

ABSTRACT
Background
We previously showed that 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD – a persistent organic pollutant) activates the aryl hydrocarbon receptor (AHR) in pancreatic islets. The AHR is known to crosstalk with hypoxia-inducible factor 1α (HIF1α) in other cell types but AHR-HIF1α crosstalk has not been previously examined in islet cells. Islet cell function is sensitive to hypoxia; we hypothesize that AHR activation by environmental pollutant(s) will interfere with the HIF1α pathway response in islets, which may be detrimental to islet cell function and survival during periods of hypoxia.
Methods
We assessed AHR-HIF1α crosstalk by treating human donor islets and stem cell-derived islets (SC-islets) with 10 nM TCDD ± 1% O2 and measuring gene expression of downstream targets of AHR (e.g. CYP1A1) and HIF1α (e.g. HMOX1).
Results
In SC-islets, co-treatment with TCDD + hypoxia consistently suppressed CYP1A1 induction compared with TCDD treatment alone. In human islets, TCDD + hypoxia co-treatment suppressed CYP1A1 induction, but only in 2 of 6 donors. Both SC-islets and human donor islets displayed hypoxia-mediated suppression of glucose-6-phosphate catalytic subunit 2 (G6PC2) expression. Glucose-stimulated insulin secretion (GSIS) in human donor islets was impaired by hypoxia exposure, but unaffected by TCDD exposure.
Conclusion
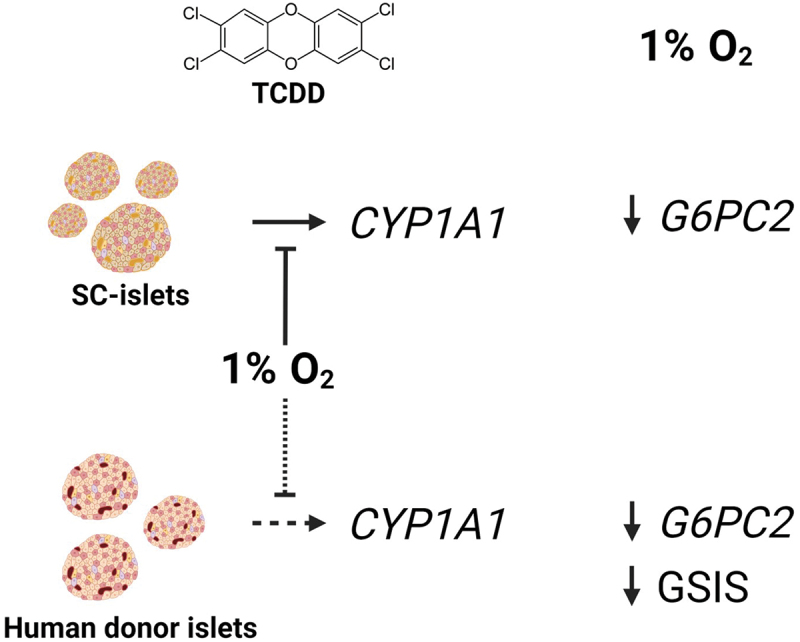
Our study shows consistent AHR-HIF1α crosstalk in SC-islets and variable crosstalk in primary human islets, depending on the donor. In both cell models, hypoxia exposure interfered with activation of the AHR pathway by TCDD but there was no evidence that AHR activation interfered with the HIF1α pathway. In summary, our data show that co-exposure to an environmental pollutant and hypoxia results in molecular crosstalk in islets.
KEYWORDS: dioxin, hypoxia, human donor islets, stem cell-derived islets (SC-islets), CYP1A1, G6PC2
GRAPHICAL ABSTRACT
1. Introduction
Diabetes prevalence is increasing rapidly on a global scale. In 2021, 537 million adults worldwide were estimated to be living with diabetes and the incidence rate is projected to increase by 46% by 20451. Type 2 diabetes (T2D) typically develops through myriad factors causing β-cell death or dysfunction, alongside peripheral insulin resistance2–4. Traditional risk factors for T2D include genetics, diet, and lifestyle. However, emerging research implicates exposure to man-made persistent organic pollutants (POPs) as an additional risk factor for T2D incidence5.
POPs are globally distributed, resulting in ubiquitous human exposure6. Dioxins and dioxin-like compounds are a class of POPs characterized by a molecular structure of chlorinated benzene rings and a high affinity for the aryl hydrocarbon receptor (AHR)7,8. Although serum concentrations of dioxins are decreasing globally9, epidemiology studies have reported positive associations between dioxin exposure and T2D incidence in diverse populations with lipid-adjusted serum dioxin concentrations ranging from 0.3 to 3388 ppt5,10–14. The most toxic and widely studied dioxin is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Although the mechanism linking TCDD exposure and T2D incidence remains unclear, our laboratory has shown that TCDD exposure to mice in vivo and to mouse or human islets ex vivo upregulates cytochrome P450 1A1 (Cyp1a1/CYP1A1) expression in islet cells15–17, an indication of AHR activation. The effects of TCDD on glucose tolerance, plasma insulin levels, and insulin sensitivity were abolished in β-cell specific Ahr knockout mice, suggesting that the AHR pathway in β-cells is mediating the adverse metabolic effects of TCDD18.
The AHR is a class I bHLH/PAS protein associated with cellular and xenobiotic metabolism19–24. Cytosolic AHR is inactive, but when bound by a ligand AHR translocates to the nucleus where the ligand-AHR complex dimerizes to the class II bHLH/PAS protein, aryl hydrocarbon nucleus translocator (ARNT). The AHR-ARNT complex recognizes and binds to xenobiotic response element (XRE) promoter sequences25–27, leading to upregulation of CYP1A1 enzymes, which are required for the oxidation and subsequent excretion of xenobiotics23,28. ARNT is a promiscuous Class II protein that shows diverse protein–protein interactions29–31, including dimerization with the Class I bHLH/PAS protein hypoxia-inducible factor 1α (HIF1α)30–32. During periods of insufficient oxygen availability (i.e., hypoxia, < 5% O2) stable HIF1α protein translocates into the nucleus and heterodimerizes with ARNT. The HIF1α-ARNT transcription complex is recognized by hypoxia response element (HRE) promotor sequences, leading to induction of genes associated with glucose metabolism, cell survival, vascular tone, and vascular growth27,33–35.
Competitive binding of AHR and HIF1α to ARNT has been reported in epithelial lung cells, lymphocytes, breast cancer cells, adipocytes, and hepatocytes27,32,36–38. In hepatocytes and hepatoma cells exposure to hypoxia or hypoxia-mimetics consistently interferes with expression of AHR gene targets, but exposure to AHR ligands does not consistently interfere with induction of HIF1α gene targets27,36,39–41. For example, mouse hepatoma cells simultaneously exposed to hypoxia and an AHR ligand showed a 30% reduction in the AHR target Cyp1a1, but no change in the HIF1α target Vegf compared with cells exposed to normoxia and the AHR ligand40. Crosstalk between AHR and HIF1α pathways has not been reported in pancreatic islets. We hypothesized that AHR-HIF1α crosstalk likely occurs in human islets, similar to hepatocytes, where HIF1α pathway activation interferes with the AHR pathway; we also speculate that the reverse may occur in islets—i.e., an AHR ligand interfering with HIF1α activation.
There are numerous scenarios where pancreatic islets are exposed to periods of hypoxic stress and need to mount a HIF1α response. Pancreatic islets normally experience oxygen partial pressure (pO2) of ~ 31–37 mmHg (4–5% O2) in vivo42 and ~25 mmHg (3% O2) at the islet surface when cultured in vitro43. Tightly regulated low physiological pO2 is required for pancreatic development and has been reported in subpopulations of mature pancreatic islets34,43. However, sustained glucotoxic and/or lipotoxic stress results in pathological hypoxia ( < 2% O2) that shifts cellular metabolism from aerobic to anaerobic and severely impairs glucose stimulated insulin secretion (GSIS)44. In islet transplantation, a robust HIF1α-driven response is critical for graft revascularization and the success of transplantation45,46. It is important to understand how environmental factors, such as ubiquitous exposure to environmental AHR ligands47, may influence the ability of islets to mount an appropriate hypoxia response. Additionally, given that AhRactivation in β-cells appears to mediate the adverse metabolic effects of TCDD in mice18, it is also important to understand if hypoxic stress could interfere with adaptation of islets to AHR ligands.
Human embryonic stem cell (hESC)-derived islet-like endocrine cells (SC-islets) are a developmental research model with potential for clinical diabetes treatment48. In the present study, we use a genetically modified hESC line that expresses EGFP under control of the INSULIN promotor (INS-2A-EGFP cell line49), to generate human SC-islets in vitro. Although differentiation protocols are continually improving50–52, SC-islets are not fully mature and do not show robust GSIS comparable with primary islets53–55. An advantage of SC-islets compared to primary islets from human organ donors is that SC-islets are environmentally naïve and thus have limited biological variability between differentiation batches. In contrast, human donor islets are sourced from a diverse population with a broad range of ages, genetic backgrounds, health, lifestyles, and previous environmental exposures. While donor variability is a challenge of studying human islets, this also serves as an important opportunity to examine the range of responses encountered in human islets.
In the current study, we examine AHR-HIF1α pathway crosstalk in human islet cells by exposing primary donor islets and SC-islets to hypoxia alone, TCDD alone, or hypoxia + TCDD combined for 48 hours. We measured gene targets of AHR (e.g., CYP1A1) as an indicator of AHR activation and gene targets of HIF1α (e.g., HMOX1) as an indication of HIF1α activation56. GSIS was also measured in primary islets to determine the effect of AHR-HIF1α crosstalk on human islet function.
2. Methods
2.1. hESC differentiation and maintenance
SC-islets were derived from INS-2A-EGFP WA01 hESCs by Dr. Francis Lynn (University of British Columbia, BC, Canada) using a 27+ day differentiation protocol, as previously described52,57. INS-2A-EGFP hESCs were authenticated and tested for contamination in the Lynn lab. SC-islets were then shipped from Vancouver to Ottawa and maintained in Stage 6 media [CMRL-1066 (VWR, #CA45001–114; Radnor, PA) containing 20 g/L bovine serum albumin (Sigma-Aldrich #10775835001), 1X Glutamax (Gibco #35050079), 1% penicillin-streptomycin (Gibco #15140122), 0.5 mM pyruvate (Sigma-Aldrich #S8636–100 mL), 0.5X Insulin Transferrin-Selenium-X (Thermo Fisher #51500056), 35 nM zinc sulfate (Sigma-Aldrich #Z0251-100 G), 1 mM N-acetyl-L-cysteine (Sigma-Aldrich #A9165-5 G), 10 ug/L Heparin (Sigma-Aldrich #H3149-10KU), 1:2000 Trace elements A (Corning 25–021-CI), 1:2000 Trace Elements B (Corning 25–022-CI #MT99175CI), 10 nM T3 (Sigma, #T6397-100 MG), 0.5 μM ZM 447,439 (Cedarlane Labs, #S1103-10 MM/1 ML; Burlington, ON), and 1:2000 lipid concentrate (Fisher Scientific #11905031)]. SC-islets were maintained at 37°C, 21% O2, and 5% CO2 on an orbital shaker (106 RPM) until analyses, with media changes every two days.
SC-islets are composed of a mixture of pancreatic endocrine cells, including ~ 30–70% INS-EGFP+ cells (Supplemental Figure S1). SC-islets aged to day 50–60 are capable of showing a modest GSIS response52. Therefore, we used SC-islets aged day 50+ in our experiments. SC-islets were visually assessed for cluster integrity and GFP+ fluorescence upon arrival and were not included in this study if quality appeared poor. Each SC-islet differentiation was considered one “biological replicate” (n = 3 technical replicates per condition per differentiation; n = 3 biological replicates).
2.2. Human organ donor islet cell culture
Human islets were provided by the Alberta Diabetes Institute IsletCore at the University of Alberta (www.bcell.org/adi-isletcore) with the assistance of the Human Organ Procurement and Exchange (HOPE) program, Trillium Gift of Life Network (TGLN), and other Canadian organ procurement organizations. Islet isolation was approved by the Human Research Ethics Board at the University of Alberta (Pro00013094). All donors’ families gave informed consent for the use of pancreatic tissue in research. All research using human islets was approved by the Research Ethics Board at Carleton University.
Islet purity ranged between 75% and 90% for this study (Figure 4H). Donor islets were shipped overnight to Ottawa in CMRL media (Fisher Scientific, #11–530–037). Upon arrival, islets were visually assessed for islet integrity and transferred to low glucose DMEM media (LG-DMEM; Fisher Scientific, #11–885–084) supplemented with 10% FBS (Sigma-Aldrich #F1051-500 ML) and 1% penicillin-streptomycin (Gibco, #15140122; Billings, MT) and cultured at 37°C, 21% O2, and 5% CO2 overnight to allow for recovery. See Figure 4H for donor characteristics. Human islets from separate donors were considered as biological replicates (n = 3–5 technical replicates per condition per donor; n = 3 biological replicates per sex).
Figure 4.
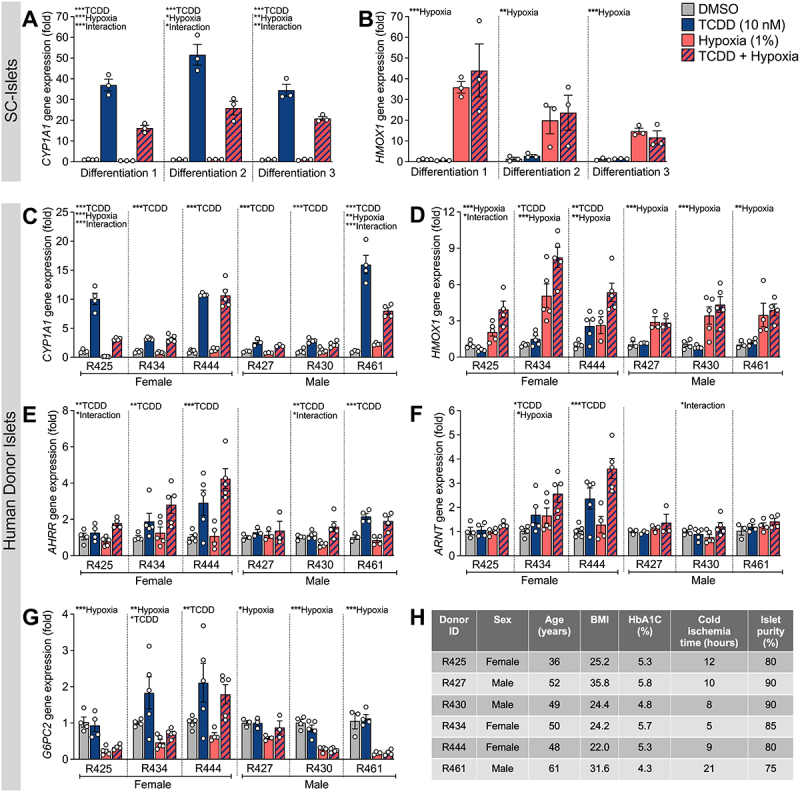
Co-treatment of human islets to TCDD + hypoxia impairs induction of CYP1A1 by TCDD in 2 of 6 donors. SC-islets and human islets were exposed to TCDD (10 nM) ± hypoxia (1% O2) for 48 hours. Gene expression was measured in SC-islets and human islets for (A,C) CYP1A1 and (B,D) HMOX1, and in human islets for (E) AHRR, (F) ARNT, and (G) G6PC2. Gene expression is relative to DMSO-treated islets. (A,B) Separate SC-islet biological replicates (i.e., different differentiations) and (C-G) separate human islet donors are shown for each gene. Data are mean ± SEM and individual data points represent technical replicates. Significance within each biological replicate was determined by a two-way ANOVA and Dunnett post-doc test. Overall treatment and interaction effects are indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001). Multiple comparison analysis showing treatment groups that are significantly different from each other are indicated by different letters (p < 0.05). (H) Human islet characteristics are summarized for each individual donor.
2.3. Experimental conditions
Two experimental paradigms were used in this study (Figure 1). First, we treated SC-islets with TCDD ± hypoxia continuously for 48 hours or pre-treated SC-islets with either TCDD alone or hypoxia alone for 24 hours prior to co-treatment with TCDD + hypoxia for 24 hours (Figure 1A). Lastly, we compared the effects of TCDD ± hypoxia co-treatment for 48 hours in SC-islets versus human donor islets (Figure 1B). A concentration of 10 nM TCDD (3,219 ppt) was selected following previous findings that 10 nM TCDD was sufficient to upregulate CYP1A1 mRNA and enzyme activity in human donor islets16. Hypoxia exposure was set at 1% O2 (7.6 mmHg O2) to mimic physiological hypoxia shown to activate the HIF1α pathway58.
Figure 1.
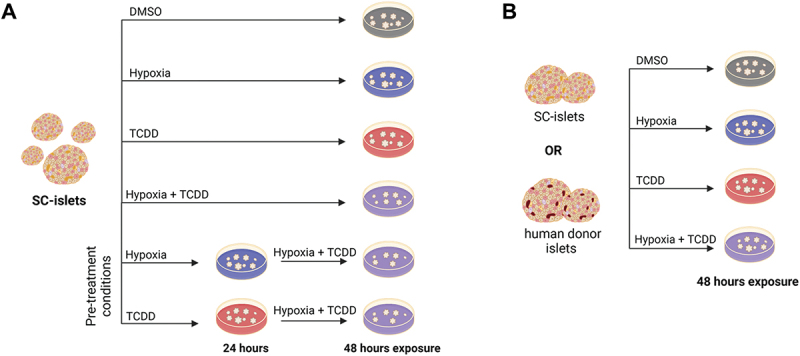
Summary of treatment conditions applied to SC-islets and human donor islets. (A) SC-islets were exposed to TCDD (10 nM) ± hypoxia (1% O2) for 48 hours continuously or to TCDD or hypoxia pre-treatment for 24 hours followed by TCDD + hypoxia co-treatment for 24 hours. (B) SC-islets or human donor islets were exposed to TCDD ± hypoxia co-treatment continuously for 48 hours. Made with BioRender.com.
SC-islets or human donor islets were hand-picked and transferred to six-well non-TC treated plates (VWR, #10861–554) to undergo the treatment condition outlined above: (i) DMSO (#276855-100 ML, Sigma-Aldrich, Oakville, ON, Canada) vehicle control; (ii) normoxia (21% O2, 5% CO2); (iii) 10 nM TCDD (#AD404SDMSO10X1ML, Accustandard, Chromatographic Specialties, Brockville, ON, Canada); (iv) hypoxia (1% O2) in a triple-gas (N2-O2-CO2) incubator (ThermoForma, Series II, 3130, Marietta, Ohio).
2.4. Quantitative real time PCR
Immediately following treatment, SC-islets or human islets were transferred to buffer RLT (#79216, Qiagen, Hilden, Germany) and stored at −80°C. mRNA was isolated using RNeasy Micro kit (#74004, Qiagen, Hilden, Germany) as per the manufacturer’s instructions. DNase treatment was performed prior to cDNA synthesis with the iScriptTM gDNA Clear cDNA Synthesis Kit (#1725035, Bio-Rad, Mississauga, ON, Canada). Real time (RT) qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, #1725271) and run on a CFX394 (Bio-Rad). PPIA was used as the reference gene. Primer efficiency ranged between 87% and 110%, and sequences are listed in Table S1. Data were analyzed using the 2ΔΔCT relative quantitation method and normalized to the DMSO vehicle control.
2.5. Glucose-stimulated insulin secretion
SC-islets displayed limited glucose responsiveness in the Bruin lab, despite being modestly glucose responsive in other labs52. Therefore, only human islets were used to measure the impact of TCDD ± hypoxia on GSIS (n = 3–5 technical replicates per donor; n = 6 biological replicates, i.e., donors). Immediately following treatment, 20 islets per condition were moved to pre-warmed (37°C) Krebs – Ringer bicarbonate buffer (KRBB) solution (115 mmol/l NaCl, 5 mmol/l KCl, 24 mmol/l NaHCO3, 2.5 mmol/L CaCl2, 1 mmol/l MgCl2, 10 mmol/l HEPES, 0.1% (wt/vol.) BSA, pH 7.4) with 2.8 mmol/L glucose (low glucose, LG) for a 60 min pre-incubation in 1.5 mL Eppendorf tubes. Supernatant was removed and replaced with 500 μL of LG KRBB for 60 min, followed by transfer to 500 μL of KRBB with 16.7 mmol/L glucose (high glucose, HG) for 60 min at 37°C. The LG KRBB and HG KRBB samples were centrifuged, and the supernatant stored at − 20°C. The GSIS assay was conducted under normoxic conditions and in the absence of TCDD, regardless of previous treatments. To measure insulin content, supernatant was removed and replaced with an acid-ethanol solution of 1.5% vol/vol HCl in 75% vol/vol ethanol at 4°C overnight and neutralized with 1 mol/L Tris base (pH 7.5) before long-term storage at − 20°C. Low and high glucose GSIS samples were diluted 1:10 in KRBB and acid-ethanol lysed samples were diluted 1:100 in KRBB prior to measurement of human insulin concentration by ELISA (#80-INSHU-CH10, ALPCO, Salem, NH, USA).
2.6. Statistics
All statistics were performed using GraphPad Prism 10.0.0 (GraphPad Software Inc., La Jolla, CA). For all analyses, p < 0.05 was considered statistically significant and any potential outliers were tested for significance using a Grubbs’ test with α = 0.05. We tested for variance between biological replicates within model using a nested one-way ANOVA. We tested for main and interaction effects using an ordinary two-way ANOVA with a Tukey’s multiple comparisons test. Comparisons between different treatment groups are visualized on graphs by showing different letters above each bar to indicate significant differences (i.e., “a” is different than “b” and “ab” is not different from either “a” or “b”). Non-parametric statistics were used in cases where the data failed normality or equal variance tests. A Spearman (rho) correlation test was used to assess correlation between donor islet quality and clinical parameters (age, BMI, HbA1c, cold ischemia time, and islet particle index) against degree of CYP1A1 induction by TCDD exposure relative to CYP1A1 induction by TCDD + hypoxia exposure. Data are presented as means ± SEM. Figure legends specify whether individual datapoints are technical or biological replicates.
3. Results
3.1. Co-exposure of SC-islets to TCDD + hypoxia impairs CYP1A1 induction by TCDD, regardless of pre-treatment conditions
To determine if hypoxia interferes with TCDD-mediated AHR activation in islets, we measured CYP1A1 and HMOX1 induction in SC-islets following exposure to TCDD ± hypoxia (Figure 1A). TCDD significantly upregulated CYP1A1 expression in SC-islets ~41-fold compared to the vehicle (Figures 2A and 3A). However, when SC-islets were co-treated with TCDD + hypoxia, the magnitude of CYP1A1 induction was suppressed to ~ 21-fold (Figures 2A and 3A). Hypoxia exposure induced HMOX1 ~24-fold in SC-islets compared to normoxia with no effect of co-treatment with TCDD (Figures 2B and 3B).
Figure 2.
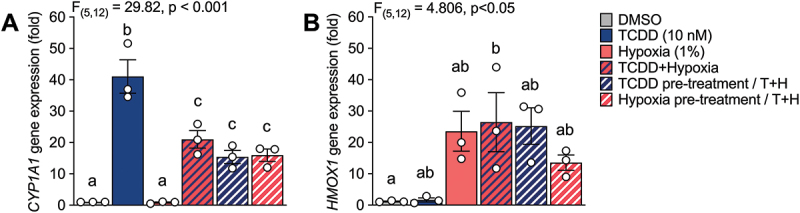
Hypoxia interfered with CYP1A1 upregulation by TCDD in SC-islets. (A) CYP1A1 and (B) HMOX1 gene expression in SC-islets co-treated with TCDD (10 nM) ± hypoxia (1% O2) for 48 hours or following either TCDD or hypoxia pre-treatment for 24 hours and TCDD + hypoxia co-treatment for 24 hours. Gene expression is relative to DMSO-treated SC-islets. Data are mean ± SEM and individual data points represent biological replicates (i.e. different differentiations). Significance was determined by a one-way ANOVA and Tukey post-hoc test. Multiple comparison analysis showing treatment groups that are significantly different from each other are indicated by different letters (p < 0.05).
Figure 3.
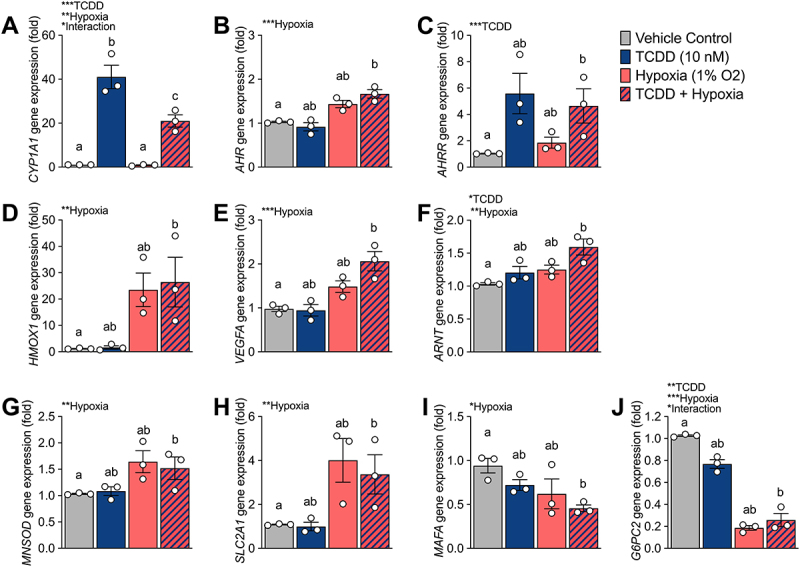
TCDD + hypoxia co-treatment causes interactive effect for CYP1A1 and G6PC2 expression in SC-islets. SC-islets were exposed to TCDD (10 nM) ± hypoxia (1% O2) for 48 hours and gene expression was measured for (A) CYP1A1, (B) AHR, (C) AHRR, (D) HMOX1, (E) VEGFA, (F) ARNT, (G) MNSOD, (H) SLC2A1, (I) MAFA, and (J) G6PC2. Gene expression is relative to DMSO-treated SC-islets. Data are mean ± SEM and individual data points represent biological replicates (i.e. different differentiations). Significance was determined by a two-way ANOVA and Tukey post-hoc test. Overall treatment and interaction effects are indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001). Multiple comparison analysis showing treatment groups that are significantly different from each other are indicated by different letters (p < 0.05).
We also investigated whether the degree of crosstalk between AHR and HIF1α in SC-islets is influenced by which pathway is activated first—i.e., a first-come, first-serve effect. We hypothesized that activation of either the AHR or HIF1α pathway before competitive co-exposure would dictate the dimerization partner of ARNT. SC-islets were pre-treated with either TCDD or hypoxia for 24 hours prior to co-treatment with TCDD + hypoxia for 24 hours (Figure 1A). We found that all TCDD + hypoxia co-treatment conditions significantly impaired the degree of CYP1A1 induction compared to treatment with TCDD alone (Figure 2A). Pre-treatment of SC-islets with either TCDD or hypoxia did not alter this effect (Figure 2A). Similarly, all hypoxia treatment conditions upregulated HMOX1 in SC-islets, irrespective of co-treatment or pre-treatment with TCDD (Figure 2B).
3.2. Co-exposure of SC-islets to TCDD + hypoxia also causes an interaction effect on G6PC2
To further explore the impact of molecular crosstalk in SC-islets, we focused on the first 4 conditions: vehicle, TCDD alone, hypoxia alone, or TCDD + hypoxia for 48-hours (Figure 1B). We measured CYP1A1 expression alongside other AHR targets (AHR, AHRR), HIF1α targets (HMOX1, VEGFA), ARNT, biomarkers of β-cell maturity (MAFA – a transcription factor required for β-cell maturity and function59, SLC2A1 – a glucose transporter required for glucose-stimulated insulin secretion60, and G6PC2 – glucose 6 phosphatase catalytic subunit 2, an islet-specific enzyme involved in the futile glucose flux with glucokinase and positively associated with fasting blood glucose levels in rodents and humans61–65, and MNSOD – an antioxidant that protects against reactive oxygen species produced by mitochondrial oxidative stress66,67(Figure 3). As shown in Figure 2A and re-plotted here, there was a significant interaction effect of TCDD + hypoxia co-treatment on CYP1A1 expression in SC-islets (Figure 3A). The AHR repressor gene, AHRR, was significantly upregulated by TCDD exposure, as expected, but unlike CYP1A1 there was no interaction effect with hypoxia (Figure 3C). Exposure of SC-islets to hypoxia significantly upregulated AHR, HMOX1, VEGFA, MNSOD, and SLC2A1 (Figure 3B,D,E,G,H) and downregulated MAFA expression (Figure 3I); none of these changes were affected by co-treatment with TCDD. Both TCDD and hypoxia exposure alone modestly upregulated ARNT expression, but there was no interaction effect on ARNT (Figure 3F). Interestingly, an unexpected interaction effect was seen for G6PC2 expression (Figure 3J). Both TCDD alone and hypoxia alone significantly downregulated G6PC2 expression compared with vehicle/normoxia control conditions (Figure 3J). Hypoxia-mediated downregulation dominated the interaction effect seen following TCDD + hypoxia co-treatment (Figure 3J).
3.3. TCDD consistently induces CYP1A1 in human donor islets but AHR-HIF1α crosstalk varies between donors
We next examined whether crosstalk between AHR and HIF1α pathways also occurs in primary human islets from deceased organ donors. Human islets from 3 male and 3 female donors were included in our study. Average donor age was 49.3 (±8.0) years, BMI was 27.2 (±5.3), and HbA1c was 5.2 (±0.6) (Figure 4H). Human donor islets were co-exposed to TCDD ± hypoxia continuously for 48 hours (Figure 1B). Unlike SC-islets, which showed consistent treatment effects across all biological replicates (i.e., different differentiations; Figure 4A,B), the effects of TCDD ± hypoxia on human islets varied substantially between donors. These variable effects are summarized in Table 1, detailed statistics are provided in Supplementary Table S2, and additional donor information can be found at humanislets.com68.
Table 1.
SC-islets and human donor islets treated with TCDD ± hypoxia show variability for gene expression changes.
| CYP1A1 | HMOX1 | AHRR | ARNT | G6PC2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell source | TCDD | Hypoxia | Crosstalk | TCDD | Hypoxia | Crosstalk | TCDD | Hypoxia | Crosstalk | TCDD | Hypoxia | Crosstalk | TCDD | Hypoxia | Crosstalk |
| SC-Islets | ↑ (41-fold) | ↑ (1.1-fold) | ✓ | - | ↑ (24-fold) | x | ↑ (6-fold) | - | x | ↑ (1.2-fold) | ↑ (1.2-fold) | x | ↓ | ↓ | ✓ |
| R425 - ♂ | ↑ (10-fold) | ↓ | ✓ | - | ↑ (2-fold) | ✓ | ↑ (1.3-fold) | - | ✓ | - | - | x | - | ↓ | x |
| R434 -♂ | ↑ (3-fold) | - | x | ↑ (1.4-fold) | ↑ (5-fold) | x | ↑ (2-fold) | - | x | ↑ (1.7-fold) | ↑ (1.7-fold) | x | ↑ (2-fold) | ↓ | x |
| R444 -♂ | ↑ (11-fold) | - | x | ↑ (2.5-fold) | ↑ (2.6-fold) | x | ↑ (3-fold) | - | x | ↑ (2-fold) | - | x | ↑ (2-fold) | - | x |
| R427 -♀ | ↑ (3-fold) | ↓ | x | - | ↑ (3-fold) | x | - | - | x | - | - | x | - | ↓ | x |
| R430 -♀ | ↑ (3-fold) | - | ✓ | - | ↑ (3-fold) | x | ↑ (1.1-fold) | - | ✓ | - | - | ✓ | - | ↓ | x |
| R461 -♀ | ↑ (15-fold) | ↑ (2-fold) | ✓ | - | ↑ (3-fold) | x | ↑ (2-fold) | - | x | - | - | x | - | ↓ | x |
A summary table of the main effects of TCDD (10 nM) ± hypoxia (1% O2) on SC-islets and human donor islets for expression of CYP1A1, HMOX1, AHRR, ARNT, and G6PC2. Bolded fold-increase indicates significance for TCDD or hypoxia treatments alone, compared with vehicle control.
TCDD exposure significantly increased CYP1A1 expression in all donors, albeit to varying degrees (2.5-fold to 15.8-fold, Figure 4C, Table 1). We saw a significant interaction effect for TCDD + hypoxia co-treatment suppressing the magnitude of CYP1A1 induction compared with TCDD alone in 2 of the 6 donors (Donors R425 and R461, Figure 4C, Table 1). There was also a trending interaction effect in Donor R430 (p = 0.0552, Figure 4C, Table 1). No interaction effect for CYP1A1 expression was found in the other donor islets co-exposed to TCDD + hypoxia (Figure 4C, Table 1).
We next correlated various islet and donor parameters (age, BMI, HbA1c, cold ischemia time, and islet particle index) against CYP1A1 fold induction change by co-exposure (i.e., CYP1A1 fold induction following TCDD relative to CYP1A1 fold induction following TCDD + hypoxia). Interestingly, cold ischemia time positively correlated with the CYP1A1 fold induction change by co-exposure (rho = 0.89, p = 0.03; Supplemental Table S3). This preliminary analysis suggests that islets with longer cold ischemia time had a greater degree of crosstalk between AHR and HIF1α pathways.
3.4. TCDD consistently upregulates AHRR expression in human islets; hypoxia consistently induces HMOX1 and downregulates G6PC2 expression in human islets
All donors showed a main effect or trend for modest HMOX1 upregulation by hypoxia (2.2-fold to 5.0-fold, Figure 4D, Table 1). A modest interaction effect by TCDD + hypoxia co-exposure augmenting HMOX1 upregulation was seen for one donor (R425, Figure 4D, Table 1).
All donors except one (Donor R427) showed a main effect for TCDD exposure upregulating AHRR expression (Figure 4E, Table 1); this is consistent with the effect seen in SC-islets (Figure 3C, Table 1). In addition, two donors (R425 and R430) showed a modest TCDD + hypoxia interaction effect for AHRR (Figure 4E, Table 1).
Changes in ARNT expression were highly variable between donors. Three donors showed no change in ARNT expression across treatments (Donors R425, R427, R461; Figure 4F, Table 1). Donor R430 showed a modest interaction effect, Donor R434 showed a main effect for both hypoxia and TCDD, and Donor R444 showed a main effect for TCDD exposure only (Figure 4F, Table 1).
All human donors except R444 showed a significant main effect for hypoxia to downregulate G6PC2 expression (Figure 4G, Table 1). Donors R434 and R444 additionally showed a main effect for G6PC2 upregulation by TCDD (Figure 4G, Table 1). TCDD-driven upregulation of G6PC2 in donor islets contrasts the modest but consistent TCDD-driven G6PC2 downregulation seen in the SC-islets (Figure 3J, Table 1). No interaction effects were found for G6PC2 expression among the donor islets, which also differs from the significant interaction effect seen in SC-islets.
3.5. Hypoxia, but not TCDD, negatively impacts donor islet GSIS
To determine whether TCDD ± hypoxia exposure impacted islet function we assessed GSIS immediately following 48-hour exposure conditions. Across all donors, TCDD did not impact basal or glucose-stimulated insulin secretion compared with vehicle control (Figure 5A,B). Exposure to hypoxia dramatically reduced insulin secretion in response to high glucose (Figure 5A,B). This effect was maintained but not amplified when cells were co-treated with hypoxia + TCDD (Figure 5A,B). Hypoxia treatment resulted in mild reduction of total insulin content in human islets, regardless of co-treatment with TCDD (Figure 5C). When insulin secretion was normalized to insulin content, there was no significant effect of treatment on GSIS (Figure 5D).
Figure 5.
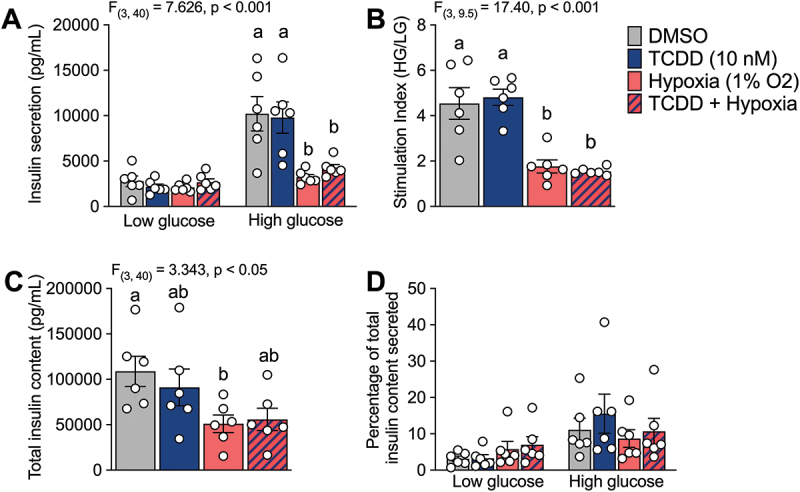
Hypoxia exposure negatively impacts glucose stimulated insulin secretion. Human islets were exposed to TCDD (10 nM) ± hypoxia (1% O2) for 48 hours and (A) insulin secretion was measured following 1 hour in low (2.8 mM) and high (16.7 mM) glucose concentrations. (B) Stimulation index is expressed as secreted insulin concentration following high glucose relative to low glucose stimulation. (C) Total insulin content in islets following acid ethanol lysis. (D) Insulin secretion following low and high glucose stimulation normalized by total insulin content. Data are mean ± SEM and individual data points represent biological replicates (i.e. different islet donors). Significance was determined with (A, D) a two-way ANOVA and Šidák post-hoc test, (B) a brown-forsythe ANOVA and Dunnett post-hoc test, and (C) a one-way ANOVA and Dunnett post-hoc test. Multiple comparison analysis showing treatment groups that are significantly different from each other are indicated by different letters (*p < 0.05).
4. Discussion
This study investigated whether there is crosstalk between AHR and HIF1α pathways in SC-islets and human donor islets. TCDD exposure consistently activated the AHR pathway, as indicated by CYP1A1 upregulation, in SC-islets and all human islet donors. Interestingly, the degree of CYP1A1 induction was more pronounced in SC-islets than human islets and varied substantially between human islet donors. Hypoxia exposure consistently interfered with TCDD-mediated CYP1A1 induction in the SC-islet model, which suggests possible crosstalk between AHR and HIF1α pathways. We also saw evidence of AHR-HIF1α crosstalk in human islets but only in 2 of 6 donors.
There are many variables that may contribute to the consistency of AHR-HIF1α crosstalk in SC-islets compared to human donor islets. First, we speculate that previous environmental exposures likely influence the sensitivity of donor islets to TCDD and/or hypoxia exposure ex vivo. SC-islets are environmentally naive whereas human islets have been exposed to a broad variety of environmental contaminants throughout their lifetime69,70, which impact baseline AHR pathway activation between donors. Furthermore, dietary habits, smoking or drug use, and other environmental factors can lead to cellular hypoxia71–73, which likely impacts baseline HIF1α pathway activation. Donor age is another important source of biological variation; the human islets used in our study were isolated from donors between 36 to 61 years of age. AHR activation and downstream effects are known to vary with age74,75 and baseline AHR and ARNT levels vary by age and tissue state76. Our sample size of human donors was insufficient to do a well-powered correlation analysis to determine which clinical parameters are driving variation in AHR-HIF1α crosstalk, but we did note a positive correlation between cold ischemia time and suppression of CYP1A1 induction by co-exposure to TCDD + hypoxia. Duration of cold ischemia time does not impact donor islet retrieval or function and does not impair transplantation in appropriately selected recipients77–79. However, these data suggest an extended period of cold ischemia may sensitize donor islets to AHR-HIF1α crosstalk ex vivo. In summary, the differences in robustness and variability of AHR-HIF1α crosstalk between SC-islets and human donor islets likely arises from a mixture of these considerations and reflect the need for caution when interpreting cell-line responses as a proxy for primary human cell biology. The variability in responses between different human donors emphasizes the need for large sample sizes in future work examining AHR-HIF1α crosstalk in human donor islets. A larger sample size is critical to ensure reproducibility and allow for appropriate stratification based on donor characteristics, such as biological sex.
One surprising finding from this study was the lack of impaired GSIS in human islets after 48-hour exposure to TCDD ex vivo. In our previous publication, we reported modestly impaired insulin secretion under high glucose conditions in TCDD-exposed islets from 5 different human donors, although this did not reach statistical significance16. Here, using 6 additional donors, the effects of TCDD on static GSIS are highly variable between donors and the overall effect does not approach statistical significance under either low or high glucose conditions. More detailed perifusion analysis would provide additional insight into the dynamics of insulin secretion (e.g., first-phase versus second-phase responses) from human islets following ex vivo TCDD exposure. Furthermore, markers of mitochondrial function, endoplasmic reticulum stress, and/or cell viability can be examined in future studies for a clearer understanding of the impact of TCDD on human islets. We also speculate that TCDD may have more pronounced effects on insulin secretion following longer-term exposure and/or exposure alongside a secondary metabolic stressor, based on findings from the epidemiology literature5 and animal studies15,17,18,,80. In summary, despite the consistency in activation of the AHR (i.e., CYP1A1 and AHRR induction) across all human islet donors tested to date, there remains a wide spectrum of functional responses to TCDD; more human islet donors are needed to fully appreciate the nuances of how TCDD impacts human islet function.
In our study, the HIF1α pathway dominated over the AHR pathway in islets, even when cells were pre-treated with TCDD for 24 hours. HIF1α pathway dominance was indicated by the lack of TCDD-mediated interference with hypoxia-mediated HMOX1 induction in both SC-islets and human islets. However, our study is limited by the absence of protein measurements. Future studies should examine protein expression, particularly stability of the AHR and HIF1α, in human islet models to strengthen our understanding of AHR-HIF1α crosstalk. The dominance of hypoxia-mediated effects was also clear in the GSIS response of human islets. Hypoxia exposure profoundly blunted the GSIS response, as expected based on previous findings81–86, and this effect was seen irrespective of co-treatment with TCDD. We speculate that ARNT binding affinity is likely driving AHR-HIF1α crosstalk in human islets. Notably, there is little to no evidence of ARNT-independent gene induction by HIF1α87–91. Gradin et al. used coimmunoprecipitation to show stabilized HIF1α has a high binding affinity with ARNT in a human hepatoma cell line. Furthermore, this group found the HIF1α pathway dominates over AHR in binding with ARNT in a rabbit reticulocyte lysate41. The exact mechanism(s) driving the HIF1α-ARNT binding dominance over AHR-ARNT in SC-islets and human donor islets warrants further investigation.
The presence of AHR-HIF1α crosstalk in SC-islets and human donor islets has interesting implications for islet transplantation. Human islets are transiently hypoxic following clinical transplantation44,46 and need to mount an efficient and robust HIF1α response to induce vascularization. Given that the AHR and HIF1α pathways both rely on access to ARNT, it is possible that sustained exposure to an AHR ligand(s) could interfere with the HIF1α-mediated hypoxia response post-transplant of either SC-islets or human donor islets. While our experiment models showed the HIF1α pathway dominating AHR-HIF1α crosstalk in islets, this was a short-term (48-hr) window of crosstalk in vitro. It remains possible that chronic exposure of patients to AHR ligands could compromise engraftment post-transplant via interfering with the HIF1α pathway. Additionally, we do not fully understand the consequences of AHR pathway activation within islets, so it is difficult to appreciate the potential impact that post-transplant hypoxia may have on islet health if HIF1α activation interferes with the AHR-mediated detoxification response in engrafted islets.
The consistent hypoxia-mediated downregulation of G6PC2 in both SC-islets and human donor islets was an unexpected and interesting finding. The consistent changes in HMOX1, CYP1A1, and AHRR expression were expected based on well-established effects of hypoxia34,44 and TCDD31, in islets and other cell types. Hypoxia exposure upregulates G6pc expression in mouse liver tissue and in a human liver cell line (HepG2)92,93, but to our knowledge the connection between hypoxia exposure and G6PC2 downregulation in human islets has not been previously described. In cancer patients and cancer cell lines G6PC was described as part of the battery of gene changes associated with the Warburg effect (aerobic glycolysis)94,95, and hypoxia-exposed rats showed G6pc downregulation in liver tissue96. In humans the G6PC2 isoform is islet-specific61, associated with autoimmunity97, and responsible for tight regulation of fasting blood glucose levels62,98–100. G6PC2 expression is downregulated in human islets from donors diagnosed with T2D, a condition associated with islet hypoxia85,101–103. We speculate that beyond regulating blood glucose levels in diabetes, downregulation of G6PC2 is protective by increasing intracellular glucose levels available for anaerobic glycolysis and ATP production necessary for insulin secretion. In support, Rahim et al. (2022) showed G6pc2 knockout improved GSIS response by modulating glycolysis in a mouse pancreatic β-cell line104. Hypoxia-mediated G6PC2 downregulation may work in concert with other hypoxia-mediated gene expression changes in the islet to promote glycolysis and suppress gluconeogenesis44,105.
5. Conclusions
In conclusion, our study is the first to show evidence for AHR-HIF1α crosstalk in islets. This crosstalk was dominated by the HIF1α pathway and was consistently observed in SC-islets. However, in human islets there was substantial biological variability in both the extent that TCDD exposure upregulated CYP1A1 expression and the presence of AHR-HIF1α crosstalk. We believe further study is warranted to examine why HIF1α dominates AHR-HIF1α crosstalk in islets, and what factors contribute to the variability seen in human donor islets. Furthermore, we believe the previously unreported finding of hypoxia-mediated G6PC2 downregulation in SC-islets and human donor islets requires further investigation.
Supplementary Material
Acknowledgments
Human islets for research were provided by the Alberta Diabetes Institute IsletCore at the University of Alberta in Edmonton (http://www.bcell.org/adi-isletcore.html) with the assistance of the Human Organ Procurement and Exchange (HOPE) program, Trillium Gift of Life Network (TGLN), and other Canadian organ procurement organizations. We thank the IsletCore staff, the islet donors, and their families.
We thank Dr. Jan Mennigan for thoughtful discussions over experimental design. We thank Dr. Ji Soo Yoon and Ekaterina Filatov for their valuable help designing figures.
The authors acknowledge that Carleton University is situated on the traditional, ancestral, and unceded territories of the Nehiyahwak, Nahikawe, Dakata, Lakota, Dene, and Metis Nations. The authors acknowledge that UBC and BC Children’s Hospital are situated on the traditional, ancestral, and unceded territories of the Coast Salish peoples—the Sḵwxwu´7mesh (Squamish), Səlı´lwətaʔ/Selilwitulh (Tsleil-Waututh), and xw məqkw əyəm (Musqueam) Nations.
Funding Statement
This research was supported by a Canadian Institutes of Health Research-Breakthrough T1D (CIHR-Breakthrough T1D) Team Grant (#ASD-173663/5-SRA-2020-1059-S-B) and Natural Sciences and Engineering Research Council (NSERC) Discovery Grants to JEB (#RGPIN-2017-06265) and WGW (#RGPIN-2017-06414). NG was supported by an NSERC Canadian Graduate Student-Doctoral (CGS-D) award and KAV was supported by an NSERC-Collaborative Research and Training Experience (CREATE) award provided by the Canadian Islet Research and Training Network (CIRTN). JEB is supported by an Early Researcher Award from the Ontario Government and a Dorothy Killam Fellowship.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Abbreviations
- AHR
Aryl hydrocarbon receptor
- AHRR
Aryl hydrocarbon receptor repressor
- ARNT
Aryl hydrocarbon nuclear translocator
- bHLH
Basic helix loop helix
- CYP1A1
Cytochrome P450 1A1
- GSIS
Glucose stimulated insulin secretion
- G6PC2
Glucose-6-phosphate catalytic subunit 2
- HIF1α
Hypoxia inducible factor 1α
- HRE
Hypoxia response element
- MAFA
V-maf musculoaponeurotic fibrosarcoma oncogene homolog A
- MNSOD
Manganese superoxide dismutase
- PAS
Period/ARNT/Sim
- POP
Persistent organic pollutant
- SC-islets
Stem cell-derived islets
- T2D
Type 2 diabetes
- TCDD
2,3,7,8 tetrachlorodibenzo-p-dioxin
- XRE
Xenobiotic response element
Author contributions
NG: Conceptualization, Methodology, Formal analysis, Investigation, Writing – Original draft, Writing – Review & editing, Visualization. JEB: Conceptualization, Methodology, Formal analysis, Resources, Writing – Review & editing, Visualization, Supervision, Funding acquisition. WGW: Conceptualization, Methodology, Resources, Writing – Review & editing. KVA: Investigation, Writing – Review & editing. FCL: Investigation, Resources, Writing – Review & editing.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19382014.2025.2526871
References
- 1.International Diabetes Federation. IDF Diabetes Atlas . 10th edition. www.diabetesatlas.org.
- 2.Weir GC, Bonner-Weir S.. Five stages of evolving-cell dysfunction during progression to diabetes Diabetes 53 Dec Suppl 3:S16–18. 2004. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- 3.Cernea S, Dobreanu M. Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochem Med (Zagreb). 2013;23(3):266–280. doi: 10.11613/BM.2013.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiasson J-L, Mi rabasa-Lhoret R. Prevention of type 2 diabetes insulin resistance and-cell function. Diabetes. 2004;53(suppl_3):S34–S38. doi: 10.2337/diabetes.53.suppl_3.S34. [DOI] [PubMed] [Google Scholar]
- 5.Gang N, Van Allen K, Villeneuve PJ, MacDonald H, Bruin JE. Sex-specific associations between type 2 diabetes incidence and exposure to dioxin and dioxin-like pollutants: a meta-analysis. Front Toxicol. 2021;3(February):1–14. doi: 10.3389/ftox.2021.685840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srogi K. Levels and congener distributions of PCDDs, PCDFs and dioxin-like PCBs in environmental and human samples: a review. Environ Chem Lett. 2008;6(1):1–28. doi: 10.1007/s10311-007-0105-2. [DOI] [Google Scholar]
- 7.Gonzalez FJ, Fernandez-Salguero P. The aryl hydrocarbon receptor. Studies using the AHR-null mice. Drug Metab Dispos. 1998;26(12):1194–1198. [PubMed] [Google Scholar]
- 8.Weiß C, Kolluri SK, Kiefer F, Göttlicher M. Complementation of ah receptor deficiency in hepatoma cells: negative feedback regulation and cell cycle control by the ah receptor. Exp Cell Res. 1996;226(1):154–163. doi: 10.1006/excr.1996.0214. [DOI] [PubMed] [Google Scholar]
- 9.Aylward LL, Hays SM. Temporal trends in TCDD body burden: decreases over three decades and implications for exposure levels. J Expo Anal Environ Epidemiol. 2002;12(5):319–328. doi: 10.1038/sj.jea.7500233. [DOI] [PubMed] [Google Scholar]
- 10.Mannetje A, Eng A, Walls C, Dryson E, Douwes J, Bertazzi P, Ryder-Lewis S, Scott D, Brooks C, McLean D, et al. Morbidity in New Zealand pesticide producers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Environ Int. 2018;110(October2017):22–31. doi: 10.1016/j.envint.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Bertazzi PA, Consonni D, Bachetti S, Rubagotti M, Baccarelli A, Zocchetti C, Pesatori AC. Health effects of dioxin exposure: a 20-year mortality study. Am J Epidemiol. 2001;153(11):1031–1044. doi: 10.1093/aje/153.11.1031. [DOI] [PubMed] [Google Scholar]
- 12.Huang CY, Wu CL, Yang YC, Chang JW, Kuo YC, Cheng YY, Wu JS, Lee CC, Guo HR. Association between dioxin and diabetes mellitus in an endemic area of exposure in Taiwan a population-based study. Med (Baltimore). 2015;94(42):e1730. doi: 10.1097/MD.0000000000001730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steenland K, Calvert G, Ketchum N, Michalek J. Dioxin and diabetes mellitus: an analysis of the combined NIOSH and Ranch hand data. Occup Environ Med. 2001;58(10):641–648. doi: 10.1136/oem.58.10.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JS, Lim HS, Cho S, Cheong HK, Lim MK. Impact of agent orange exposure among Korean Vietnam veterans. Industrial Health. 2003;41(3):149–157. doi: 10.2486/indhealth.41.149. [DOI] [PubMed] [Google Scholar]
- 15.Matteo G, Hoyeck MP, Blair HL, Zebarth J, Rick KRC, Williams A, Gagné R, Buick JK, Yauk CL, Bruin JE. Prolonged low-dose dioxin exposure impairs metabolic adaptability to high-fat diet feeding in female but not male mice. Endocrinol. 2021;162(6):1–18. doi: 10.1210/endocr/bqab050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahim M, MacFarlane EM, Matteo G, Hoyeck MP, Rick KRC, Farokhi S, Copley CM, O’Dwyer S, Bruin JE. Functional cytochrome P450 1A enzymes are induced in mouse and human islets following pollutant exposure. Diabetologia. 2020;63(1):162–178. doi: 10.1007/s00125-019-05035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoyeck MP, Blair H, Ibrahim M, Solanki S, Elsawy M, Prakash A, Rick KRC, Matteo G, O’Dwyer S, Bruin JE. Long-term metabolic consequences of acute dioxin exposure differ between male and female mice. Sci Rep. 2020;10(1):1–10. doi: 10.1038/s41598-020-57973-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoyeck MP, Ching ME, Basu L, van Allen K, Palaniyandi J, Perera I, Poleo-Giorgani E, Hanson AA, Ghorbani P, Fullerton MD, Bruin JE. The aryl hydrocarbon receptor in β-cells mediates the effects of TCDD on glucose homeostasis in mice Mol Metab. 2024;81:101893. doi: 10.1016/j.molmet.2024.101893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sondermann NC, Faßbender S, Hartung F, Hätälä AM, Rolfes KM, Vogel CFA, Haarmann-Stemmann T. Functions of the aryl hydrocarbon receptor (AHR) beyond the canonical AHR/ARNT signaling pathway. Biochem Pharmacol. 2023;208():115371. doi: 10.1016/j.bcp.2022.115371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Safe S, Jin U, Park H, Chapkin RS, Jayaraman A. Aryl hydrocarbon receptor (AHR) ligands as selective AHR modulators (SAhRms). Int J Mol Sci. 2020;21(18):6654. doi: 10.3390/ijms21186654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao N, Whitelaw ML. The emerging roles of AhR in physiology and immunity. Biochem Pharmacol. 2013;86(5):561–570. doi: 10.1016/j.bcp.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Hahn ME, Karchner SI, Merson RR. Diversity as opportunity: insights from 600 million years of AHR evolution. Curr Opin Toxicol. 2017;2(February 2017):58–71. doi: 10.1016/j.cotox.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujii-Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Bioph Res Co. 2005;338(1):311–317. doi: 10.1016/j.bbrc.2005.08.162. [DOI] [PubMed] [Google Scholar]
- 24.Bock KW. Aryl hydrocarbon receptor (AHR) functions: balancing opposing processes including inflammatory reactions. Biochem Pharmacol. 2020;178(May):114093. doi: 10.1016/j.bcp.2020.114093. [DOI] [PubMed] [Google Scholar]
- 25.Denison MS, Soshilov AA, He G, Degroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci. 2011;124(1):1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pongratz I, Mason GGF, Poellingerf L. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity. J Biol Chem. 1992;267(19):13728–13734. [PubMed] [Google Scholar]
- 27.Vorrink SU, Domann FE. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1α signaling node. Chem Biol Interact. 2014;218:82–88. doi: 10.1016/j.cbi.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Q, Baldwin KT. 2.3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J Biol Chem. 2000;275(12):8432–8438. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- 29.Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc Natl Acad Sci USA. 2003;100(11):6517–6522. doi: 10.1073/pnas.1136688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabatini PV, Lynn FC. All-encomPassing regulation of β-cells: PAS domain proteins in β-cell dysfunction and diabetes. Trends In Endocrinol & Metab. 2015;26(1):49–57. doi: 10.1016/j.tem.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 31.McIntosh BE, Hogenesch JB, Bradfield CA. Mammalian per-arnt-sim proteins in environmental adaptation. Annu Rev Physiol. 2009;72(1):625–645. doi: 10.1146/annurev-physiol-021909-135922. [DOI] [PubMed] [Google Scholar]
- 32.Zhang M, Hu Y, Yang F, Zhang J, Zhang J, Yu W, Wang M, Lv X, Li J, Bai T, et al. Interaction between AhR and HIF-1 signaling pathways mediated by ARNT/HIF-1β. BMC Pharmacol Toxicol. 2022;23(1). doi: 10.1186/s40360-022-00564-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev Cell. 2009;17(6):755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15(4):678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nie M, Blankenship AL, Giesy JP. Interactions between aryl hydrocarbon receptor (AhR) and hypoxia signaling pathways. Environ Toxicol Pharmacol. 2001;10(1–2):17–27. doi: 10.1016/S1382-6689(01)00065-5. [DOI] [PubMed] [Google Scholar]
- 37.Calahorra J, Martínez-Lara E, Granadino-Roldán JM, Martí JM, Cañuelo A, Blanco S, Oliver FJ, Siles E. Crosstalk between hydroxytyrosol, a major olive oil phenol, and HIF-1 in MCF-7 breast cancer cells. Sci Rep. 2020;10(1). doi: 10.1038/s41598-020-63417-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Amine Z, Mauger JF, Imbeault P. CYP1A1, VEGFA and adipokine responses of human adipocytes co-exposed to PCB126 and hypoxia. Cells. 2022;11(15):2282. doi: 10.3390/cells11152282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan WK, Yao G, Gu YZ, Bradfield CA. Cross-talk between the aryl hydrocarbon receptor and hypoxia inducible factor signaling pathways: demonstration of competition and compensation. J Biol Chem. 1999;274(17):12115–12123. doi: 10.1074/jbc.274.17.12115. [DOI] [PubMed] [Google Scholar]
- 40.Gassmann M, Kvietikova I, Rolfs A, Wenger RH. Oxygen- and dioxin-regulated gene expression in mouse hepatoma cells. Kidney Int. 1997;51(2):567–574. doi: 10.1038/ki.1997.81. [DOI] [PubMed] [Google Scholar]
- 41.Gradin K, McGuire J, Wenger RH, Kvietikova I, Whitelaw ML, Toftgård R, Tora L, Gassmann M, Poellinger L. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the arnt transcription factor. Mol Cell Biol. 1996;16(10):5221–5231. doi: 10.1128/mcb.16.10.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlsson P-O, Liss P, Andersson A, Jansson L. Measurements of oxygen tension in native and transplanted rat pancreatic islets. Diabetes. 1998;47(7):1027–1032. doi: 10.2337/diabetes.47.7.1027. [DOI] [PubMed] [Google Scholar]
- 43.Olsson R, Carlsson PO. A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes. 2011;60(8):2068–2075. doi: 10.2337/db09-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cantley J, Grey ST, Maxwell PH, Withers DJ. The hypoxia response pathway and β-cell function. Diabetes Obes Metab. 2010;12(SUPPL. 2):159–167. doi: 10.1111/j.1463-1326.2010.01276.x. [DOI] [PubMed] [Google Scholar]
- 45.Miao G, Ostrowski RP, Mace J, Hough J, Hopper A, Peverini R, Chinnock R, Zhang J, Hathout E. Dynamic production of hypoxia-inducible factor-1α in early transplanted islets. Am J Transpl. 2006;6(11):2636–2643. doi: 10.1111/j.1600-6143.2006.01541.x. [DOI] [PubMed] [Google Scholar]
- 46.Cantley J, Walters SN, Jung MH, Weinberg A, Cowley MJ, Whitworth TP, Kaplan W, Hawthorne WJ, O’Connell PJ, Weir G, et al. A preexistent hypoxic gene signature predicts impaired islet graft function and glucose homeostasis. Cell Transplant. 2013;22(11):2147–2159. doi: 10.3727/096368912X658728. [DOI] [PubMed] [Google Scholar]
- 47.Li L, Chen C, Li D, Breivik K, Abbasi G, Li YF. What do we know about the production and release of persistent organic pollutants in the global environment?. Environ Sci: Adv. 2022;2(1):55–68. doi: 10.1039/d2va00145d. [DOI] [Google Scholar]
- 48.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32(11):1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 49.Novakovsky G, Sasaki S, Fornes O, Omur ME, Huang H, Bayly CL, Zhang D, Lim N, Cherkasov A, Pavlidis P, et al. In silico discovery of small molecules for efficient stem cell differentiation into definitive endoderm. Stem Cell Rep. 2023;18(3):765–781. doi: 10.1016/j.stemcr.2023.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, Lan LM, Nguyen VQ, Giacometti S, et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol. 2019;21(2):263–274. doi: 10.1038/s41556-018-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis JC, Ryaboshapkina M, Kenty JH, Eser Ö, Menon P, Melton S, Song DA. IAPP Marks mono-hormonal stem-cell derived β cells that maintain stable insulin production in vitro and in vivo. bioRxiv Preprint Apr 11 2024.04.10.587726. 2024; doi: 10.1101/2024.04.10.587726. [DOI] [Google Scholar]
- 52.Mar S, Filatov E, Sasaki S, Mojibian M, Zhang D, Yang A, Yang A, Nian C, Lynn FC. Tracking insulin- and glucagon-expressing cells in vitro and in vivo using a double-reporter human embryonic stem cell line. Diabetes. 2024;74(2):1–36. doi: 10.2337/db24-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, Millman JR. Acquisition of dynamic function in human stem cell-derived β cells. Stem Cell Rep. 2019;12(2):351–365. doi: 10.1016/j.stemcr.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hrvatin S, O’Donnell CW, Deng F, Millman JR, Pagliuca FW, DiIorio P, Rezania A, Gifford DK, Melton DA. Differentiated human stem cells resemble fetal, not adult, β cells. Proc Natl Acad Sci USA. 2014;111(8):3038–3043. doi: 10.1073/pnas.1400709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruin JE, Erener S, Vela J, Hu X, Johnson JD, Kurata HT, Lynn FC, Piret JM, Asadi A, Rezania A, et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014;12(1):194–208. doi: 10.1016/j.scr.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 56.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AMK. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272(9):5375–5381. doi: 10.1074/jbc.272.9.5375. [DOI] [PubMed] [Google Scholar]
- 57.Castro-Gutierrez R, Alkanani A, Mathews CE, Michels A, Russ HA. Protecting stem cell derived pancreatic beta-like cells from diabetogenic T cell recognition. Front Endocrinol. 2021;12:12. doi: 10.3389/fendo.2021.707881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carreau A, Hafny-Rahbi B, El Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15(6):1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishimura W, Iwasa H, Tumurkhuu M. Role of the transcription factor MAFA in the maintenance of pancreatic β-cells. Int J Mol Sci. 2022;23(9):4478. doi: 10.3390/ijms23094478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berger C, Zdzieblo D. Glucose transporters in pancreatic islets Pflugers Arch 472 9 1249–1272. 2020. doi: 10.1007/s00424-020-02383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bosma KJ, Rahim M, Singh K, Goleva SB, Wall ML, Xia J, Syring KE, Oeser JK, Poffenberger G, McGuinness OP, et al. Pancreatic islet beta cell-specific deletion of G6pc2 reduces fasting blood glucose. J Mol Endocrinol. 2020;64(4):235–248. doi: 10.1530/JME-20-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Brien RM. Moving on from GWAS: functional studies on the G6PC2 gene implicated in the regulation of fasting blood glucose. Curr Diab Rep. 2013;13(6):768–777. doi: 10.1007/s11892-013-0422-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem. 2009;284(43):29241–29245. doi: 10.1074/jbc.R109.025544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balboa D, Barsby T, Lithovius V, Saarimäki-Vire J, Omar-Hmeadi M, Dyachok O, Montaser H, Lund PE, Yang M, Ibrahim H, et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat Biotechnol. 2022;40(7):1042–1055. doi: 10.1038/s41587-022-01219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR. Single-cell transcriptome profiling reveals β cell maturation in stem cell-derived islets after transplantation. Cell Rep. 2020;32(8):108067. doi: 10.1016/j.celrep.2020.108067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pardo M, Tirosh O. Protective signalling effect of manganese superoxide dismutase in hypoxia-reoxygenation of hepatocytes. Free Radic Res. 2009;43(12):1225–1239. [DOI] [PubMed] [Google Scholar]
- 67.Kaewpila S, Venkataraman S, Buettner GR, Oberley LW. Manganese superoxide dismutase modulates hypoxia-inducible factor-1α induction via superoxide. Cancer Res. 2008;68(8):2781–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ewald JD, Lu Y, Ellis CE, Worton J, Kolic J, Sasaki S, Zhang D, dos Santos T, Spigelman AF, Bautista A, et al. HumanIslets.com: Improving accessibility, integration, and usability of human research islet data. Cell Metab. 37 (1) : 7–11 doi: 10.1016/j.cmet.2024.09.001. 2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nichols BR, Hentz KL, Aylward L, Hays SM, Lamb JC. Age-specific reference ranges for polychlorinated biphenyls (PCB) based on the NHANES 2001-2002 survey. J Toxicol Environ Health a - Part A: Current Issues. 2007;70(21):1873–1877. doi: 10.1080/15287390701457688. [DOI] [PubMed] [Google Scholar]
- 70.Hue O, Marcotte J, Berrigan F, Simoneau M, Doré J, Marceau P, Marceau S, Tremblay A, Teasdale N. Plasma concentration of organochlorine compounds is associated with age and not obesity. Chemosphere. 2007;67(7):1463–1467. doi: 10.1016/j.chemosphere.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 71.Fricker M, Goggins BJ, Mateer S, Jones B, Kim RY, Gellatly SL, Jarnicki AG, Powell N, Oliver BG, Radford-Smith G, et al. Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction. JCI Insight. 2018;3(3). doi: 10.1172/jci.insight.94040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Linden MA, Pincu Y, Martin SA, Woods JA, Baynard T. Moderate exercise training provides modest protection against adipose tissue inflammatory gene expression in response to high-fat feeding. Physiol Rep. 2014;2(7):e12071. doi: 10.14814/phy2.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jalali Z, Khademalhosseini M, Soltani N, Esmaeili Nadimi A. Smoking, alcohol and opioids effect on coronary microcirculation: an update overview. BMC Cardiovasc Disord. 2021;21(1). doi: 10.1186/s12872-021-01990-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salminen A. Mutual antagonism between aryl hydrocarbon receptor and hypoxia-inducible factor-1α (AhR/HIF-1α) signaling: impact on the aging process. Cellular Signalling. 2022;99(July):110445. doi: 10.1016/j.cellsig.2022.110445. [DOI] [PubMed] [Google Scholar]
- 75.Salminen A. Aryl hydrocarbon receptor (AhR) reveals evidence of antagonistic pleiotropy in the regulation of the aging process. Cellular And Mol Life Sci. 2022;79(9):1–22. doi: 10.1007/s00018-022-04520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pitt JA, Feng L, Abbott BD, Schmid J, Batt RE, Costich TG, Koury ST, Bofinger DP. Expression of AhR and ARNT mRNA in cultured human endometrial explants exposed to TCDD. Tox Sci. 62(2): 289–298. 2001. doi: 10.1093/toxsci/62.2.289. [DOI] [PubMed] [Google Scholar]
- 77.Lyon JG, Carr ALJ, Smith NP, Marfil-Garza B, Spigelman AF, Bautista A, O’Gorman D, Kin T, Shapiro AJ, Senior PA, MacDonald PE. Human research islet cell culture outcomes at the Alberta diabetes Institute IsletCore. Islets. 2024;16(1). doi: 10.1080/19382014.2024.2385510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lyon J, Manning Fox JE, Spigelman AF, Kim R, Smith N, O’Gorman D, Kin T, Shapiro AMJ, Rajotte RV, MacDonald PE. Research-focused isolation of human islets from donors with and without diabetes at the Alberta diabetes Institute IsletCore. Endocrinology. 2016;157(2):560–569. doi: 10.1210/en.2015-1562. [DOI] [PubMed] [Google Scholar]
- 79.Wassmer CH, Perrier Q, Combescure C, Pernin N, Parnaud G, Cottet-Dumoulin D, Brioudes E, Bellofatto K, Lebreton F, Berishvili E, et al. Impact of ischemia time on islet isolation success and posttransplantation outcomes: a retrospective study of 452 pancreas isolations. Am J Transpl. 2021;21(4):1493–1502. doi: 10.1111/ajt.16320. [DOI] [PubMed] [Google Scholar]
- 80.Hoyeck MP, Merhi RC, Blair HL, Spencer CD, Payant MA, Martin Alfonso DI, Zhang M, Matteo G, Chee MJ, Bruin JE. Female mice exposed to low doses of dioxin during pregnancy and lactation have increased susceptibility to diet-induced obesity and diabetes. Mol Metab. 2020;42(October):101104. doi: 10.1016/j.molmet.2020.101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Puri S, Cano DA, Hebrok M. A role for von hippel-lindau protein in pancreatic β-cell function. Diabetes. 2009;58(2):433–441. doi: 10.2337/db08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cantley J, Selman C, Shukla D, Abramov AY, Forstreuter F, Esteban MA, Claret M, Lingard SJ, Clements M, Harten SK, et al. Deletion of the Von Hippel-Lindau gene in pancreatic β cells impairs glucose homeostasis in mice. J Clin Invest. 2009;119(1):125–135. doi: 10.1172/JCI26934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zehetner J, Danzer C, Collins S, Eckhardt K, Gerber PA, Ballschmieter P, Galvanovskis J, Shimomura K, Ashcroft FM, Thorens B, et al. pVHL is a regulator of glucose metabolism and insulin secretion in pancreatic β cells. Genes Dev. 2008;22(22):3135–3146. doi: 10.1101/gad.496908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gerber PA, Bellomo EA, Hodson DJ, Meur G, Solomou A, Mitchell RK, Hollinshead M, Chimienti F, Bosco D, Hughes SJ, et al. Hypoxia lowers SLC30A8/ZnT8 expression and free cytosolic Zn2+ in pancreatic beta cells. Diabetologia. 2014;57(8):1635–1644. doi: 10.1007/s00125-014-3266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26(10):501–518. doi: 10.1089/ars.2016.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Komatsu H, Cook C, Wang CH, Medrano L, Lin H, Kandeel F, Tai YC, Mullen Y, Bencharit S. Oxygen environment and islet size are the primary limiting factors of isolated pancreatic islet survival. PLOS ONE. 2017;12(8):e0183780. doi: 10.1371/journal.pone.0183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han Y, Yang K, Proweller A, Zhou G, Jain MK, Ramirez-Bergeron DL. Inhibition of ARNT severely compromises endothelial cell viability and function in response to moderate hypoxia. Angiogenesis. 2012;15(3):409–420. doi: 10.1007/s10456-012-9269-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Salceda S, Beck I, Caro J. Absolute requirement of aryl hydrocarbon receptor nuclear translocator protein for gene activation by hypoxia. Archiv Biochem Biophys. 1996;334(2):389–394. doi: 10.1006/abbi.1996.0469. [DOI] [PubMed] [Google Scholar]
- 89.Wood SM, Gleadle JM, Pugh CW, Hankinson O, Ratcliffe PJ. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxie induction of gene expression: studies in arnt-deficient cells. J Biol Chem. 1996;271(25):15117–15123. doi: 10.1074/jbc.271.25.15117. [DOI] [PubMed] [Google Scholar]
- 90.Chilov D, Camenisch G, Kvietikova I, Ziegler U, Gassman M, Wenger RH. Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1alpha. J Cell Sci. 1999;112(8):1203–1212. doi: 10.1242/jcs.112.8.1203. [DOI] [PubMed] [Google Scholar]
- 91.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386(6623):403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 92.Gautier-Stein A, Soty M, Chilloux J, Zitoun C, Rajas F, Mithieux G. Glucotoxicity induces glucose-6-phosphatase catalytic unit expression by acting on the interaction of HIF-1α with CREB-binding protein. Diabetes. 2012;61(10):2451–2460. doi: 10.2337/db11-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Määttä J, Sissala N, Dimova EY, Serpi R, Moore LG, Koivunen P. Hypoxia causes reductions in birth weight by altering maternal glucose and lipid metabolism. Sci Rep. 2018;8(1). doi: 10.1038/s41598-018-31908-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang C, Wang M, Ji F, Peng Y, Wang B, Zhao J, Wu J, Zhao H. A novel glucose metabolism-related gene signature for overall survival prediction in patients with glioblastoma. biomed Res Int. 2021;2021(1):2021. doi: 10.1155/2021/8872977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanders E, Diehl S. Analysis and interpretation of transcriptomic data obtained from extended Warburg effect genes in patients with clear cell renal cell carcinoma. Oncoscience. 2015;2(2):151–186. doi: 10.18632/oncoscience.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hara Y, Watanabe N. Changes in expression of genes related to glucose metabolism in liver and skeletal muscle of rats exposed to acute hypoxia. Heliyon. 2020;6(7):e04334. doi: 10.1016/j.heliyon.2020.e04334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dogra RS, Vaidyanathan P, Prabakar KR, Marshall KE, Hutton JC, Pugliese A. Alternative splicing of G6PC2, the gene coding for the islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), results in differential expression in human thymus and spleen compared with pancreas. Diabetologia. 2006;49(5):953–957. doi: 10.1007/s00125-006-0185-8. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y, Martin CC, Oeser JK, Sarkar S, McGuinness OP, Hutton JC, O’Brien RM. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia. 2007;50(4):774–778. doi: 10.1007/s00125-006-0564-1. [DOI] [PubMed] [Google Scholar]
- 99.Chen JW, Wang SL, Liao PC, Chen HY, Ko YC, Lee CC. Relationship between insulin sensitivity and exposure to dioxins and polychlorinated biphenyls in pregnant women. Environ Res. 2008;107(2):245–253. doi: 10.1016/j.envres.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 100.Chen W-M, Erdos MR, Jackson AU, Saxena R, Sanna S, Silver KD, Timpson NJ, Hansen T, Orrù M, Grazia Piras M, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest. 2008; doi: 10.1172/jci34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen K, Zhang J, Huang Y, Tian X, Yang Y, Dong A. Single-cell RNA-seq transcriptomic landscape of human and mouse islets and pathological alterations of diabetes. iScience. 2022;25(11):105366. doi: 10.1016/j.isci.2022.105366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Catrina SB, Zheng X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia. 2021;64(4):709–716. doi: 10.1007/s00125-021-05380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kolic J, Sun WG, Cen HH, Ewald JD, Rogalski JC, Sasaki S, Sun H, Rajesh V, Xia YH, Moravcova R, et al. Proteomic predictors of individualized nutrient-specific insulin secretion in health and disease. Cell Metab. 2024;36(7):1619–1633.e5. doi: 10.1016/j.cmet.2024.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rahim M, Nakhe AY, Banerjee DR, Overway EM, Bosma KJ, Rosch JC, Oeser JK, Wang B, Lippmann ES, Jacobson DA, et al. Glucose-6-phosphatase catalytic subunit 2 negatively regulates glucose oxidation and insulin secretion in pancreatic β-cells. J Biol Chem. 2022;298(4):101729. doi: 10.1016/j.jbc.2022.101729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu N, Cai X, Liu T, Zou J, Wang L, Wang G, Liu Y, Ding X, Zhang B, Sun P, et al. Hypoxia-inducible factor-1α mediates the expression of mature β cell-disallowed genes in hypoxia-induced β cell dedifferentiation. Biochem Biophys Res Commun. 2020;523(2):382–388. doi: 10.1016/j.bbrc.2019.12.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.