

Abstract
In budding yeast, exit from mitosis is achieved by inactivation of
Cdc28/Clb2 activity. Although it is not clear at present how
mitotic exit is triggered, a growing body of evidence suggests that the
Tem1 GTPase plays a critical role in mediating this pathway and that
Bfa1 and Bub2 constitute a two-component GTPase-activating protein to
negatively regulate Tem1. Here, we have demonstrated that introduction
of bfa1Δ suppresses the growth defects associated with
the cdc5–1 mutation significantly better than that of
bub2Δ, suggesting that Bfa1 may have a previously
uncharacterized role in this pathway. Overexpression of
BFA1 efficiently arrested the cell cycle at postanaphase
even in the absence of BUB2, whereas overexpression of
BUB2 weakly induced mitotic arrest only in the presence
of BFA1. Coimmunoprecipitation and in
vitro binding studies indicate that Bfa1 binds strongly to Tem1
independently of Bub2. Provision of GDP+AlF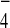 , which
mimics the GTPase transition state, enhanced the Bub2-Tem1 interaction
both in vitro and in vivo. Interestingly,
introduction of bfa1Δ, but not bub2Δ,
greatly increased the interaction between Tem1 and Cdc15, a step that
is thought to be critical for activating the mitotic exit network. Our
data suggest that, in addition to its role as a putative, two-component
GTPase-activating protein with Bub2, Bfa1 also can play a role in the
regulation of mitotic exit by directly inhibiting the interaction
between Tem1 and Cdc15 even in the absence of Bub2.
, which
mimics the GTPase transition state, enhanced the Bub2-Tem1 interaction
both in vitro and in vivo. Interestingly,
introduction of bfa1Δ, but not bub2Δ,
greatly increased the interaction between Tem1 and Cdc15, a step that
is thought to be critical for activating the mitotic exit network. Our
data suggest that, in addition to its role as a putative, two-component
GTPase-activating protein with Bub2, Bfa1 also can play a role in the
regulation of mitotic exit by directly inhibiting the interaction
between Tem1 and Cdc15 even in the absence of Bub2.
Keywords: Cdc15 ‖two-component GAP‖budding yeast
In budding yeast, entry into mitosis is achieved by activation of the mitotic cyclin-dependent kinase Cdc28, whereas exit from mitosis requires Cdc28 inactivation. It is widely appreciated that mitotic exit is a prerequisite step to initiate cytokinesis. Thus, regulation of mitotic exit is critical to ensure precise transmission of genetic and cytosolic materials into two dividing cells. Although it is not clear at the molecular level how exit from mitosis is triggered, various genetic and biochemical analyses have suggested that the small GTPase Tem1 likely may regulate Cdc15 (1, 2), which, in turn, phosphorylates and activates the Dbf2/Mob1 protein complex (3, 4). Activation of Tem1 ultimately leads to the release of Cdc14 from the nucleolus (5–7), which inactivates Cdc28 activity by dephosphorylating the Cdh1/Hct1 of anaphase-promoting complex (APC) to stimulate APC-dependent degradation of mitotic cyclins. In addition, Cdc14 is also shown to dephosphorylate and stabilize the Cdc28 inhibitor, Sic1, and its transcription factor, Swi5 (8). Dephosphorylation of Swi5 has been shown to be important for translocation of Swi5 into the nucleus, where it can promote induction of SIC1 (9, 10).
Both genetic and sequence database analyses suggest that Tem1, Lte1, Bfa1, and Bub2 may function at the proximal end of this signaling pathway (11). Lte1 encodes a putative GTP/GDP exchange factor (12). Bfa1 and Bub2 are closely related to Byr4 and Cdc16, respectively, in Schizosaccharomyces pombe, which constitutes a two-component GTPase-activating protein (GAP) for the Spg1 GTPase (13), suggesting that Bfa1 and Bub2 also may form a two-component GAP for the Tem1 GTPase. Both Bub2 and Cdc16 are similar to GAPs for Ypt GTPases in yeast and Rab GTPases in mammals, whereas Bfa1 and Byr4 are not related to any known proteins. Bfa1 and Bub2 bind Tem1 in vivo at the cytoplasmic side of spindle pole body (SPB), whereas the Lte1 is associated with the cortex of the bud (14). This spatial separation was suggested to be an important mechanism of preventing untimely activation of mitotic exit in the absence of nuclear division (7, 14). Bfa1 and Bub2 participate in the spindle-position checkpoint by monitoring the migration of the SPB into the bud (7, 14, 15). In addition, recent reports suggest that the Bub2 pathway monitors defects in cytoplasmic microtubule structures (16) and that Bfa1 and Bub2 may function as a universal checkpoint in response to various defects such as DNA damage or spindle misorientation (17). Thus, negative regulation of mitotic exit by Bfa1 and Bub2 is likely to be important to prevent Tem1-mediated mitotic exit before completion of anaphase B.
In this report, we show that Bfa1 directly binds to Tem1 and regulates the Tem1–Cdc15 interaction even in the absence of Bub2. Together with its suggested role as a two-component GAP with Bub2 (2, 17, 18), our data suggest that Bfa1 may play a dual role as an important negative regulator of mitotic exit by participating in both Bub2-independent regulation of Tem1–Cdc15 interaction and Bub2-dependent regulation of the Tem1 GTPase activity.
Materials and Methods
Plasmid Construction.
To generate the pET21b-BFA1 (pKL1151) and pET21b-BUB2 (pKL1149) plasmids, a DNA fragment containing the entire ORF of each gene was PCR-amplified by using strain S288C genomic DNA as a template. The obtained fragments were digested with either BamHI and SalI for BFA1 or EcoRI and XhoI for BUB2 (the two enzyme sites for each ORF included at the 5′ and 3′ primers flanking the coding sequence of each gene) and then inserted between the corresponding sites in pET21b bacterial expression vector (Novagen). Both pET21b-BFA1 and pET21b-BUB2 constructs express recombinant proteins fused with both N-terminal T7 and C-terminal 6×His epitope tags. The entire ORFs of BFA1 and BUB2 were sequenced and confirmed to be identical to those in the Saccharomyces Genome Database (Stanford University, Stanford, CA). GST-Tem1 (19) was kindly provided by A. Toh-E (University of Tokyo). To express BFA1 or BUB2 under the control of GAL1 promoter, the DNA fragments obtained from pKL1151 or pKL1149 by digesting with XbaI and PstI were ligated with a YCp33-derived integration vector. The resulting plasmids, pKL1313 and pKL1315, were digested with either EcoRV or NcoI to achieve targeted integration of GAL1-T7-BFA1–6His or GAL1-T7-BUB2–6His at the URA3 locus.
Strain Construction.
All yeast strains used in this study are in a W303–1A genetic background. Strains obtained from other sources were crossed with a wild-type W303–1A at least four times to convert them into the W303–1A genetic background. Null strains generated by one-step gene disruption were confirmed by PCR or Southern hybridization analyses (data not shown). Yeast cell cultures and transformations were carried out by standard methods (20). A bfa1Δ∷his5+ mutation was introduced into strain KLY1546 by a one-step gene-disruption method described previously (21). Strains expressing BFA1-HA∷his5+ or BUB2-HA∷his5+ all were generated as above with the pFA6a-3HA-his5+MX6 module (21). Strain KLY2612 expressing CDC15-HA was generated by a targeted integration at the LEU2 locus with pRS305-CDC15-HA construct (a gift of D. O. Morgan, Univ. of California, San Francisco). To express either GAL1-T7-BFA1–6His or GAL1-T7-BUB2–6His, cells were cultured under induction conditions, as described (22), and then fixed with 3.7% formaldehyde at the indicated time points. DNA was stained with 4′,6′-diamidino-2-phenylindole and observed under a fluorescent microscope.
Preparation of Recombinant Proteins and in Vitro Protein–Protein Interactions.
Recombinant T7-Bfa1–6His, T7-Bub2–6His proteins were expressed in
Escherichia coli strain BL21 DE3 and partially purified with
Ni-NTA column (Qiagen) according to the manufacturer's protocol. GST
(glutathionine S-transferase) or GST-Tem1 (a gift of A.
Toh-E) bound to glutathione-Sepharose beads was incubated with
recombinant Bfa1 or Bub2 proteins at 4°C for 1 h in PBS
supplemented with 5 mM MgCl2 + 0.05% Nonidet
P-40. GDP and guanosine 5′-[γ-thio]triphosphate (GTP[γS]) were
supplemented at the final concentration of 100 μM, whereas aluminum
fluoride (AlF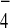 ) (Sigma) was supplemented at 110 μM.
The resin was washed with the binding buffer five times, and bound
proteins were eluted with SDS-sample buffer and analyzed by
immunoblotting with an anti-T7 antibody (Novagen) and an anti-GST
antibody (CLONTECH).
) (Sigma) was supplemented at 110 μM.
The resin was washed with the binding buffer five times, and bound
proteins were eluted with SDS-sample buffer and analyzed by
immunoblotting with an anti-T7 antibody (Novagen) and an anti-GST
antibody (CLONTECH).
Immunoprecipitation and Immunoblot Analyses.
Yeast cells were lysed in TED buffer [40 mM Tris⋅Cl, pH 7.5/0.25 mM EDTA/1 mM DTT/1 mM AEBSF (Pefabloc; Boehringer Mannheim)/10 μg/ml pepstatin A/10 μg/ml leupeptin/10 μg/ml aprotinin] with an equal volume of glass beads (Sigma). The obtained lysates were spun at 2,000 × g for 2 min to remove unbroken cells and beads. The resulting supernatants were considered total cellular lysates. For immunoprecipitation, total cellular lysates were subjected to further centrifugation at 15,000 × g for 30 min to further clarify the extracts. The resulting supernatants (S15) were diluted to 1 ml with TBSN buffer [20 mM Tris⋅Cl, pH 8.0/150 mM NaCl/0.5% Nonidet P-40/5 mM EGTA/1.5 mM EDTA/0.5 mM Na3VO4/20 mM p-nitrophenyl phosphate (PNPP)] before incubation with an anti-myc antibody (Zymed) and a control mouse IgG (Sigma). Protein G-Sepharose 4B (Zymed) was added, and incubation was continued for an additional 1 h to precipitate the antibody. Immunoblot analyses were carried out with anti-HA, anti-GST (CLONTECH), and anti-T7 antibodies (Novagen), as described (23).
Results
Loss of BFA1 or BUB2 Alleviates Mitotic Exit Defect Associated with the cdc5–1 Mutation.
Although it is not known whether Bfa1 and Bub2 directly regulate the activity of the Tem1 GTPase, various genetic data suggest that they are likely to be involved in the mitotic exit pathway (7, 14, 17, 24, 25). To directly examine whether Bfa1 and Bub2 genetically interact with other components in the pathway, we introduced either bfa1Δ or bub2Δ into the temperature-sensitive cdc5–1 mutant, which has been shown to be defective in mitotic exit at restrictive temperature (26, 27). The cdc5–1 bfa1Δ or cdc5–1 bub2Δ double mutants grew well at 35°C, whereas the cdc5–1 mutant did not (Fig. 1A). Examination of these mutants under various temperatures revealed that the cdc5–1 bfa1Δ mutant grew better than the cdc5–1 bub2Δ mutant. The cdc5–1 bfa1Δ bub2Δ triple mutants grew at a degree similar to the cdc5–1 bfa1Δ double mutant (Fig. 1A). To confirm the growth differences between the cdc5–1 bfa1Δ and the cdc5–1 bub2Δ mutants at elevated temperatures, we transformed the cdc5–1 bfa1Δ bub2Δ triple mutants with a centromeric plasmid containing BFA1 or BUB2. Consistent with the above observation, provision of BFA1 inhibited the growth of the triple mutant more strongly than that of BUB2 (Fig. 1B). As expected, provision of both the BFA1 and BUB2 plasmids resulted in a more robust, temperature-sensitive growth defect under various conditions examined (Fig. 1B). These data indicate that Bfa1 is a more potent inhibitor of the mitotic exit defect associated with the cdc5–1 mutation than is Bub2. This observation further suggests that, in addition to their proposed role as a two-component GAP with Bub2, Bfa1 also may have an additional role in regulating exit from mitosis. In contrast, introduction of either bfa1Δ or bub2Δ into the cdc16–1 mutant failed to suppress its growth defect at the nonpermissive temperature (data not shown). Because Cdc16 is a component of APC and has an essential role in the metaphase-to-anaphase transition, these observations support the notion that Bfa1 and Bub2 specifically inhibit mitotic exit.
Figure 1.

(A) Introduction of a bfa1Δ suppresses the temperature-sensitive cdc5–1 growth defect better than that of a bub2Δ. Various mutant strains cultured overnight were serially diluted and spotted onto YEP-glucose plates and then incubated for 2 days at the indicated temperatures. 1, KLY2156 (cdc5–1); 2, KLY2454 (cdc5–1 bfa1Δ); 3, KLY2398 (cdc5–1 bub2Δ); 4, KLY2630 (cdc5–1 bfa1Δ bub2Δ). (B) To confirm the better growth phenotype of the cdc5–1 mutant by bfa1Δ than by bub2Δ, strain KLY2630 (cdc5–1 bfa1Δ bub2Δ) was transformed with a centromeric BFA1 or BUB2 plasmid or with both. Transformants were cultured in appropriate selective liquid medium overnight, serially diluted, and spotted onto SDM-URA (pRS316 vector, pRS316-BUB2), SDM-LEU (pRS315-BFA1), or SDM-LEU-URA (pRS315-BFA1 + pRS316-BUB2) plates. Plates were incubated at the indicated temperatures for 3 days. 1, KLY2630 + pRS316 vector; 2, KLY2630 + pRS315-BFA1; 3, KLY2630 + pRS316-BUB2; 4, KLY2630 + pRS315-BFA1+ pRS316-BUB2.
Overexpression of Bfa1, but Not Bub2, Results in a Potent Inhibition of Mitotic Exit.
To investigate whether Bfa1 and Bub2 are mutually required for their mitotic functions, we examined the effect of BFA1 or BUB2 overexpression in the presence or absence of BUB2 or BFA1, respectively. To this end, GAL1-T7-BFA1 or GAL1-T7-BUB2 was stably integrated into the genome of various TEM1-myc strains and then expressed upon shifting the cultures to YEP-galactose medium. All of the strains grew well in the absence of BFA1 or BUB2 expression on YEP-glucose medium. In the presence (KLY2243) or absence (KLY3032) of BUB2, cells overexpressing BFA1 did not grow and exhibited a large-budded morphology with fully elongated spindles (Fig. 2A and data not shown). However, overexpression of BUB2 partially inhibited cell growth only in the presence of BFA1 (Fig. 2A) and induced a chained cell phenotype (data not shown), suggestive of a defect in cytokinesis. In a wild-type TEM1 background, the GAL1-T7-BUB2-dependent growth inhibition was negligible even in the presence of BFA1, whereas the GAL1-T7-BFA1-dependent inhibition remained unchanged even in the absence of BUB2 (data not shown). The complete inhibition of cell growth achieved by GAL1-BFA1 appeared to be a result of a direct inhibition of the mitotic exit pathway, because provision of multiple copies of TEM1 or CDC15 into either KLY2243 or KLY3032 fully reversed this phenotype. In addition, provision of a dominant allele of CDC14 (CDC14TAB6–1), which already is known to bypass the requirement of TEM1 function (28), efficiently cured this growth defect (Fig. 2B). However, provision of multiple copies of CDC5 did not rescue this defect significantly (Fig. 2B). A recent report suggested that Cdc5 functions upstream of Bfa1 and contributes to the phosphorylation of the latter (29). Thus, our observation suggests that overexpression of CDC5 was not likely to be sufficient to overcome the mitotic exit inhibition imposed by GAL1-BFA1 expression.
Figure 2.

Overexpression of BFA1 under GAL1 promoter control induces a drastic mitotic arrest. (A) Wild-type and various mutant cells bearing either GAL1-T7-BFA1–6His or GAL1-T7-BUB2–6His were streaked onto either YEP-glucose or YEP-galactose plate as indicated and incubated for 3 days. Overexpression of BFA1 completely inhibited the cell growth, whereas overexpression of BUB2 inhibited it modestly only in the presence of Bfa1. 1, KLY2170 (TEM1-myc); 2, KLY2243 (TEM1-myc GAL1-BFA1); 3, KLY3032 (TEM1-myc GAL1-BFA1 bub2Δ); 4, KLY2244 (TEM1-myc GAL1-BUB2); 5, KLY3044 (TEM1-myc GAL1-BUB2 bfa1Δ). (B) Reversal of the BFA1-induced mitotic arrest by overexpression of TEM1 and CDC15, but not by CDC5. Strain KLY2243 (GAL1-BFA1) was transformed with various constructs. The obtained transformants were streaked onto YEP-galactose to examine their cell growth. Wild-type cells were also cultured as a comparison for cell growth under these conditions. 1, Wild-type KLY1546; 2, KLY2243 (TEM1-myc GAL1-BFA1) transformed with pRS424-TEM1; 3, KLY2243 transformed with pRS424-CDC15; 4, KLY2243 transformed with pRS424-CDC14TAB6–1 (25); 5, KLY2243 transformed with YEp351-CDC5; 6, KLY2243 transformed with pRS424 vector. (C) To closely monitor the arresting phenotype upon induction of either GAL1-BFA1 or GAL1-BUB2, samples were taken at the indicated time points after shifting the cultures to YEP-galactose medium. The fixed cells were subjected to DNA staining with 4′,6′-diamidino-2-phenylindole and then observed under a fluorescent microscope to determine the fraction of cells with separated sister chromatids. (Inset) The levels of either Bfa1 or Bub2 protein expressed under the induction conditions were determined by immunoblotting with an anti-T7 antibody. 1 and ▴, KLY2170 (TEM1-myc); 2 and ■, KLY2243 (TEM1-myc GAL1-BFA1); 3 and □, KLY3032 (TEM1-myc GAL1-BFA1 bub2Δ); 4 and ●, KLY2244 (TEM1-myc GAL1-BUB2); 5 and ○, KLY3044 (TEM1-myc GAL1-BUB2 bfa1Δ).
To quantify the efficiency of mitotic arrest achieved by GAL1-BFA1 or GAL1-BUB2 overexpression, various TEM1-myc strains were harvested at the indicated time points after induction and the fraction of cells arrested at postanaphase were determined. Approximately 99% of cells (n = 310) were arrested with a postanaphase nuclei morphology after inducing GAL1-BFA1 for 3 h (Fig. 2C). In contrast, overexpression of GAL1-BUB2 induced a postanaphase arrest with much slower kinetics than that observed with GAL1-BFA1 (Fig. 2C). Interestingly, loss of BUB2 did not influence the inhibitory capacity of GAL1-BFA1, whereas loss of BFA1 abolished the mild inhibitory capacity of GAL1-BUB2 (Fig. 2 A and C). These observations suggest that Bfa1 does not require Bub2 to induce mitotic inhibition, whereas Bub2 requires Bfa1 to achieve it. Under these conditions, both GAL1-BFA1 and GAL1-BUB2 were expressed at similar levels (Fig. 2C Inset).
Bfa1 Interacts with Tem1 Independently of Bub2 both in Vivo and in Vitro.
Previous studies have shown a physical interaction between Bfa1 and
Bub2 with Tem1 (14, 29) but failed to address the binding nature of
Bfa1 and Bub2 to Tem1. The strong inhibition of mitotic exit achieved
by GAL1-BFA1 in the absence of BUB2 suggests that
Bfa1 may bind directly to Tem1 and participate in regulation of mitotic
exit without Bub2. To investigate this possibility and to determine the
relative binding affinities of Bfa1 and Bub2 to Tem1, strain KLY2705,
which expresses Bfa1-HA, Bub2-HA, and Tem1-myc under
endogenous promoter controls, was used. Consistent with
previous observations (14, 29), immunoprecipitation of Tem1-myc with
anti-myc antibody, but not with a mouse IgG control,
coimmunoprecipitated both Bfa1-HA and Bub2-HA. However, the amount of
Bfa1 associating with Tem1 was reproducibly severalfold greater than
that of Bub2 (Fig. 3A). GAPs
function by stabilizing the GTPase transition state.
AlF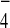 has been shown to promote formation of the
GTPase transition state by mimicking the γ-phosphate of GTP (30).
Several classes of small GTPases can stably interact with their
respective GAPs by forming a high-affinity complex with GDP-bound
GTPases in the presence of AlF
has been shown to promote formation of the
GTPase transition state by mimicking the γ-phosphate of GTP (30).
Several classes of small GTPases can stably interact with their
respective GAPs by forming a high-affinity complex with GDP-bound
GTPases in the presence of AlF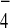 (31–33). Thus, we
examined possible interactions between Tem1 and Bfa1 or Tem1 and Bub2
in the presence of GDP+AlF
(31–33). Thus, we
examined possible interactions between Tem1 and Bfa1 or Tem1 and Bub2
in the presence of GDP+AlF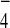 . Provision of
GDP+AlF
. Provision of
GDP+AlF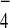 reproducibly enhanced interaction between
Tem1 and Bub2 severalfold, whereas it decreased the interaction between
Tem1 and Bfa1 under the same conditions (Fig. 3B). In
contrast, provision of GDP alone did not influence these interactions
(Fig. 3B). These data suggest that Bub2 binding to Tem1 is
favored in the transition state of the Tem1 GTPase.
reproducibly enhanced interaction between
Tem1 and Bub2 severalfold, whereas it decreased the interaction between
Tem1 and Bfa1 under the same conditions (Fig. 3B). In
contrast, provision of GDP alone did not influence these interactions
(Fig. 3B). These data suggest that Bub2 binding to Tem1 is
favored in the transition state of the Tem1 GTPase.
Figure 3.

Differential interactions of Bfa1 and Bub2 to Tem1. (A)
Bfa1 interacts tightly with Tem1 in vivo. To determine
their relative affinities to Tem1-myc, both BFA1 and
BUB2 were C-terminally tagged with a HA epitope and
expressed under endogenous promoter control. Associated
proteins in anti-myc immunoprecipitates were separated by
SDS/PAGE. After the proteins were transferred onto
polyvinylidene difluoride membrane, proteins interacting with anti-HA
or anti-myc antibodies were detected by immunoblotting. Recombinant HA
proteins were detected with anti-HA immunoblotting, whereas Tem1-myc
was detected with an anti-myc antibody. Immunoprecipitation of Tem1-myc
with an anti-myc antibody from S15 fractions coprecipitated Bfa1-HA and
weakly coprecipitated Bub2-HA. Asterisks indicate a nonspecific,
cross-reacting protein with an anti-HA antibody. BFA1-HA
BUB2-HA, strain KLY2705; BFA1-HA, strain
KLY2701; BUB2-HA, strain KLY2703; Input, 10% of S15
subjected to an anti-myc immunoprecipitation; anti-myc, anti-myc
immunoprecipitation; control Ig, control mouse IgG immunoprecipitation.
(B) Enhanced Tem1-Bub2 interaction in the presence of
GDP+AlF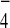 . To examine the effect of
GDP+AlF
. To examine the effect of
GDP+AlF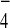 in Tem1-Bfa1-Bub2 interactions,
immunoprecipitation of Tem1-myc was carried out with S15 fraction of
strain KLY2705 in TBSN (0.1% Nonidet P-40) supplemented with either
GDP alone or GDP+AlF
in Tem1-Bfa1-Bub2 interactions,
immunoprecipitation of Tem1-myc was carried out with S15 fraction of
strain KLY2705 in TBSN (0.1% Nonidet P-40) supplemented with either
GDP alone or GDP+AlF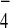 . Coprecipitated Bfa1-HA and
Bub2-HA were detected with an anti-HA antibody. To determine the amount
of Tem1-myc immunoprecipitated, the same membrane was subjected to
anti-myc immunoblotting. Composites are assembled from the same
exposure. Anti-myc, anti-myc immunoprecipitation; control Ig, control
mouse IgG immunoprecipitation; Input, 5% of S15. (C)
Interaction between Bfa1 and Tem1 does not require Bub2. To determine
association of Bfa1-HA and Bub2-HA with Tem1-myc, anti-myc
immunoprecipitation was carried out with S15 fractions prepared from
various strains under the same conditions as in A.
BFA1-HA BUB2-HA, strain KLY2705; BFA1-HA,
strain KLY2701; BFA1-HA bub2Δ, strain KLY3061;
BUB2-HA, strain KLY2703; BUB2-HA bfa1Δ,
strain KLY3065.
. Coprecipitated Bfa1-HA and
Bub2-HA were detected with an anti-HA antibody. To determine the amount
of Tem1-myc immunoprecipitated, the same membrane was subjected to
anti-myc immunoblotting. Composites are assembled from the same
exposure. Anti-myc, anti-myc immunoprecipitation; control Ig, control
mouse IgG immunoprecipitation; Input, 5% of S15. (C)
Interaction between Bfa1 and Tem1 does not require Bub2. To determine
association of Bfa1-HA and Bub2-HA with Tem1-myc, anti-myc
immunoprecipitation was carried out with S15 fractions prepared from
various strains under the same conditions as in A.
BFA1-HA BUB2-HA, strain KLY2705; BFA1-HA,
strain KLY2701; BFA1-HA bub2Δ, strain KLY3061;
BUB2-HA, strain KLY2703; BUB2-HA bfa1Δ,
strain KLY3065.
The observed differential binding of Bfa1 and Bub2 to Tem1 under various conditions suggests that formation of Tem1-Bfa1-Bub2 complex is likely to be dynamic in vivo. Thus, we examined whether the interactions between Tem1 and Bfa1 or Tem1 and Bub2 required Bub2 or Bfa1, respectively. In exponentially growing cells, introduction of bub2Δ did not influence the level of Bfa1 associating with Tem1, suggesting that Bub2 is not required for the Bfa1-Tem1 interaction under these conditions (Fig. 3C, lane 3). Introduction of bfa1Δ slightly decreased the level of Bub2 coprecipitating with Tem1 but did not abolish this interaction (Fig. 3C, lane 5). This weak Bub2-Tem1 interaction in the bfa1Δ background was detected reproducibly in Tem1-myc but not in control IgG immunoprecipitations (data not shown). These observations suggest that a fraction of the Bub2-Tem1 interaction may occur independently of Bfa1. The loss of BFA1 or BUB2 function did not influence the expression levels of Bub2 and Bfa1, respectively (Fig. 3C).
To investigate further the binding nature of Bfa1 and Bub2 to Tem1,
in vitro binding studies were carried out by using GST-Tem1
as a ligand. Bfa1 tightly binds to GST-Tem1 under various conditions
examined (Fig. 4). However, unlike the
two-component Byr4/Cdc16 of S. pombe, in which
binding of Cdc16 to Spg1 depends on the presence of Byr4 (34), Bub2
also directly binds to Tem1 under these conditions. Consistent with the
in vivo coimmunoprecipitation experiments, provision of
GDP+AlF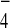 , but not GDP or GTP[γS], significantly
increased the interaction between Tem1 and Bub2. Because the major
function of GAP proteins is to stabilize the transition state of their
respective GTPases, these data strongly suggest that Bub2 is the GAP
for the Tem1 GTPase. In addition, provision of both Bfa1 and Bub2
appeared to enhance the Tem1-Bfa1 and Tem1-Bub2 interactions (Fig. 4),
suggesting that Tem1, Bfa1, and Bub2 may form a heterotrimeric complex.
This observation further suggests that the interactions among these
proteins are not mutually exclusive in vitro. Neither Bfa1
nor Bub2 interacted with GST under these conditions (control lane).
However, unlike in vivo coimmunoprecipitation experiments,
provision of GDP+AlF
, but not GDP or GTP[γS], significantly
increased the interaction between Tem1 and Bub2. Because the major
function of GAP proteins is to stabilize the transition state of their
respective GTPases, these data strongly suggest that Bub2 is the GAP
for the Tem1 GTPase. In addition, provision of both Bfa1 and Bub2
appeared to enhance the Tem1-Bfa1 and Tem1-Bub2 interactions (Fig. 4),
suggesting that Tem1, Bfa1, and Bub2 may form a heterotrimeric complex.
This observation further suggests that the interactions among these
proteins are not mutually exclusive in vitro. Neither Bfa1
nor Bub2 interacted with GST under these conditions (control lane).
However, unlike in vivo coimmunoprecipitation experiments,
provision of GDP+AlF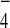 did not diminish the
interaction between Tem1 and Bfa1. Whether this apparent discrepancy is
due to the presence of other components in the mitotic exit network
in vivo, such as Lte1, is not yet understood.
did not diminish the
interaction between Tem1 and Bfa1. Whether this apparent discrepancy is
due to the presence of other components in the mitotic exit network
in vivo, such as Lte1, is not yet understood.
Figure 4.

Bfa1 or Bub2 directly interacts with Tem1 in vitro. To
investigate interactions among recombinant Bfa1, Bub2, and Tem1
proteins under various conditions, partially purified 0.5 μg each of
T7-Bfa1 and T7-Bub2 proteins was added into reaction tubes containing
either bead-bound GST or GST-Tem1, which is supplemented with GDP,
GTP[γS], or GDP + AlF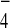 . Proteins associating with
either GST-Tem1 or GST were precipitated and detected by immunoblotting
with an anti-T7 antibody. The same membrane subsequently was blotted
with an anti-GST antibody to detect the precipitated GST and GST-Tem1
fusion protein. Control GST blot was assembled with GST-Tem1 blot from
the same exposure, and the migration difference does not reflect the
actual mobility differences of these two proteins in the gel. Input,
15% of the proteins used in the binding assays; control, GST control
with both Bfa1 and Bub2 in the presence of GDP+AlF
. Proteins associating with
either GST-Tem1 or GST were precipitated and detected by immunoblotting
with an anti-T7 antibody. The same membrane subsequently was blotted
with an anti-GST antibody to detect the precipitated GST and GST-Tem1
fusion protein. Control GST blot was assembled with GST-Tem1 blot from
the same exposure, and the migration difference does not reflect the
actual mobility differences of these two proteins in the gel. Input,
15% of the proteins used in the binding assays; control, GST control
with both Bfa1 and Bub2 in the presence of GDP+AlF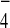 .
GDP and GTP[γS] were supplemented at the final concentration of 100
μM, whereas AlF
.
GDP and GTP[γS] were supplemented at the final concentration of 100
μM, whereas AlF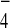 was supplemented at 110 μM.
Bfa1, T7-Bfa1-His-6 recombinant protein; Bub2, T7-Bub2-His-6
recombinant protein.
was supplemented at 110 μM.
Bfa1, T7-Bfa1-His-6 recombinant protein; Bub2, T7-Bub2-His-6
recombinant protein.
Loss of BFA1 Leads to an Enhanced Association Between Tem1 and Cdc15.
A growing body of evidence suggests that the interaction between Tem1 and Cdc15 is likely to be a critical step for relaying mitotic exit signals during the normal cell cycle (1, 2). Our data show that bfa1Δ can facilitate the mitotic exit of the cdc5–1 bub2Δ at elevated temperatures, suggesting that bfa1Δ can contribute additionally to mitotic exit in the absence of Bub2. Because Bfa1 binds tightly to Tem1 and overexpression of BFA1 can potently inhibit mitotic exit in the absence of BUB2, we reasoned that Bfa1 might directly contribute to the interaction between Tem1 and Cdc15. To test this possibility, we investigated whether introduction of either a bfa1Δ or a bub2Δ mutation influences the interaction between these two proteins. In exponentially growing cells, a small fraction of Cdc15 was associated with Tem1 immunoprecipitates. Introduction of bfa1Δ greatly enhanced the interaction between Cdc15 and Tem1, whereas introduction of bub2Δ slightly enhanced it. Deletion of both bfa1 and bub2 resulted in a similar degree of association much like the deletion of bfa1 alone (Fig. 5). These observations suggest that during the normal cell cycle, Bfa1 may exert an inhibitory role by interfering with Tem1-Cdc15 interaction, regardless of the presence of Bub2. Similar results were obtained by using the cells treated with 15 μg/ml nocodazole for 3 h (data not shown), suggesting that Tem1-Cdc15 interaction also is likely to be critical under spindle checkpoint-activation conditions. Taken together, these observations suggest that Bfa1 negatively regulates the interaction between Tem1 and Cdc15 even in the absence of Bub2.
Figure 5.

Loss of BFA1 enhances the interaction between Tem1 and Cdc15. To determine the effect of Bfa1 or Bub2 in the interaction between Cdc15-HA and Tem1-myc, Tem1-myc was immunoprecipitated from S15 fractions prepared from various strains as described in Fig. 3. Cdc15-HA and Tem1-myc proteins were detected with anti-HA and anti-myc antibodies, respectively. Asterisk denotes a cross-reacting protein with an anti-HA antibody. Lanes: 1, strain KLY2170 (TEM1-myc); 2, strain KLY2612 (CDC15-HA); 3, strain KLY2614 (TEM1-myc CDC15-HA); 4, strain KLY2623 (TEM1-myc CDC15-HA bub2Δ); 5, strain KLY2625 (TEM1-myc CDC15-HA bfa1Δ); 6, strain KLY2627 (TEM1-myc CDC15-HA bub2Δ bfa1Δ).
Discussion
In S. pombe, both genetic and biochemical studies have shown that Byr4 and Cdc16 negatively regulate the Spg1 GTPase (13), which mediates the septum formation pathway. Cdc16 binds to Spg1 and exhibits GAP activity only in the presence of Byr4 in vitro (34), suggesting that Byr4 and Cdc16 form a two-component GAP and down-regulate the Spg1 GTPase (13, 35).
Bub2 has high similarity with Cdc16 throughout the entire amino acid sequence, and Bfa1 also has a limited homology with Byr4 in the C-terminal region, suggesting that Bfa1 and Bub2 may negatively regulate the Tem1 GTPase-mediated mitotic exit network in S. cerevisiae. Our genetic analyses have shown that bfa1Δ suppresses the mitotic exit defect associated with the cdc5–1 mutation better than that of bub2Δ. However, these observations differ somewhat from the previous report in which a bfa1Δ bub2Δ double mutant exhibited a spindle checkpoint-activated cell cycle delay similar to each of the single mutants in the presence of nocodazole (25). It is possible that slower mitotic progression in the cdc5–1 mutant at elevated temperatures may have allowed us to discern differences among mutants.
It has been shown that Bfa1 and Bub2 are mutually required for their
localization at the SPB and that both Bfa1 and Bub2 are required for
Tem1 localization to the SPB during interphase and early mitosis (14).
Bfa1 and Bub2 have been shown to physically associate with each other
throughout the cell cycle and bind to Tem1 during mitosis and early
G1 (29). These data suggest that Bfa1 and Bub2
may form a heterodimeric complex that is critical for their subcellular
localization and also for the interaction with and localization of Tem1
at the SPB. However, our in vivo pull-down and in
vitro binding studies clearly have demonstrated that precipitation
of Tem1 strongly coprecipitated Bfa1, but not Bub2. Provision of
GDP+AlF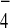 enhanced the in vivo Tem1-Bub2
interaction, whereas it decreased the Bfa1-Tem1 interaction under the
same conditions. In addition, both in vitro and in
vivo, Bfa1 or Bub2 appear to interact with Tem1 in the absence of
Bub2 or Bfa1, respectively. These observations suggest that
interactions between the Tem1-Bfa1-Bub2 proteins are likely to be
highly dynamic in vivo.
enhanced the in vivo Tem1-Bub2
interaction, whereas it decreased the Bfa1-Tem1 interaction under the
same conditions. In addition, both in vitro and in
vivo, Bfa1 or Bub2 appear to interact with Tem1 in the absence of
Bub2 or Bfa1, respectively. These observations suggest that
interactions between the Tem1-Bfa1-Bub2 proteins are likely to be
highly dynamic in vivo.
Several lines of evidence suggest that, independently of Bub2, Bfa1 may be involved directly in regulating the interaction between Tem1 and Cdc15, perhaps as a core component of the Tem1-Cdc15 complex. First, coimmunoprecipitation and in vitro binding studies revealed tight interaction between Tem1 and Bfa1 even in the bub2Δ background. Second, overexpression of BFA1 inhibited mitotic exit in a Bub2-independent manner, which was fully reversed by overexpression of Tem1 or Cdc15. This observation suggests that Bfa1 may directly prevent Tem1 from interacting with its effector(s). Third, regardless of the presence of Bub2, loss of BFA1 greatly enhanced the interaction between Tem1 and Cdc15. Thus, the observation that bfa1Δ suppresses the cdc5–1 growth defect better than bub2Δ at elevated temperatures is likely to be attributable to the additional role of Bfa1 in negatively regulating the Tem1-Cdc15 interaction.
Our data suggest that, in addition to its suggested role as a two-component GAP with Bub2, Bfa1 provides an additional step to prevent premature mitotic exit by directly binding to Tem1 and interfering with the Tem1-Cdc15 interaction. Thus, how the interaction between Tem1 and Bfa1 is regulated is likely to be a key issue as to how the mitotic exit pathway is triggered. Recent biochemical analyses suggest that Cdc5 functions upstream of Tem1 (2), similar to the ability of Plo1 to activate the Spg1 pathway in S. pombe (36). The mitotic exit defect associated with the cdc5–1, but not with the tem1–3, cdc15–2, or cdc14–1 mutation, was suppressed efficiently by the provision of the bfa1Δ or bub2Δ mutation (data not shown), suggesting that Cdc5 functions upstream of Bfa1 and Bub2 or in a parallel pathway distinct from that with Tem1, Cdc15, and Cdc14. In addition, provision of multiple copies of CDC5 did not suppress the mitotic arrest induced by overexpression of BFA1 (Fig. 2B), whereas it could efficiently rescue the arrest induced by overexpression of BUB2 (H. Ro and K.S.L., unpublished data). These data suggest that overexpression of BFA1, but not BUB2, can effectively block a Cdc5-dependent step in mitotic exit. Recently, Hu et al. (37) have demonstrated that Cdc5 directly phosphorylates Bfa1, and this phosphorylation diminishes the interaction between Tem1 and Bfa1. Thus, the dissociation of Bfa1 and also the Bfa1-dependent Bub2 fraction from Tem1 likely may be critical to augment interaction between Tem1 and Cdc15 and to activate the mitotic exit network.
Acknowledgments
We are grateful to Orna Cohen-Fix and Philip R. Lee for critical reading of this manuscript and two anonymous reviewers for suggesting valuable comments for improvement. We also thank Chong J. Park and Dan Ilkovitch for technical support, Simonetta Piatti (Biologia dei Microrganismi, Milan) for the bub2Δ strain, Sue Jaspersen and David O. Morgan (Univ. of California, San Francisco) for providing the Cdc15-HA and Cdc15-HA-7A constructs, and Akio Toh-E (Univ. of Tokyo) for providing GST-TEM1 expression construct.
Abbreviations
- APC
anaphase-promoting complex
- GAP
GTPase-activating protein
- SPB
spindle pole body
- GST
glutathionine S-transferase
References
- 1.Asakawa K, Yoshida S, Otake F, Toh-E A. Genetics. 2001;157:1437–1450. doi: 10.1093/genetics/157.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee S E, Frenz L M, Wells N J, Johnson A L, Johnston L H. Curr Biol. 2001;11:784–788. doi: 10.1016/s0960-9822(01)00228-7. [DOI] [PubMed] [Google Scholar]
- 3.Mah A S, Jang J, Deshaies R J. Proc Natl Acad Sci USA. 2001;98:7325–7330. doi: 10.1073/pnas.141098998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visintin R, Amon A. Mol Biol Cell. 2001;12:2961–2974. doi: 10.1091/mbc.12.10.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visintin R, Hwang E S, Amon A. Nature (London) 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- 6.Shou W, Seol J H, Shevchenko A, Baskerville C, Moazed D, Chen Z W, Jang J, Shevchenko A, Charbonneau H, Deshaies R J. Cell. 1999;97:233–244. doi: 10.1016/s0092-8674(00)80733-3. [DOI] [PubMed] [Google Scholar]
- 7.Bardin A J, Visintin R, Amon A. Cell. 2000;102:21–31. doi: 10.1016/s0092-8674(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 8.Visintin R, Craig K, Hwang E S, Prinz S, Tyers M, Amon A. Mol Cell. 1998;2:709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- 9.Knapp D, Bhoite L, Stillman D J, Nasmyth K. Mol Cell Biol. 1996;16:5701–5707. doi: 10.1128/mcb.16.10.5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- 11.Morgan D O. Nat Cell Biol. 1999;1:E47–E53. doi: 10.1038/10039. [DOI] [PubMed] [Google Scholar]
- 12.Shirayama M, Matsui Y, Tanaka K T, Toh-E A. Yeast. 1994;10:451–461. doi: 10.1002/yea.320100404. [DOI] [PubMed] [Google Scholar]
- 13.Furge K A, Wong K, Armstrong J, Balasubramanian M, Albright C F. Curr Biol. 1998;8:947–954. doi: 10.1016/s0960-9822(98)70394-x. [DOI] [PubMed] [Google Scholar]
- 14.Pereira G, Hofken T, Grindlay J, Manson C, Schiebel E. Mol Cell. 2000;6:1–10. [PubMed] [Google Scholar]
- 15.Fraschini R, Formenti E, Lucchini G, Piatti S. J Cell Biol. 1999;145:979–991. doi: 10.1083/jcb.145.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daum J R, Gomez-Ospina N, Winey M, Burke D J. Curr Biol. 2000;10:1375–1378. doi: 10.1016/s0960-9822(00)00780-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Hu F, Elledge S J. Curr Biol. 2000;10:1379–1382. doi: 10.1016/s0960-9822(00)00779-x. [DOI] [PubMed] [Google Scholar]
- 18.Hoyt M A. Cell. 2000;102:267–270. doi: 10.1016/s0092-8674(00)00031-3. [DOI] [PubMed] [Google Scholar]
- 19.Shirayama M, Matsui Y, Toh-E A. Mol Cell Biol. 1994;14:7476–7482. doi: 10.1128/mcb.14.11.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherman F, Fink G R, Hicks J B. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 21.Longtine M S, McKenzie A, Demarini D J, Shah N G, Wach A, Brachat A, Philippsen P, Pringle J R. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 22.Lee K S, Erikson R L. Mol Cell Biol. 1997;17:3408–3417. doi: 10.1128/mcb.17.6.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song S, Grenfell T Z, Garfield S, Erikson R L, Lee K S. Mol Cell Biol. 2000;20:286–298. doi: 10.1128/mcb.20.1.286-298.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fesquet D, Fitzpatrick P J, Johnson A L, Kramer K M, Toyn J H, Johnston L H. EMBO J. 1999;18:2424–2434. doi: 10.1093/emboj/18.9.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li R. Proc Natl Acad Sci USA. 1999;96:4989–4994. doi: 10.1073/pnas.96.9.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charles J, Jaspersen S, Tinker-Kulberg R, Hwang L, Szidon A, Morgan D. Curr Biol. 1998;8:497–507. doi: 10.1016/s0960-9822(98)70201-5. [DOI] [PubMed] [Google Scholar]
- 27.Shirayama M, Zachariae W, Ciosk R, Nasmyth K. EMBO J. 1998;17:1336–1349. doi: 10.1093/emboj/17.5.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shou W, Sakamoto K M, Keener J, Morimoto K W, Traverso E E, Azzam R, Hoppe G J, Renny-Feldman R M, DeModena J, Moazed D, et al. Mol Cell. 2001;8:45–55. doi: 10.1016/s1097-2765(01)00291-x. [DOI] [PubMed] [Google Scholar]
- 29.Lee S E, Jensen S, Frenz L M, Johnson A L, Fesquet D, Johnston L H. J Cell Sci. 2001;114:2345–2354. doi: 10.1242/jcs.114.12.2345. [DOI] [PubMed] [Google Scholar]
- 30.Bigay J, Deterre P, Pfister C, Chabre M. FEBS Lett. 1985;191:181–185. doi: 10.1016/0014-5793(85)80004-1. [DOI] [PubMed] [Google Scholar]
- 31.Ahmadian M R, Mittal R, Hall A, Wittinghofer A. FEBS Lett. 1997;408:315–318. doi: 10.1016/s0014-5793(97)00422-5. [DOI] [PubMed] [Google Scholar]
- 32.Mittal R, Ahmadian M R, Goody R S, Wittinghofer A. Science. 1996;273:115–117. doi: 10.1126/science.273.5271.115. [DOI] [PubMed] [Google Scholar]
- 33.Vincent S, Brouns M, Hart M J, Settleman J. Proc Natl Acad Sci USA. 1998;95:2210–2215. doi: 10.1073/pnas.95.5.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li C, Furge K A, Cheng Q C, Albright C F. J Biol Chem. 2000;275:14381–14387. doi: 10.1074/jbc.275.19.14381. [DOI] [PubMed] [Google Scholar]
- 35.Furge K A, Cheng Q C, Jwa M, Shin S, Song K, Albright C F. J Biol Chem. 1999;274:11339–11343. doi: 10.1074/jbc.274.16.11339. [DOI] [PubMed] [Google Scholar]
- 36.Mulvihill D P, Petersen J, Ohkura H, Glover D M, Hagan I M. Mol Biol Cell. 1999;10:2771–2785. doi: 10.1091/mbc.10.8.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu F, Wang Y, Liu D, Li Y, Qin J, Elledge S J. Cell. 2001;107:655–665. doi: 10.1016/s0092-8674(01)00580-3. [DOI] [PubMed] [Google Scholar]