight precursor, profilaggrin, which is subsequently processed. The diffused bands correspond to the multiple products of this processing. Keratin 5 is constitutively expressed in keratinocytes in culture irrespective of growing or differentiating conditions and is used as equal loading control. (B) Similar suppression of keratinocyte differentiation marker expression by p21 and TSG101. Primary keratinocytes were uninfected (low calcium medium, LC) or infected with Ad-GFP, a recombinant Ad expressing full-length p21 or Ad-TSG101 at 100 MOI. Attached (A) and spontaneously detached (D) keratinocytes at 24 h after infection were analyzed by immunoblotting with antibodies against the indicated differentiation marker proteins. (C) Differential effects on differentiation marker expression by Ad-TSG101 on wild-type versus p21-deficient keratinocytes. Primary keratinocytes from p21+/+ and p21−/− mice of the same genetic background (Sencar) were infected with Ad-GFP and Ad-TSG101 at 50 MOI, and spontaneously detached cells were analyzed for differentiation marker expression as described for B.
Increased TSG101 Expression Leads to p21-Dependent Inhibition of Growth and Cyclin/CDK Activity.
The p21 protein is destabilized selectively in differentiating, as opposed to proliferating, keratinocytes (6). In the latter cells, increased TSG101 expression did not affect either p21 transcription (Fig. 2B) or protein level (Fig. 2A Right). However, increased TSG101 expression in proliferating keratinocytes resulted in dramatic p21-dependent effects on DNA synthesis; infection with the Ad-TSG101 even at low multiplicity strongly inhibited DNA synthesis in wild-type keratinocytes and suppressed it in p21−/− cells only at a high MOI and to a very limited extent, i.e., >10–20 times less than in wild-type cells (Fig. 4A). In parallel with the observed inhibition of DNA synthesis, TSG101 overexpression suppressed CDK2 kinase activity in wild-type keratinocytes to a much greater extent than in p21−/− cells (Fig. 4B). Immunoblotting of CDK2 or cyclin E immunoprecipitates from wild-type cells with anti-TSG101 antibodies showed that even a relatively small increase in TSG101 protein levels by Ad-TSG101 infection at low multiplicity resulted in the association of this protein with cyclin/CDK complexes in the presence of p21. However, in p21−/− cells, association of TSG101 with cyclin/CDK complexes was detected only in cells transfected with large amounts of a TSG101-expressing construct (Fig. 4C).
Figure 4.

TSG101 causes growth suppression and cyclin/CDK inhibition in keratinocytes in a p21-dependent manner. (A) Differential sensitivity of wild-type versus p21-deficient keratinocytes to TSG101-induced growth suppression. Primary keratinocytes derived from either wild-type or p21 null mice were infected with Ad-GFP and Ad-TSG101 at the indicated MOI. DNA synthesis was measured at 24 h after infection in triplicate wells by a 1-h [3H]thymidine incorporation assay. Values are expressed as percentages of uninfected controls. The SD is indicated. Similar results were obtained in two other independent experiments. (B) Inhibition of CDK2 kinase activity by TSG101. Primary keratinocytes from wild-type or p21−/− mice were infected with Ad-GFP or Ad-TSG101 at the indicated MOI. NI, nonimmune control. Total cell extracts (300 μg) were immunoprecipitated (IP) with anti-CDK2 antibodies and tested for in vitro CDK2 kinase activity by using histone H1 as substrate. A fraction of the immunoprecipitates was immunoblotted with antibodies against CDK2 for normalization. Results were quantified by densitometric scanning of the autoradiographs and expressed as relative arbitrary units to either wild-type (WT) or p21−/− (KO) controls after normalizing for CDK2 protein amounts (Right). (C) Recruitment of TSG101 to cyclin/CDK complexes depending on p21 function. Primary keratinocytes from wild-type or p21−/− mice were infected with Ad-GFP or Ad-TSG101 at the indicated MOI. Total cell extracts (2 mg) were immunoprecipitated with antibodies against CDK2 or cyclin E (CycE) and immunoblotted with antibodies against TSG101, p21, p27, cyclin E, and CDK2 as indicated.
Suppression of Endogenous TSG101 Expression Causes a Doubling of the Fraction of Keratinocytes in the S Phase of the Cell Cycle.
To learn whether endogenous TSG101 also affects cell-cycle control, we transfected a construct containing TSG101 cDNA in the antisense direction into growing keratinocytes together with an expression vector for RFP as a marker for transfected cells. The fraction of cells in S phase was found to increase from 20% in the control to 40% in the antisense TSG101-expressing cells, as assessed by BrdUrd labeling for various amounts of time (Fig. 5A). To confirm that the observed increase in the fraction of cells in the S phase of the cell cycle resulted from interference with expression of endogenous TSG101 protein, similarly cotransfected cells were purified for RFP expression by fluorescence-activated cell sorter separation. Immunoblotting of cell extracts with anti-TSG101 antibodies showed that expression of the endogenous protein was suppressed strongly in the cells transfected with the antisense TSG101 cDNA vector, whereas the amount of the unrelated RasGAP protein was not changed (Fig. 5B). Down-modulation of TSG101 had no effect on p21 protein levels in growing keratinocytes, which is consistent with the observation that TSG101-mediated stabilization of p21 occurs only in differentiating cells. In parallel with our functional BrdUrd-labeling results, phosphorylation of p105-Rb, which is mediated directly by cyclin/CDK and is required for the G1/S transition in the cell cycle (24), was increased in cells transfected with the antisense TSG101 cDNA while total p105-Rb expression remained constant (Fig. 5C).
Figure 5.

Suppression of endogenous TSG101 expression causes a doubling of the fraction of keratinocytes in the S phase of the cell cycle. (A) Growing keratinocytes were transfected with either an antisense (AS) TSG101 cDNA vector or an empty vector control together with an expression vector for RFP. Cells were labeled with BrdUrd for the indicated times (h) before the termination of the experiment (total 72 h after transfection). Cells were fixed in 3% paraformaldehyde and stained subsequently with anti-BrdUrd antibodies as described. Labeling indexes are expressed as percentages of BrdUrd-positive cells that also were RFP-positive. In each case, a minimum of 120 cells from six independent fields was counted. Similar results were obtained in two other independent experiments. (B) Suppression of endogenous TSG101 expression by antisense TSG101 cDNA. Keratinocytes were transfected with either an antisense TSG101 cDNA vector or an empty vector control together with an expression vector for RFP. Transfected RFP-positive cells were purified by sorting at 72 h after transfection. Cell extracts were analyzed by sequential immunoblotting with antibodies against TSG101, p21, and RasGAP as an unrelated internal control. (C) Increased phosphorylation of the p105-Rb protein in keratinocytes expressing antisense TSG101. The same cell extracts as shown in B were analyzed by immunoblotting with phospho-specific antibodies against Rb (Upper) or antibodies recognizing the total control Rb protein (Lower). Similar results were obtained in two other independent experiments.
Discussion
Earlier work has shown that p21 plays a dual role in keratinocyte self-renewal and differentiation; induction of p21 expression is one of the earliest cell-cycle regulatory events that contributes to differentiation-associated growth arrest, and down-modulation of the p21 protein is required for the later stages of differentiation (4, 6). We have shown here that the TSG101 protein has effects in keratinocytes that resemble those of p21 and depend on p21 function.
Stabilization of the p21 protein by TSG101 in both HEK293F cells and differentiating keratinocytes is associated with, but is not necessarily the direct result of, interaction between TSG101 and p21. Based on sequence analysis, it has been suggested that TSG101 is a ubiquitin-conjugating enzyme E2 variant (14), and the UBC-like domain has been implicated in TSG101-mediated stabilization of the MDM2 protein (18) as well as in other cellular functions involving ubiquitination (16, 17). Stabilization of the p21 protein by TSG101 is accompanied by only limited interference with p21 ubiquitination, consistent with the fact that p21, although a substrate for ubiquitination, also can undergo proteasome-mediated degradation by a ubiquitin-independent mechanism (23). A recent report indicated that p21 degradation can be mediated by a direct interaction of this protein with the C8 α-subunit of the 20S proteasome (25), and an attractive possibility is that this interaction is prevented by TSG101. Besides direct binding, TSG101 also may stabilize p21 through a more indirect mechanism involving, for instance, ubiquitin-mediated endosomal sorting pathways (15–17). It is important to stress, however, that TSG101 does not function as a general determinant of p21 stability in keratinocytes but counteracts selectively the destabilization of the p21 protein that accompanies differentiation. Although the molecular basis for the selective destabilization of p21 in terminally differentiating keratinocytes remains to be established, endogenous levels of the TSG101 protein were also found to decrease in these cells, suggesting that this coordinate down-modulation is important for the late stages of differentiation. In fact, increased TSG101 expression under these conditions suppresses differentiation in a p21-dependent manner and with similar effects as direct p21 overexpression. An attractive possibility is that the ability of TSG101 and p21 to suppress differentiation is mediated by specific modulation of gene expression. In fact, both p21 (26, 27) and TSG101 (28, 29) have been implicated in transcription control by interaction with the coactivator p300, and they may be part of same complexes.
In proliferating keratinocytes, increased TSG101 expression does not affect p21 levels but causes significant growth inhibition that is largely p21-dependent. Our data are consistent with a model whereby p21 is required for the efficient recruitment of TSG101 to cyclin/CDK complexes in a manner that parallels the previously demonstrated ability of p21 to promote cyclin D/CDK4 complex formation (30). As for these complexes (31), the requirement of p21 for TSG101/cyclin/CDK association is not absolute, because such association was observed at high TSG101 expression levels even in the absence of p21 expression. One possible compensatory mechanism is provided by the association of TSG101 with p27, which we found to be increased in the cyclin/CDK complexes recovered from p21−/− keratinocytes. Although a single molecule of p21 can suppress cyclin E/CDK2 complex activity effectively, the on/off rate of p21 is such that at any given time a fraction of these complexes is free of p21 and active (32). Detailed in vitro binding studies will be required to determine whether this dynamic equilibrium is under TSG101 control.
Stable NIH 3T3 transformants showing either TSG101 deficiency or overproduction exhibit a neoplastic phenotype (7), and loss of the TSG101 gene during embryonal development results in the cessation of proliferation at embryonic day 6.5 (11). These complex effects may be attributable, at least in part, to a secondary chain of events triggered by altered TSG101 function. We have provided here an initial analysis of the “acute” effects of suppression of TSG101 expression in growth of normal primary cells. Consistent with the effects of increased TSG101 expression, suppression of endogenous TSG101 expression caused a doubling of the fraction of keratinocytes in the S phase of the cell cycle with a corresponding increase of p105-Rb phosphorylation. Absence of p21 results in a similar increase of cycling keratinocytes (4) and, in vivo, in increased sensitivity to skin carcinogenesis (5, 33, 34). Increased incidence of mammary carcinomas in p21-deficient mice has been reported also (35). An attractive possibility for future studies is that TSG101 may participate in at least some of these p21 tumor-suppressive functions.
Acknowledgments
We are grateful to Ms. A. Rajadurai for skillful technical help and Drs. C. Brisken and C. Missero for critical reading of the manuscript. This work was supported in part by National Institutes of Health Grants AR39190, CA16038, and CA73796 (to G.P.D.) and the Cutaneous Biology Research Center through the Massachusetts General Hospital/Shiseido Company agreement. S.N.C. received support from a Helmut Horten Foundation Award from the California Breast Cancer Research Program and from the National Foundation for Cancer Research.
Abbreviations
- CDK
cyclin-dependent kinase
- CKI
CDK inhibitor
- PCNA
proliferating cell nuclear antigen
- GFP
green fluorescent protein
- RFP
red fluorescent protein
- MOI
multiplicity/multiplicities of infection
- Ad
adenovirus
References
- 1.Sherr C J, Roberts J M. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 2.Dotto G P. Biochim Biophys Acta. 2000;1471:M43–M56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 3.Missero C, Calautti E, Eckner R, Chin J, Tsai L H, Livingston D M, Dotto G P. Proc Natl Acad Sci USA. 1995;92:5451–5455. doi: 10.1073/pnas.92.12.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Missero C, Di Cunto F, Kiyokawa H, Koff A, Dotto G P. Genes Dev. 1996;10:3065–3075. doi: 10.1101/gad.10.23.3065. [DOI] [PubMed] [Google Scholar]
- 5.Topley G I, Okuyama R, Gonzales J G, Conti C, Dotto G P. Proc Natl Acad Sci USA. 1999;96:9089–9094. doi: 10.1073/pnas.96.16.9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Cunto F, Topley G, Calautti E, Hsiao J, Ong L, Seth P K, Dotto G P. Science. 1998;280:1069–1072. doi: 10.1126/science.280.5366.1069. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Cohen S N. Cell. 1996;85:319–329. doi: 10.1016/s0092-8674(00)81111-3. [DOI] [PubMed] [Google Scholar]
- 8.Lee M P, Feinberg A P. Cancer Res. 1997;57:3131–3134. [PubMed] [Google Scholar]
- 9.Gayther S A, Barski P, Batley S J, Li L, de Foy K A, Cohen S N, Ponder B A, Caldas C. Oncogene. 1997;15:2119–2126. doi: 10.1038/sj.onc.1201591. [DOI] [PubMed] [Google Scholar]
- 10.Oh Y, Proctor M L, Fan Y H, Su L K, Hong W K, Fong K M, Sekido Y S, Gazdar A F, Minna J D, Mao L. Oncogene. 1998;17:1141–1148. doi: 10.1038/sj.onc.1202029. [DOI] [PubMed] [Google Scholar]
- 11.Ruland J, Sirard C, Elia A, MacPherson D, Wakeham A, Li L, Luis De La Pompa J, Cohen S N, Mak T W. Proc Natl Acad Sci USA. 2001;98:1859–1864. doi: 10.1073/pnas.98.4.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng G H, Lih C J, Cohen S N. Cancer Res. 2000;60:1736–1741. [PubMed] [Google Scholar]
- 13.Xie W, Li L, Cohen S N. Proc Natl Acad Sci USA. 1998;95:1595–1600. doi: 10.1073/pnas.95.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koonin E V, Abagyan R A. Nat Genet. 1997;16:330–331. doi: 10.1038/ng0897-330. [DOI] [PubMed] [Google Scholar]
- 15.Bishop N, Woodman P. J Biol Chem. 2001;276:11735–11742. doi: 10.1074/jbc.M009863200. [DOI] [PubMed] [Google Scholar]
- 16.Katzmann D J, Babst M, Emr S D. Cell. 2001;106:145–155. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- 17.Garrus J E, von Schwedler U K, Pornillos O W, Morham S G, Zavitz K H, Wang H E, Wettstein D A, Stray K M, Cote M, Rich R L, Myszka D G, Sundquist W I. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Liao J, Ruland J, Mak T W, Cohen S N. Proc Natl Acad Sci USA. 2001;98:1619–1624. doi: 10.1073/pnas.98.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vidal M, Brachmann R K, Fattaey A, Harlow E, Boeke J D. Proc Natl Acad Sci USA. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He T C, Zhou S, da Costa L T, Yu J, Kinzler K W, Vogelstein B. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dotto G P. Crit Rev Oral Biol Med. 1999;10:442–457. doi: 10.1177/10454411990100040201. [DOI] [PubMed] [Google Scholar]
- 22.Maki C G, Howley P M. Mol Cell Biol. 1997;17:355–363. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheaff R J, Singer J D, Swanger J, Smitherman M, Roberts J M, Clurman B E. Mol Cell. 2000;5:403–410. doi: 10.1016/s1097-2765(00)80435-9. [DOI] [PubMed] [Google Scholar]
- 24.Hatakeyama M, Weinberg R A. Prog Cell Cycle Res. 1995;1:9–19. doi: 10.1007/978-1-4615-1809-9_2. [DOI] [PubMed] [Google Scholar]
- 25.Touitou R, Richardson J, Bose S, Nakanishi M, Rivett J, Allday M J. EMBO J. 2001;20:2367–2375. doi: 10.1093/emboj/20.10.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkins N D, Felzien L K, Betts J C, Leung K, Beach D H, Nabel G J. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 27.Snowden A W, Anderson L A, Webster G A, Perkins N D. Mol Cell Biol. 2000;20:2676–2686. doi: 10.1128/mcb.20.8.2676-2686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe M, Yanagi Y, Masuhiro Y, Yano T, Yoshikawa H, Yanagisawa J, Kato S. Biochem Biophys Res Commun. 1998;245:900–905. doi: 10.1006/bbrc.1998.8547. [DOI] [PubMed] [Google Scholar]
- 29.Sun Z, Pan J, Hope W X, Cohen S N, Balk S P. Cancer. 1999;86:689–696. doi: 10.1002/(sici)1097-0142(19990815)86:4<689::aid-cncr19>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 30.LaBaer J, Garrett M D, Stevenson L F, Slingerland J M, Sandhu C, Chou H S, Fattaey A, Harlow E. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 31.Cheng M, Olivier P, Diehl J A, Fero M, Roussel M F, Roberts J M, Sherr C J. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hengst L, Gopfert U, Lashuel H A, Reed S I. Genes Dev. 1998;12:3882–3888. doi: 10.1101/gad.12.24.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Philipp J, Vo K, Gurley K E, Seidel K, Kemp C J. Oncogene. 1999;18:4689–4698. doi: 10.1038/sj.onc.1202840. [DOI] [PubMed] [Google Scholar]
- 34.Weinberg W C, Fernandez-Salas E, Morgan D L, Shalizi A, Mirosh E, Stanulis E, Deng C, Hennings H, Yuspa S H. Cancer Res. 1999;59:2050–2054. [PubMed] [Google Scholar]
- 35.Adnane J, Jackson R J, Nicosia S V, Cantor A B, Pledger W J, Sebti S M. Oncogene. 2000;19:5338–5347. doi: 10.1038/sj.onc.1203956. [DOI] [PubMed] [Google Scholar]