

Abstract
Factor VII (FVII) deficiency is a rare bleeding disorder with a prevalence of approximately 1:300,000–500,000 individuals. We explored whether adeno-associated virus (AAV)-mediated gene therapy can achieve durable and functional expression of human FVII (hFVII) in vivo. Wild-type hFVII (hFVIIwt) and a naturally occurring splice variant designated hFVII(-22) (GenBank: NM_019616.4) were expressed under the control of the Apolipoprotein E-derived hepatic locus control region and the α1-anti-trypsin promoter. Expression cassettes were packaged in either recombinant AAV5 (rAAV5) or recombinant AAV8 (rAAV8). For both hFVIIwt and hFVII(-22), 5- to 20-fold higher plasma levels of hFVII could be obtained when rAAV8 was used as a vector as opposed to rAAV5. Interestingly, hFVII levels obtained by employing rAAV8 expressing the hFVII(-22) cDNA variant were approximately 10 times higher than those obtained using rAAV8 expressing hFVIIwt. Based on these results, we generated an rAAV8-based gene therapy vector encoding hFVII(-22) and evaluated long-term expression in vivo. Employing a vector dose of 0.8 × 1012 genome copies (gc)/kg, we observed 48 weeks of functional hFVII expression which peaked at 16 IU/mL and stabilized at 7 IU/mL. These results support the pre-clinical development of AAV8-mediated delivery employing the splice-variant hFVII(-22) for patients suffering from FVII deficiency.
Keywords: gene therapy, FVII deficiency, rare bleeding disorders, liver-directed gene therapy, augmentation therapy, AAV8, AAV5
Graphical abstract
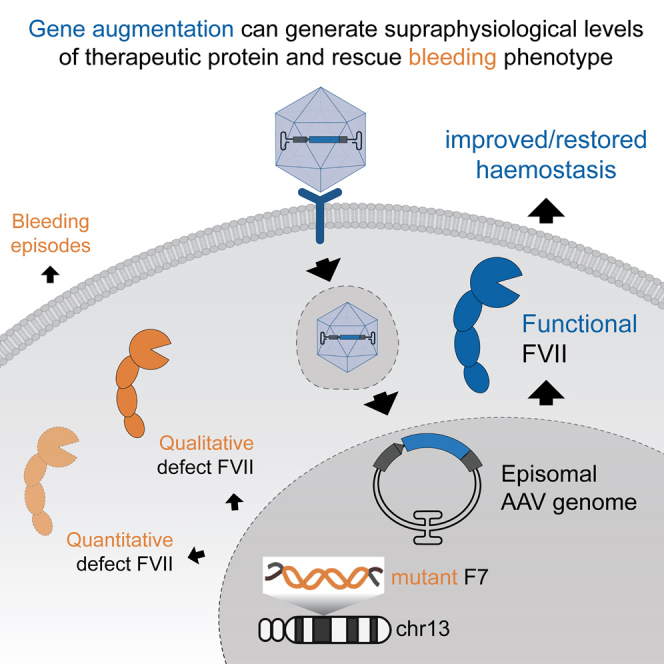
D’Amico and colleagues showed that intravenous injection of AAV8 vector carrying an optimized human FVII transgene results in sustained supraphysiological plasma levels of human FVII in wild-type mice with no adverse events detected after 48 weeks. This result justifies further pre-clinical study for gene therapy for FVII deficiency.
Introduction
Factor VII (FVII) deficiency is a rare autosomal recessive bleeding disorder with an estimated prevalence of 1 in every 300,000–500,000 individuals.1,2 Severe FVII deficiency is caused by homozygous or compound heterozygous mutations in F7 (13q34), which result in low or undetectable plasma FVII levels.3 FVII concentration in normal human plasma is extremely low (1.5–3 μg/mL), and, according to the International Registry of Factor VII deficiency and the Seven Treatment Evaluation Registry, an FVII level of less than or equal to 26% of normal4 indicate potentially significant deficiency and levels <5% are associated with a severe bleeding phenotype.5 However, in contrast to other bleeding disorders, such as hemophilia A/B, bleeding phenotypes in FVII deficiency are poorly correlated with FVII activity levels, and it is not unusual to encounter patients classified as severe who do not report regular bleeding episodes.2
Currently, the best therapeutic approach to treat FVII deficiency is prophylaxis regimes of either 30 μg/kg dose of recombinant activated FVII (rFVIIa) three times a week or 20–40 mg/kg of plasma-derived FVII two to three times a week for long-term prophylaxis.6,7,8 The need for regular repeated administration is due to the fact that FVII has the shortest half-life of the coagulation factors (about 5 h), which is only modestly longer than the half-life of FVIIa (about 2 h).5 Dose intervals are even shorter in children, for whom plasma clearance is more rapid than in adults (3–4 h).6
Gene therapy can eliminate the need for recurrent prophylactic FVII administration by providing sustained, endogenous high-level coagulation factor production and prevent bleeding with limited safety concerns.5 This approach is particularly effective for monogenic diseases, including many bleeding disorders like hemophilia A/B and FVII deficiencies, for which symptoms are typically associated with loss of function for a single protein.5 Notably, FVII is structurally and functionally related to human Factor IX (FIX), for which successful gene therapy has been developed9 and is currently undergoing several clinical trials10,11,12 with at least two products, etranacogene dezaparvocvec and fidanacogene elaparvovec, receiving marketing authorization.13 Restoring plasma FVII levels above 5% is predicted to effectively protect patients from bleeding symptoms and is a feasible target, considering that recent gene therapy trails for hemophilia A/B demonstrated sustained functional protein levels above 5% for many years after treatment and significant reductions in bleeding events.5
Given the rarity of FVII deficiency, it is still quite challenging to perform prospective pre-clinical studies to evaluate the feasibility of novel treatments, such as gene therapy. However, some preliminary studies on the application of adeno-associated viruses (AAVs) for liver-directed FVII gene transfer in large animal models have produced promising results. Administration of an AAV8 vector encoding canine FVII to FVII-deficient male dogs at a dose of 6 × 1011 genome copies (gc)/kg resulted in sustained and stable FVII levels up to 15% of normal with no reported side effects for over 1 year post-injection.14 In another study, administration of 4 × 1011 gc/kg of a self-complementary AAV5 vector encoding human FVII (hFVII) to adult non-human primates (NHPs) resulted in stable FVII levels of approximately 7.2% of normal up to 6 months post-injection.15 Another study conducted in an FVII-deficient mouse model in which an AAV8 vector encoding for mouse FVII (mFVII) was delivered at 35 days of life also showed significant improvement in the survival rate of treated versus untreated animals for at least 65 days after administration.15 This was an important finding, given that more than 20% of patients with severe FVII deficiency have intracranial hemorrhage, the majority of which occurs in the first few weeks of life to be associated with high morbidity and mortality.16
The aim of the present study is to provide further evidence that AAV-directed gene therapy is a viable approach to treat FVII deficiency. First of all, we conducted a proof-of-concept study in which we directly compare the efficacies of recombinant AAV9 (rAAV9), AAV5 (rAAV5), and AAV8 (rAAV5) for delivering an FVII transcript in wild-type mice. Secondly, we optimized both the expression cassette and the cDNA sequence of the therapeutic protein to improve efficacy. Finally, in line with current understanding of the impact of 5′-cytosine-phosphate-guanine-3’ (CpG) content in long-term expression of rAAV-encoded transgenes,17,18,19,20,21,22,23,24 we generated two CpG-deprived FVII variants to evaluate the effect of reducing the CpG content on plasma FVII levels and protein activity.
Results
Liver transduction is more effective with rAAV8 compared to rAAV5 or rAAV9
Biodistribution analysis of rAAV5, rAAV8, and rAAV9 in wild-type C57BL/6 mice was performed using a pAAV-chicken beta-actin (CAG)-luciferase construct delivered by tail vein injection at a dose of 1.6 × 1013 gc/kg (Figure 1A). At weeks 1, 2, 4, 6, and 10 post-injection, bioluminescence measured in the In Vivo Imaging System (IVIS, Xenogen) showed that rAAV9 was detected in multiple organs, whereas both rAAV5 and rAAV8 showed more concentrated signal in the liver (Figure 1B), which is in agreement with previous findings.25 Quantification of transgene expression confirmed that liver transduction was significantly higher for rAAV8 and rAAV9, compared to rAAV5 (Figure 1C). Moreover, rAAV5-mediated luciferase expression declined over time, whereas expression in rAAV8- and rAAV9-transduced animals stabilized over time (Figure 1C). Based on their preferential transgene expression in the liver, the rAAV5 and rAAV8 capsids were selected for subsequent experiments.
Figure 1.

In vivo biodistribution of AAV5, AAV8, and AAV9
(A) CAG-luciferase construct was packaged into rAAV5, rAAV8, or rAAV9 to test which wild-type serotype will target the liver more efficiently at a dose of 1.6 × 1012 gc/kg in wild-type C57BL/6 mice (n = 5/group). (B) Representative individual pictures of the IVIS measurement taken at 1, 2, 4, 6, and 10 weeks post-injection. Animals were subjected to IVIS measurements after intraperitoneal injection of D-Luciferin. The AAV-mediated expressed transgene luciferase is detected by the emission of photons (scale bar is 1 × 106 photons/cm2/s/sr within the ROI. (C) Quantified radiance has been transformed into logarithmic scale and presented as means (SD). Transformed data were analyzed by two-way ANOVA with Tuckey’s multiple comparisons test. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Liver transduction of human FVII is higher with rAAV8 than rAAV5 even at 10-fold lower dose
A second cohort of wild-type C57BL/6 mice underwent tail vein injection of rAAV8 or rAAV5 encoding for wild-type human FVII (hFVIIwt) at doses of 1.6 × 1012 gc/kg and 1.6 × 1013 gc/kg (Figure 2A). All human FVII (hFVII) transcripts used in this study were under the control of the well-characterized Apolipoprotein E (ApoE)-derived hepatic locus control region (HCR)-α1-anti-trypsin (ApoEHCR-hAAT) promoter,26 hereafter referred to as hAAT promoter, which produced long-lasting stable expression using both rAAV5 and rAAV8 capsids (Figure S1).
Figure 2.

Evaluation of AAV5- versus AAV8-mediated FVII expression
(A) rAAV5 was packaged with either hFVIIwt or cohFVII(-22) under the control of the hAAT promoter and injected at a dose of 1.6 × 1013 gc/kg; while rAAV8 was packaged with the same constructs and injected at a dose of 1.6 × 1012 gc/kg instead; (B) hFVIIwt levels in plasma of male C57BL/6 mice delivered by either rAAV5 or rAAV8; (C) human cohFVII(-22) levels in plasma of male C57BL/6 mice delivered by either rAAV5 or rAAV8. In both rAAV5- and rAAV8-mediated expression of hFVII recombinant protein, the hFVIIwt was outperformed by the shorter hFVII(-22) transcript. Data are presented as means (SD). See also Figure S2. Data were analyzed by Student’s t test. ∗∗∗∗p < 0.0001.
rAAV8 produced significantly better transduction in the liver at 10 weeks after injection, compared to rAAV5. hFVII plasma levels were also higher for the rAAV8-hAAT-hFVIIwt (3.0 ± 0.6 U/mL) compared to the rAAV5-hAAT-hFVIIwt group (0.1 ± 0.1 U/mL) (Figure 2B).
We also studied whether the naturally occurring shortened version of hFVII, harboring a 22 amino acid deletion in the propeptide ([hFVII(-22)]; GenBank: NM_019616.4), would result in the same levels of transgene expression as hFVIIwt. For these experiments, we adopted the fully CpG-depleted codon-optimized version of hFVII(-22) developed by Binny et al.,15 cohFVII(-22). Tail vein injection was performed in two additional groups of animals to deliver either rAAV8-hAAT-cohFVII(-22) or rAAV5-hAAT-cohFVII(-22) at the same conditions used for the hFVIIwt constructs (Figure 2A). Comparison of circulating hFVII plasma levels showed that, again, transgene expression levels delivered by rAAV8 were higher, with 23.4 ± 9.7 U/mL of cohFVII(-22) detected at week 8 post-injection as opposed to only 0.9 ± 1.2 U/mL for transgene delivered by rAAV5 (Figure 2C). For the different conditions, stable levels of hFVII expression were observed for up to 32 weeks (Figures 2B and 2C). Notably, all antigen measurements fell within the detection range of the hFVII-specific enzyme-linked immunosorbent assay (ELISA), as described in the materials and methods section.
Overall, these results show that rAAV8 transduces mouse liver more efficiently than rAAV5. In addition, significantly higher plasma levels of hFVII are obtained by using the splice variant lacking 22 amino acids of the hFVII propeptide regardless of the delivery capsid (Figure S2).
rAAV8 preferentially transduces the liver while rAAV5 also transduces lung tissue
The biodistribution for the aforementioned combinations of rAAV5 and rAAV8 vectors with hFVIIwt or cohFVII(-22) transgenes was determined based on vector copy numbers in the lung, spleen, kidney, and liver at 32 weeks following administration of rAAV5 either encoding hFVIIwt or cohFVII(-22) (Figure 3). Most of rAAV5 copies were found in the lungs and liver, and relatively high copy numbers were also found in the kidney and spleen (Figure 3, left panel). rAAV8 harboring the same payloads was primarily found in the liver, with much lower levels in the kidney, spleen, and lungs (Figure 3, right panel). Muscle transduction was not detected. Interestingly, the presence of genomic copies in the liver was roughly 10 times higher for rAAV8-mediated delivery than rAAV5-mediated delivery (Figure S3). In summary, in wild-type mice, rAAV5 preferentially transduces the lungs and, to a lesser extent, the liver, whereas rAAV8 mainly transduces the liver.
Figure 3.

Biodistribution of AAV5 and AAV8 in mice tissues
The chart summarized the relative distribution of both rAAV5 vectors and rAAV8 vectors copies per transduced organ detected by qPCR. For extended dataset, see also Figure S3.
AAV-delivery of a codon-optimized hFVII construct results in long-term, stable in vivo expression of active hFVII
A third cohort of 24 wild-type C57BL/6 mice underwent tail vein injection to perform long-term evaluation of codon-optimized hFVII delivered by rAAV8: hAAT-RBD-hFVIIwt vs. hAAT-hFVIIwt and hAAT-RBD-hFVII(-22) vs. hAAT-cohFVII(-22) (Figure 4).
Figure 4.

Long-term expression using AAV8 capsid
(A) AAV8 capsid was packaged with either FVIIwt and coFVII(-22) or two alternative codon-optimized versions, RBD-FVIIwt and RBD-FVII(-22), under the control of the hAAT promoter. Injection was performed at a dose of 0.8 × 1012 gc/kg in wild-type C57BL/6 mice (n = 6/group). (B) Plasma levels of FVII after blood collection up to 48 weeks post-injection. No difference was observed between FVIIwt and the correspondent codon-optimized version RBD-FVIIwt as well as between coFVII(-22) and its codon-optimized version RBD-FVII(-22); however, both RBD-FVII(-22) and coFVII(-22) overperformed FVII(wt) and RBD-FVII(wt). For extended dataset, see also Figure S5. (C) FVII activity assay is used to test the same samples used for plasma antigen level. FVII activity assay is directly proportional to the amount of active protein present in the plasma of the injected mice. Dotted black line corresponds to mouse FVII threshold level (1 U/mL). Transcripts derived from FVII(-22) variants were confirmed to overperform FVIIwt variants. For extended dataset, see also Figure S6. Graphs show group means (SD); data were analyzed by two-way ANOVA with Tuckey’s multiple comparisons test. ns, non-significant; ∗∗∗∗p < 0.0001.
Two novel codon-optimized versions, RBD-hFVIIwt and RBD-hFVII(-22), were designed to reduce the overall CpG content of the hFVII cDNA. In addition, specific 5′and 3′ UTR elements were added to the mRNA to further promote expression (Figure S4). Mice were injected with 0.8 × 1012 gc/kg of rAAV8 capsid encoding for RBD-hFVIIwt RBD-hFVII(-22), hFVIIwt, or cohFVII(-22) under the control of the same hAAT promoter (Figure 4A). Based on the results of the hFVII-specific ELISA assay, there were no significant differences in hFVII antigen levels between RBD-hFVIIwt and hFVIIwt or cohFVII(-22) and RBD-hFVII(-22) transgenes (Figure 4B). In particular, at 4 weeks post-injection, both cohFVII(-22) and RBD-hFVII(-22) resulted in similar antigen levels (16.6 ± 6.7 U/mL and 12.9 ± 4.3 U/mL, respectively). Furthermore, these antigen levels stabilized at 7.0 ± 2.7 U/mL for cohFVII(-22) and 4.5 ± 2.0 U/mL for RBD-hFVII(-22) at 48 weeks post-injection (Figure 4B). Moreover, for rAAV8-hAAT-hFVIIwt and rAAV8-hAAT-RBD-hFVIIwt, plasma hFVII levels were approximately 10-fold lower than the encoding codon-optimized hFVII(-22) variants (Figures 4B and S5).
We also determined FVII activity levels in plasma samples from these mice (Figure 4C). The FVII chromogenic assay does not distinguish between mouse FVII (mFVII) and hFVII. Nevertheless, FVII activity levels were consistent with the results of the hFVII antigen ELISA (Figure 4B). Levels as high as 15–20 U/mL were achieved for rAAV8 expressing cohFVII(-22) and RBD-FVII(-22), similar to the FVII antigen data (Figure S6). Our data show that long-term expression of biologically active hFVII(-22) under the control of the hAAT promoter can be achieved using rAAV8 delivery. No statistically significant differences were observed between codon-optimized and no-codon-optimized variants.
Dose-response evaluation confirms AAV-mediated gene therapy is dependent on the dosage of virus infused
A fourth cohort of 24 wild-type C57BL/6 mice was used to perform a dosing study to try to identify the most optimal therapeutic window in mice. An rAAV8 vector encoding for the hAAT-RBD-FVII(-22) expression cassette was administered intravenously into the tail vein at 0.3 × 1011, 0.8 × 1011, 1.6 × 1011, or 0.8 × 1012 gc/kg (Figure 5A). We chose these doses to reflect the FVII levels we observed previously (Figure 4B). Mice infused with 0.8 × 1012 gc/kg showed the highest FVII levels, which were comparable to our previous observations (Figure 5B). Wild-type mice injected with the dose of 1.6 × 1011 gc/kg showed FVII levels of 1 U/mL corresponding to physiological levels, while lower levels of hFVII antigen were observed at both 0.8 × 1011 and 0.3 × 1011 gc/kg (Figure 5B, right zoomed-in view). As expected, our results indicate that hFVII levels obtained are dependent on the dosage of rAAV infused. Dosages of 0.8 × 1012 and 1.6 × 1011 gc/kg may give rise to hFVII levels that are therapeutically effective.
Figure 5.

In vivo dosing study
(A) To identify the optimal dosage, hAAT-RBD-FVII(-22) was packaged into AAV8. Injections were performed at a concentration of 0.3 × 1011, 0.8 × 1011, 1.6 × 1011, or 0.8 × 1012 gc/kg in wild-type C57BL/6 mice (n = 5–6/group); (B) FVII antigen level detected after injection at 0.3 × 1011, 0.8 × 1011, 1.6 × 1011, or 0.8 × 1012 gc/kg or 1.6 × 1011, 0.8 × 1011, and 0.3 × 1011 gc/kg. Graphs show group means (SD); data were analyzed by two-way ANOVA with Tuckey’s multiple comparisons test. ns, non-significant; ∗∗∗∗p < 0.0001.
Long-term in vivo exposure to AAV capsids induces the production of anti-capsid antibodies, in contrast to PBS-injected control mice
Anti-AAV8 antibodies were measured in the plasma of mice from the long-term study (Figure 4) to evaluate whether an immune response could explain the sharp decline in hFVII levels observed between weeks 4 and 8 post-injection (Figure 4B). Plasma samples collected at 6, 12, 24, and 48 weeks were used and compared to those from PBS-injected control mice of the same background. Notably, anti-AAV8 antibodies were detected at all tested time points in treated animals, whereas PBS-injected controls remained negative. The anti-capsid immune response showed a tendency to decline over time, with a significant reduction observed between 6 and 48 weeks (Figure 6). These results indicate the presence of a sustained immune response against the AAV8 capsid that gradually diminishes over time.
Figure 6.

Anti-AAV8 antibodies in treated samples
The bar graph showed plasma levels of anti-AAV8 antibodies in mice injected with rAAV8 capsids encoding for FVIIwt, coFVII(-22) RBD-FVIIwt, and RBD-FVII(-22), under the control of the hAAT promoter, after 6, 12, and 48 weeks post-injection as well as PBS-injected animals (n = 5–6/group). Graphs show group means (SD); data were analyzed by one-way ANOVA with Tuckey’s multiple comparisons test; ∗p < 0.05.
Discussion
FVII deficiency is a rare bleeding disorder caused by mutations in F7, resulting in a mild to severe bleeding phenotypes. Clinical manifestation of this disease is extremely heterogeneous, with about one-third of non-severe patients remaining asymptomatic throughout their lives and patients classified as severe often showing lower-than-expected tendencies for bleeding events. Their wide range of clinical manifestations implies that factors other than FVII, such as tissue factor, von Willebrand factor, or platelets levels, also influence bleeding risk.2,3 As a result, prophylactic treatments are usually only recommended for patients with history of severe bleeding episodes in either the central nervous system or gastro-intestinal tract.3 Treatment involves either administration of recombinant FVIIa concentrate, plasma-derived FVII concentrate, FVII-containing prothrombin complex concentrate, or plasma.8 However, the short half-life of these products necessitate lifelong administration of multiple injections per week, impacting patients’ quality of life.5,6,7 A single therapeutic infusion with long-lasting effects such as gene therapy would bring great benefits to FVII-deficient patients. Previous studies using a canine model for FVII deficiency have demonstrated that AAV8-mediated gene therapy can produce supraphysiological FVII levels for over 2.7 years.14 Moreover, an independent study in rhesus macaques reported restored FVII levels of 10%–20% following AAV-mediated gene therapy, further emphasizing the feasibility of this technology for treating FVII deficiency.15
In this study, we provide further evidence that gene therapy is a viable option for treating FVII deficiency and justify further pre-clinical testing. Our goal was not to evaluate the effects of AAV-directed gene transfer on bleeding phenotype, which has already been documented,15 but to focus on investigating the stability and long-term efficacy of different AAV vectors, as well as transgene codon-optimization strategies, an area that remains largely unexplored. Firstly, we directly compared the effectiveness of rAAV5, rAAV8, and rAAV9 vectors for liver-directed gene therapy. In clinical studies, AAV8 is used most frequently for bleeding disorders, AAV9 for lysosomal storage disorders, and AAV5 for multiple other disorders that require gene transfer in the liver.27 Consistent with previous reports,28 rAAV9 showed less-specific distribution throughout the body, while rAAV5 and rAAV8 both resulted in effective liver transduction. Both rAAV5 and rAAV8 have been successfully used for hemophilia A and B gene therapy.13,18 However, rAAV5 has lower transduction efficiency and is usually administered at doses between 200 and 600 × 1011 gc/kg, compared to rAAV8, which is used at a dose between 6 and 120 × 1011 gc/kg.29 To compensate for this difference in transduction efficiency, we injected rAAV5 at a 10-fold higher dose than rAAV8, and, yet, rAAV8-mediated delivery resulted in higher FVII levels. Vector genome quantification via qPCR-based biodistribution assays revealed that rAAV5 mainly targeted the lung and, to a lesser extent, the liver, spleen, and kidney, whereas rAAV8 was detected mostly in the liver. This result seems to contradict luciferase data in which both rAAV8 and rAAV5 are mainly detected in the liver, which may result from differences in the efficiency of tissue transduction versus protein translation in different tissues. Our results suggest that AAV vectors may efficiently transduce the lung. Nevertheless, the lack of luminescent signal observed suggests that, despite the presence of a strong CAG promoter, target cells for AAV5 in the lung do not efficiently produce the luciferase protein as the liver. It is therefore important to note that luciferase detection only provides a rough estimate of AAV distribution, which is biased by the translational efficiency of a specific tissue, whereas vector genome detection offers a more accurate measure of the amount of viral genome uptake per tissue. These data highlight the importance of targeting tissue-specific promoters to prevent off-target expression, which could cause unwanted side effects in tissues where normal endogenous expression of the protein is absent or low. The prominent lung targeting of rAAV5 is not entirely surprising, since the putative main receptor of AAV5, 2,3-N-linked sialic acid, is expressed on the apical surface of airway epithelial cells and makes this capsid a good candidate for gene delivery to the respiratory tract.30,31,32 A similar outcome was also observed by Mattar and coworkers when AAV8 and AAV5 tropism was compared in NHPs.33
It is interesting to note that, in terms of capsid structure, the AAV5 capsid is the most phylogenetically distinct vector serotype, and, as such, it is also the capsid for which the general population has the lowest prevalence of neutralizing antibodies (Nabs).34 The presence of Nabs can potentially inhibit the transduction of target cells by rAAV vectors, leading to the exclusion of patients with high circulating Nab concentrations from clinical trials.35 However, anti-AAV5 Nab titers are low in seropositive individuals,36,37 and, as a result, this capsid is expected to be suitable for a greater proportion of patients.12,38,39 Nevertheless, low-titer antibodies can potentially exacerbate toxicities linked to administration of high vector doses.40 Considering the lower transduction efficiency of AAV5, we therefore chose AAV8 to further assess the clinical feasibility FVII gene therapy.
While the selection of a delivery vector is essential to maximize the level of gene expression, the sequence of the therapeutic DNA cassette is probably equally as important for determining therapeutic potential. This is exemplified by the success of hemophilia B clinical trials of the FIX-Padua variant, for which a single point-mutation resulted in a hyperactive FIX protein with 8-fold higher expression than wild-type FIX.41 A recent report described another FIX variant harboring three amino acid substitutions, which significantly outperformed the FIX-Padua variant.42 In our study, we compared two alternative mRNA transcripts encoding hFVII: wild-type hFVII ([FVIIwt]; GenBank: NM_000131.4) encoding the longest isoform composed of 466 amino acid residues including a 1–20 amino acid secretory leader (signal-peptide) and 21–60 amino acid propeptide, and an FVII transcript lacking 22 amino acids in exon 2, encoding residues 22–43 in the propeptide ([FVII(-22)]; GenBank: NM_019616.4)43 but resulting in an identical mature FVII protein to FVIIwt. The propeptide region of FVII and other vitamin K-dependent proteins is needed for the carboxylation of N-terminal gamma-carboxyglutamic acid-rich domain, an essential post-translational modification required for phospholipid binding.44,45,46 Deletion of 22 amino acids from this region does not result in a loss of function, and our results are in line with this statement, as plasma of mice infused with rAAV8 encoding for hFVII variants lacking these 22 amino acids showed matching activity and antigen levels with FVII(wt). hFVII(-22) is the major transcript in healthy individuals, and deletion of residues 22–43 has no impact function, positioning it as a prime candidate for clinical development of FVII gene therapy.43 Our results clearly indicated that hFVII(-22) produced significant higher levels of circulating active FVII compared to wild-type FVII, regardless of the capsid used. This result confirmed what was previously observed in an elegant study by Binny and coworkers.15
Prior hemophilia gene therapy trials also highlight the impact of codon optimization on transgene efficiency. In the AAV8-based FIX-Padua BAX335 phase 1/2 study, a significant loss of transgene expression was observed, which was linked to the adopted codon-optimization strategy causing an accidental CpG enrichment.18 A similar story was observed for DTX101, an AAVrh10-expressing a codon-optimized FIX gene with no CpG reduction, resulting in a loss of FIX expression.21 In all these cases, the algorithms used for codon optimization were originally designed to enhance protein expression. However, this approach inadvertently led to the introduction of clusters of CpG motifs into the open reading frame of the transgene coding sequence.17 Since CpGs are known as innate pathogen-associated molecular patterns that trigger adaptive effector function, particularly Toll-like receptor 9 pathways,47 it is wise to implement codon-optimization strategies that focus on depleting CpG content rather than increasing it.17 Interestingly, it has also been suggested that complete CpG removal may be as detrimental as having high CpG content, as this may cause protein misfolding due to altered translational kinetics.48 These results further highlight the importance of balancing transgene sequence optimization aimed at improving gene expression with modifications that reinforce the safety and durability of the final product. With this goal, we evaluated the impact on duration of expression for different codon-optimization strategies: the coFVII(-22) variant reported by Binny et al., with full CpG motif depletion,15 and RBD-FVII(-22), an FVII(-22) variant that retains some CpG motifs. Both transcripts showed similar long-term, stable expression, as well as full biological activity that matched plasma antigen levels. These findings suggest that neither partial nor complete CpG motif removal compromises the biological activity or duration of AAV-mediated hFVII expression.
A major concern for AAV8 gene therapy is to consider the prevalence of AAV8 Nabs that may impact efficacy of the therapeutic approach. In the general population, AAV8 Nabs fall in the range of approximately 30%–45% in the general population, including patients with hemophilia.49 We assume that the prevalence will be similar for FVII-deficient patients. Treatment of patients with pre-existing AAV8 antibodies may potentially be preceded by B cell-depleting strategies such as rituximab or immunoglobulin G (IgG) cleaving proteases like IdeS, which are likely to provide a window for the administration of AAV8 in patients with pre-existing antibodies.50 Our current data suggest that AAV8-treated mice develop an anti-AAV8 antibody response, which may have contributed to the initial reduction in transgene expression observed between 4 and 8 weeks post-injection. However, we also observed that hFVII levels subsequently stabilized and remained consistent throughout the 48-week duration of the experiment. Interestingly, the anti-capsid immune response showed a tendency to decline by week 48. We speculate that this sustained expression may be attributable to episomal persistence of the vector, with a subset of successfully transduced cells responsible for long-term expression, whereas the decline in anti-capsid antibodies may reflect the clearance of the vector from the circulation. If neutralizing activity had remained persistent, a continued decline in hFVII levels over time would have been expected along with the maintenance or even increase of Nabs. Furthermore, although we cannot rule out the presence of anti-hFVII antibodies, our data support the notion that, at the very least, a potential immune response against the transgene does not interfere with long-term expression of functional FVII. While these findings do not constitute a comprehensive immunogenicity assessment, and further analysis would be needed to evaluate potential anti-hFVII effects on protein clearance and half-life, they offer preliminary insights and further support the feasibility of this gene therapy approach for FVII deficiency.
An important requirement for assessing the feasibility of new gene therapy approaches is accurate evaluation of dosing, to avoid supraphysiological protein levels and remain within the therapeutic window. This is particularly relevant given that thrombotic events have been reported in 3%–4% of patients with FVII deficiency2 and deep vein thrombosis has also been documented in recombinant FVIIa replacement therapy.51,52 In a clinical gene therapy study for hemophilia B, using the hyperactive FIX-Padua transgene, a patient reported serious health problems due to strongly elevated FIX activity levels (>200%).53 Moreover, a phase 3 clinical trial for hemophilia A gene therapy was put on hold due to several patients achieving FVIII levels >150%, necessitating prophylactic anti-coagulation therapy.53 Our unique combination of components, including the common hAAT promoter and AAV8 capsid, resulted in peak coFVII(-22) and RBD-FVII(-22) expression at 2–4 weeks, with plasma FVII concentrations up to 20-fold higher than normal physiological levels. Supraphysiological FVII levels (770% of normal) were also observed in a canine model for FVII deficiency, which employed an AAV8-based vector at a dosage of 4.95 × 1013 gc/kg.14 As a first step toward further pre-clinical testing, we carried out a dosing study to identify the minimal effective dose needed to elicit FVII levels inside a therapeutic window of 1%–150% of normal. Our results showed that optimal levels between 50% and 150% of normal levels can be achieved in mice with doses between 0.8 × 1012 and 1.6 × 1011 gc/kg. It is known that AAV8 transduces mouse hepatocytes better than NHPs and human hepatocytes, and we therefore expect that higher virus titers will be needed to reach therapeutic levels in humans.54
While our findings provide valuable insights, several limitations must be acknowledged. First, the study was conducted in wild-type C57BL/6 mice rather than an FVII-deficient model. Although we demonstrated efficient production and activity of the hFVII transgene, we did not evaluate its ability to correct a bleeding phenotype or assess potential inflammatory responses, which could lead to hepatocellular damage. To address these gaps, studies in large animal models are recommended, as species-specific differences in FVII activity may limit the extrapolation of hFVII efficacy relative to endogenous FVII. In vivo analysis is further complicated by the limited availability of suitable animal models that accurately recapitulate FVII deficiency in humans. While FVII-deficient mouse models exist, they often suffer from severe health complications, rendering them unsuitable for long-term studies. Second, we assumed successful vector-mediated transduction of hepatocytes based on qPCR targeting the poly A (pA) tail of the expression cassette. Although quantification using the FVII coding sequence or promoter region would have provided greater specificity, our conclusion was supported by the well-established knowledge that both AAV5 and AAV8 efficiently transduce mouse hepatocytes, thereby justifying our assumption. Finally, a more comprehensive analysis should be performed to fully assess the safety profile of an AAV-based gene therapy for FVII deficiency. Although our preliminary data showed that treated animals developed antibodies against AAV8, we cannot exclude that low levels of anti-FVII antibodies develop in response to AAV8-mediated gene delivery. We implied the absence of transgene Nabs based on the concordance between hFVII activity and antigen levels; however, this assumption requires further validation, particularly considering that an immune response to the transgene product may also impact other factors such as clearance, protein half-life, and inflammatory responses. Future pre-clinical development should include a more comprehensive assessment of the immunogenicity and side effects of both the transgene and the vector capsid.
In conclusion, our study builds upon previous findings to further demonstrate the feasibility of AAV-based gene therapy. We present novel data on CpG depletion, serotype-specific biodistribution, and anti-capsid immunogenicity, and we show that AAV-based gene therapy can provide durable and functional expression of hFVII in wild-type mice with no adverse events observed. Overall, we provided new insights that may support continued pre-clinical investigation of this therapeutic approach.
Materials and methods
Plasmid construction and rAAV production
The hFVII cDNAs were cloned into a vector backbone derived from the plasmid Adeno-Associated Virus (pAAV)-GFP plasmid described by Gray J et al.,55 which was customized with the inclusion of a multiple cloning site to allow the substitution of the characterized ApoEHCR-hAAT (hAAT) promoter from Miao et al.26 and the hFVII cDNA sequence in between the AAV2 inverted terminal repeats (ITRs). The custom pAAV plasmid (Vector Biolabs, Malvern, USA) was used as backbone for all the plasmids adopted in this paper (Figure S7A). Moreover, each construct included in this study contains the bovine growth hormone poly A (pA) tail.
The pAAV-hAAT-hFVIIwt was based on hFVII cDNA (GenBank: NM_000131.4), while the pAAV-hAAT-cohFVII(-22) was derived from the most abundant transcript, (GenBank: NM_019616.4) which encodes a hFVII variant that lacks 22 amino acids in the propeptide.43 The latter cDNA sequence was codon optimized and previously employed in a self-complementary (scAAV) gene transfer approach.15 Two codon-optimized hFVII versions were generated in this study using a proprietary codon-optimization tool (RiboPro, Oss, The Netherlands) and inserted into the pAAV backbone encoding the hAAT promoter. These plasmids were designated pAAV-hAAT-RBD-hFVIIwt and pAAV-hAAT-RBD-hFVII(-22), respectively. The sequence of the optimized hFVII cDNA’s is listed in Figure S4.
The pAAV-CAG-luciferase construct used to test the capsid in vivo biodistribution study was obtained in a similar manner, by substituting the original promoter present in the pAAV-GFP backbone with the ubiquitous CAG promoter and the coding sequence with the luciferase gene (Figure S7B).
rAAV stocks were produced as described previously.56 Briefly, for rAAV5 production, pAAV encoding the therapeutic cassette was co-transfected into HEK293T with pDP5,57 encoding the Rep/Cap and helper proteins (PlasmidFactory, Ref.PF435) using poly-ethylenimine (PEI; 1 mg/mL; Polysciences Europe, Germany) in a ratio PEI:pDNA of 3:1. For rAAV8 production, pAAV encoding the therapeutic cassette was co-transfected into HEK293T with pAAV2/8 encoding for the capsid proteins of AAV8 and the ITR from AAV2 (Addgene #112864) and the helper plasmid pAdDeltaF6 (Addgene #112867) in a ratio PEI:pDNA of 3:1. A similar approached was used for rAAV9 production, with the only exception that, in this case, plasmid pAAV2/9n encoding for the capsid proteins of AAV9 and the ITR from AAV2 (Addgene #112865) was used instead.
Medium of HEK293T cells was refreshed with Iscove’s modified Eagle’s medium, supplemented with 10% fetal calf serum, glutamine, and penicillin/streptomycin (Gibco) 1 hour before transfection and the day after. Three days after transfection, cells were lysed in lysis buffer (50 mM Tris-HCl pH 8.5, 150 mM NaCl, 0.1% Triton X-100, and 2.5 mM MgCl2). Lysates containing rAAV vectors were treated with DNase and purified using an iodixanol gradient as previously described.58 Subsequently, rAAVs were collected from the gradient, diluted with Dulbecco's Phosphate-Buffered Saline (DPBS), and concentrated using 100 kD Amicon Ultra-15 filtration units (Millipore, Amsterdam, The Netherlands) with a final buffer exchange of at least three volumes of DPBS (Gibco) supplemented with 5% sucrose. Finally, all rAAVs were stocked at −80°C until use.
The resulting rAAV preparations were titrated using real-time qPCR with serial dilutions of a linearized plasmid as a standard curve and primers directed toward the pA tail of the transgene cassette (forward: 5′-GCCTTCTAGTTGCCAGCCAT-3’; reverse: 5′-GGCACCTTCCAGGGTCAAG-3′). Titers were calculated from the obtained cycle threshold (Ct) values and expressed as genomic copies per milliliter (gc/mL).
Animal experiments
All animal procedures were approved by the local animal welfare committee and in accordance with European guidelines (86/609/EEC). Young adult wild-type C57BL/6 mice (6–8 weeks old) were given injections into the tail vein using a restrainer. A total amount of 200 μL of viral vector solution was injected per animal. The number of mice to be analyzed has been set at 5–6 per group based on similar studies performed in our group.59,60
To evaluate biodistribution of rAAV8, rAAV5, and rAAV9 capsid, luciferase bioluminescence was measured using the IVIS as previously described.61 Briefly, mice were anesthetized using isoflurane (∼3%–5% in air for induction), and anesthesia was maintained with 1%–3% isoflurane. Right before each measurement, the abdomen of the animals was shaved to remove hair and 200 μL of 50 mM D-luciferin-solution was injected intraperitoneally. Mice were then placed in prone position and imaged in a field of 12.7 × 12.7 cm in the IVIS system. Before each luminescent measurement, a grayscale photographic image was taken with auto-exposure settings on which a pseudo-color representation of the luminescent signal was superimposed. For each measurement, a region of interest (ROI) of a fixed size was drawn, and it was manually centered over the area with the highest luminescent signal. The average radiance per ROI for each animal and each time point was used for further analysis. The amount of luciferase expression is then expressed as photons per cm2 per second.
To monitor FVII levels in plasma, blood samples were obtained at week 1 and 2 post-injection and every 2 weeks thenceforward by mandibular puncture in conscious mice, using a 20G needle. Blood drops were collected in Eppendorf tubes. To avoid clotting, tubes containing 1/10th volume of 3.8% tri-sodium-citrate were used. Blood samples were centrifuged for 10 min at 5,000 rpm, and plasma was carefully pipetted into clean tubes and stored at −20°C until further use. At the end of the experiment, animals were euthanized by an overdose of pentobarbital, and the following tissues were collected: spleen, liver, muscle, lung, and kidney. Selected tissues samples were snap frozen and kept at −80°C until further processing.
Regarding the different dosing regimen adopted in this study, they are the result of practical experience. For our initial evaluation, we relied on previous data suggesting that 1.6 × 1013 gc/kg would be a suitable starting dose. However, the luciferase signal captured by the IVIS system appeared oversaturated, particularly for AAV8 (Figure 1). Consequently, for the subsequent experiment, we decided to reduce the AAV8 dose to 1.6 × 1012 gc/kg. We did not alter the AAV5 dose, as this capsid had not performed as well as AAV8. Results from the second experiment (Figure 2) confirmed AAV8’s superior performance compared to AAV5 but also showed supraphysiological levels of FVII (around 20 U/mL), indicating that even lower doses could be effective. As a result, the long-term experiment shown in Figure 4 was conducted using a dose of 1.6 × 1012 gc/kg.
FVII assays
hFVII plasma concentration was measured by a hFVII-specific sandwich ELISA as previously described.15 Briefly, 1:500 goat polyclonal anti-hFVII Ab was used as a primary antibody (AF2338, R&D Systems), with 1:2,000 sheep anti-hFVII (SAFVII-horseradish peroxidase [HRP], Affinity Biologicals) as a secondary and detection antibody. Human plasma obtained from a pool of at least 10 healthy blood donors was used as a standard. The concentration of hFVII in the plasma standard was determined with reference to the current World Health Organization international standard guidelines for FVII products (Sanquin Diagnostics). To make sure that this assay does not have cross-reactivity with mouse FVII, detection was performed on plasma from uninjected normal mice as a negative control. Furthermore, to account for any variation in matrix effects from the murine plasma, we performed a spiked experiment in which purified hFVII was spiked into mouse plasma from uninjected mice and compared with mouse plasma only (same genetic background). No signal was observed in mouse-plasma-only samples, while spiked hFVII was recovered for ≥90%, suggesting very limited interference from the different matrices (Table S1).
The ELISA was further characterized by calculating the low limit of quantification (LLQ) and upper limit of quantification (ULQ) of the hFVII-specific sandwich ELISA, which was based on the following two parameters: (1) accuracy, which represents the closeness of measured values to the true concentration, usually considered acceptable if within a range of 80%–120%, and (2) precision, which is the degree of variability among replicate measurements (also described as coefficient of variation), usually considered acceptable if equal or minor to 5%. Based on these criteria, our assay showed that the LLQ is in the range of 0.003–0.006 U/mL while the ULQ is in the range of 0.2–0.1 U/mL (Table S2).
FVII activity in mouse plasma was assayed using the BIOPHEN FVII chromogenic assay (Ref 221304, Hyphen BioMed) according to the manufacturer’s instructions.
Anti-AAV8 detection
Anti-AAV8 antibody concentrations in plasma were measured using an in-house-developed sandwich ELISA. Briefly, plates were coated overnight with 5 × 1010 cp/mL of rAAV8, produced as previously reported.56 A 1:5,000 dilution of HRP-conjugated anti-mouse IgG (NXA931, GE Healthcare) was used as both the primary and detection antibody. The ADK8 anti-AAV8 mouse monoclonal antibody (610160, ProGen) served as the standard. Plasma from PBS-injected mice was included as a negative control. To confirm assay specificity and rule out non-specific binding to the plate material, the sandwich ELISA assay was repeated in the absence of coating, plasma material, or secondary antibody. No absorbance signal was detected in any of these conditions, confirming that the ELISA setup was specific and free from background reactivity (see Table S3).
Vector DNA copy number per cell
In order to determine viral vector copy numbers in tissues, mice were sacrificed and samples of liver, spleen, kidney, lung, and muscles were collected. Genomic DNA was isolated using the DNeasy Blood & Tissue kit (QIAGEN) following the manufacturer’s protocol. The vector copy numbers in tissue were measured using the same qPCR primers adopted for AAV production titering as described earlier. We used 100 ng of genomic DNA as starting material, and serial dilutions were analyzed for vector genome quantification.
Statistical analysis
All data are represented as mean and standard deviation (SD). Comparison between AAV5 versus AAV8 in Figure 2 was performed using the Student’s t test. Two-way ANOVA with Tuckey’s multiple comparisons test was used to compare the mean value effect of each time point in the different groups in Figures 1, 4, and 5. One-way ANOVA with Tuckey’s multiple comparisons test was used to compare the mean value effect of each time point in the different groups in Figure 6. Data were considered significant at p < 0.05. Symbols indicate p values for significance designated as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Data availability
The data that support the findings of this study are available from the corresponding author, J. Voorberg, upon reasonable request.
Acknowledgments
This research was funded by the LSH-TKI (Health∼Holland) project RBD-CURE (project number LSHM20029). Tissue distribution experiments were performed by Rinske Helmhout.
Author contributions
A.D., B.B., J. Verhaagen, D.G.M.M., and J. Voorberg designed the study. A.D., B.B., and J.v.d.H. performed experiments and analyzed the data. P.H.P.K. designed and optimized FVII-specific ELISA. F.d.W. supported data analysis. B.H. performed animal injections. A.D. wrote the paper and prepared artwork files. All the authors approved the final version of the manuscript.
Declaration of interests
A.D., B.B., A.v.A., J. Verhaagen, D.G.M.M., and J. Voorberg are listed as inventors on a patent application on the expression of therapeutic genes, including FVII, for gene therapy applications.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2025.101523.
Supplemental information
References
- 1.Franchini M., Marano G., Pupella S., Vaglio S., Masiello F., Veropalumbo E., Piccinini V., Pati I., Catalano L., Liumbruno G.M. Rare congenital bleeding disorders. Ann. Transl. Med. 2018;6:331. doi: 10.21037/atm.2018.08.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sevenet P.-O., Kaczor D.A., Depasse F. Factor VII Deficiency: From Basics to Clinical Laboratory Diagnosis and Patient Management. Clin. Appl. Thromb. Hemost. 2017;23:703–710. doi: 10.1177/1076029616670257. [DOI] [PubMed] [Google Scholar]
- 3.Napolitano M., Siragusa S., Mariani G. Factor VII Deficiency: Clinical Phenotype, Genotype and Therapy. J. Clin. Med. 2017;6 doi: 10.3390/jcm6040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson K.S. An overview of inherited factor VII deficiency. Transfus. Apher. Sci. 2019;58:569–571. doi: 10.1016/j.transci.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Arruda V.R., Weber J., Samelson-Jones B.J. Gene Therapy for Inherited Bleeding Disorders. Semin. Thromb. Hemost. 2021;47:161–173. doi: 10.1055/s-0041-1722862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berrettini M., Mariani G., Schiavoni M., Rocino A., Di Paolantonio T., Longo G., Morfini M. Pharmacokinetic evaluation of recombinant, activated factor VII in patients with inherited factor VII deficiency. Haematologica. 2001;86:640–645. [PubMed] [Google Scholar]
- 7.Menegatti M., Peyvandi F. Treatment of rare factor deficiencies other than hemophilia. Blood. 2019;133:415–424. doi: 10.1182/blood-2018-06-820738. [DOI] [PubMed] [Google Scholar]
- 8.Mariani G., Napolitano M., Dolce A., Pérez Garrido R., Batorova A., Karimi M., Platokouki H., Auerswald G., Bertrand A.-M., Di Minno G., et al. Replacement therapy for bleeding episodes in factor VII deficiency. A prospective evaluation. Thromb. Haemost. 2013;109:238–247. doi: 10.1160/TH12-07-0476. [DOI] [PubMed] [Google Scholar]
- 9.Pipe S.W., Leebeek F.W.G., Recht M., Key N.S., Castaman G., Miesbach W., Lattimore S., Peerlinck K., Van der Valk P., Coppens M., et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023;388:706–718. doi: 10.1056/NEJMoa2211644. [DOI] [PubMed] [Google Scholar]
- 10.Swystun L.L., Lillicrap D. Gene Therapy for Coagulation Disorders. Circ. Res. 2016;118:1443–1452. doi: 10.1161/CIRCRESAHA.115.307015. [DOI] [PubMed] [Google Scholar]
- 11.Nathwani A.C., Tuddenham E.G.D., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C., et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H., et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Northington M.W., Rice S.E., Holmes A.L., Watts Alexander C.S. Gene-ius at work: Hemophilia B treatment enters a new era. Am. J. Health Syst. Pharm. 2025 doi: 10.1093/ajhp/zxaf005. [DOI] [PubMed] [Google Scholar]
- 14.Marcos-Contreras O.A., Smith S.M., Bellinger D.A., Raymer R.A., Merricks E., Faella A., Pavani G., Zhou S., Nichols T.C., High K.A., Margaritis P. Sustained correction of FVII deficiency in dogs using AAV-mediated expression of zymogen FVII. Blood. 2016;127:565–571. doi: 10.1182/blood-2015-09-671420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binny C., McIntosh J., Della Peruta M., Kymalainen H., Tuddenham E.G.D., Buckley S.M.K., Waddington S.N., McVey J.H., Spence Y., Morton C.L., et al. AAV-mediated gene transfer in the perinatal period results in expression of FVII at levels that protect against fatal spontaneous hemorrhage. Blood. 2012;119:957–966. doi: 10.1182/blood-2011-09-377630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kashyap R., Alim M., Chandra D., Singh R. Recurrent seizures in a neonate with intracranial hemorrhage and congenital factor VII deficiency: A missed diagnosis. J. Clin. Neonatol. 2020;9:300. doi: 10.4103/jcn.JCN_67_20. [DOI] [Google Scholar]
- 17.Wright J.F. Codon Modification and PAMPs in Clinical AAV Vectors: The Tortoise or the Hare? Mol. Ther. 2020;28:701–703. doi: 10.1016/j.ymthe.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konkle B.A., Walsh C.E., Escobar M.A., Josephson N.C., Young G., von Drygalski A., McPhee S.W.J., Samulski R.J., Bilic I., de la Rosa M., et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137:763–774. doi: 10.1182/blood.2019004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faust S.M., Bell P., Cutler B.J., Ashley S.N., Zhu Y., Rabinowitz J.E., Wilson J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Investig. 2013;123:2994–3001. doi: 10.1172/JCI68205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiang Z., Kurupati R.K., Li Y., Kuranda K., Zhou X., Mingozzi F., High K.A., Ertl H.C.J. The Effect of CpG Sequences on Capsid-Specific CD8+ T Cell Responses to AAV Vector Gene Transfer. Mol. Ther. 2020;28:771–783. doi: 10.1016/j.ymthe.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muhuri M., Levy D.I., Schulz M., McCarty D., Gao G. Durability of transgene expression after rAAV gene therapy. Mol. Ther. 2022;30:1364–1380. doi: 10.1016/j.ymthe.2022.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martino A.T., Markusic D.M. Immune Response Mechanisms against AAV Vectors in Animal Models. Mol. Ther. Methods Clin. Dev. 2020;17:198–208. doi: 10.1016/j.omtm.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rapti K., Grimm D. Adeno-Associated Viruses (AAV) and Host Immunity - A Race Between the Hare and the Hedgehog. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.753467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright J.F. Quantification of CpG Motifs in rAAV Genomes: Avoiding the Toll. Mol. Ther. 2020;28:1756–1758. doi: 10.1016/j.ymthe.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westhaus A., Cabanes-Creus M., Rybicki A., Baltazar G., Navarro R.G., Zhu E., Drouyer M., Knight M., Albu R.F., Ng B.H., et al. High-Throughput In Vitro, Ex Vivo, and In Vivo Screen of Adeno-Associated Virus Vectors Based on Physical and Functional Transduction. Hum. Gene Ther. 2020;31:575–589. doi: 10.1089/hum.2019.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao C.H., Ohashi K., Patijn G.A., Meuse L., Ye X., Thompson A.R., Kay M.A. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol. Ther. 2000;1:522–532. doi: 10.1006/mthe.2000.0075. [DOI] [PubMed] [Google Scholar]
- 27.Au H.K.E., Isalan M., Mielcarek M. Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 2021;8 doi: 10.3389/fmed.2021.809118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 29.Samelson-Jones B.J., George L.A. Adeno-Associated Virus Gene Therapy for Hemophilia. Annu. Rev. Med. 2023;74:231–247. doi: 10.1146/annurev-med-043021-033013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halbert C.L., Standaert T.A., Wilson C.B., Miller A.D. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 1998;72:9795–9805. doi: 10.1128/JVI.72.12.9795-9805.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiorini J.A., Kim F., Yang L., Kotin R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999;73:1309–1319. doi: 10.1128/JVI.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walters R.W., Yi S.M., Keshavjee S., Brown K.E., Welsh M.J., Chiorini J.A., Zabner J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 2001;276:20610–20616. doi: 10.1074/jbc.M101559200. [DOI] [PubMed] [Google Scholar]
- 33.Mattar C.N.Z., Nathwani A.C., Waddington S.N., Dighe N., Kaeppel C., Nowrouzi A., Mcintosh J., Johana N.B., Ogden B., Fisk N.M., et al. Stable human FIX expression after 0.9G intrauterine gene transfer of self-complementary adeno-associated viral vector 5 and 8 in macaques. Mol. Ther. 2011;19:1950–1960. doi: 10.1038/mt.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pipe S., Leebeek F.W.G., Ferreira V., Sawyer E.K., Pasi J. Clinical Considerations for Capsid Choice in the Development of Liver-Targeted AAV-Based Gene Transfer. Mol. Ther. Methods Clin. Dev. 2019;15:170–178. doi: 10.1016/j.omtm.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulz M., Levy D.I., Petropoulos C.J., Bashirians G., Winburn I., Mahn M., Somanathan S., Cheng S.H., Byrne B.J. Binding and neutralizing anti-AAV antibodies: Detection and implications for rAAV-mediated gene therapy. Mol. Ther. 2023;31:616–630. doi: 10.1016/j.ymthe.2023.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C., Narkbunnam N., Samulski R.J., Asokan A., Hu G., Jacobson L.J., Manco-Johnson M.J., Monahan P.E., Joint Outcome Study Investigators Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012;19:288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- 37.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 38.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 39.Majowicz A., Nijmeijer B., Lampen M.H., Spronck L., de Haan M., Petry H., van Deventer S.J., Meyer C., Tangelder M., Ferreira V. Therapeutic hFIX Activity Achieved after Single AAV5-hFIX Treatment in Hemophilia B Patients and NHPs with Pre-existing Anti-AAV5 NABs. Mol. Ther. Methods Clin. Dev. 2019;14:27–36. doi: 10.1016/j.omtm.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kishimoto T.K., Samulski R.J. Addressing high dose AAV toxicity - “one and done” or “slower and lower”. Expert Opin. Biol. Ther. 2022;22:1067–1071. doi: 10.1080/14712598.2022.2060737. [DOI] [PubMed] [Google Scholar]
- 41.VandenDriessche T., Chuah M.K. Hyperactive Factor IX Padua: A Game-Changer for Hemophilia Gene Therapy. Mol. Ther. 2018;26:14–16. doi: 10.1016/j.ymthe.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nair N., De Wolf D., Nguyen P.A., Pham Q.H., Samara-Kuko E., Landau J., Blouse G.E., Chuah M.K., VandenDriessche T. Gene therapy for hemophilia B using CB 2679d-GT: a novel factor IX variant with higher potency than factor IX Padua. Blood. 2021;137:2902–2906. doi: 10.1182/blood.2020006005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giansily-Blaizot M., Rallapalli P.M., Perkins S.J., Kemball-Cook G., Hampshire D.J., Gomez K., Ludlam C.A., McVey J.H. The EAHAD blood coagulation factor VII variant database. Hum. Mutat. 2020;41:1209–1219. doi: 10.1002/humu.24025. [DOI] [PubMed] [Google Scholar]
- 44.Hao Z., Jin D.-Y., Stafford D.W., Tie J.-K. Vitamin K-dependent carboxylation of coagulation factors: insights from a cell-based functional study. Haematologica. 2020;105:2164–2173. doi: 10.3324/haematol.2019.229047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ayombil F., Camire R.M. Insights into vitamin K-dependent carboxylation: home field advantage. Haematologica. 2020;105:1996–1998. doi: 10.3324/haematol.2020.253690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higgins-Gruber S.L., Mutucumarana V.P., Lin P.-J., Jorgenson J.W., Stafford D.W., Straight D.L. Effect of vitamin K-dependent protein precursor propeptide, vitamin K hydroquinone, and glutamate substrate binding on the structure and function of {gamma}-glutamyl carboxylase. J. Biol. Chem. 2010;285:31502–31508. doi: 10.1074/jbc.M110.143297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newton K., Dixit V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mauro V.P., Chappell S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014;20:604–613. doi: 10.1016/j.molmed.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pabinger I., Ayash-Rashkovsky M., Escobar M., Konkle B.A., Mingot-Castellano M.E., Mullins E.S., Negrier C., Pan L., Rajavel K., Yan B., Chapin J. Multicenter assessment and longitudinal study of the prevalence of antibodies and related adaptive immune responses to AAV in adult males with hemophilia. Gene Ther. 2024;31:273–284. doi: 10.1038/s41434-024-00441-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leborgne C., Barbon E., Alexander J.M., Hanby H., Delignat S., Cohen D.M., Collaud F., Muraleetharan S., Lupo D., Silverberg J., et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020;26:1096–1101. doi: 10.1038/s41591-020-0911-7. [DOI] [PubMed] [Google Scholar]
- 51.Margaritis P., High K.A. Gene therapy in haemophilia--going for cure? Haemophilia. 2010;16:24–28. doi: 10.1111/j.1365-2516.2010.02256.x. [DOI] [PubMed] [Google Scholar]
- 52.Ramdass S.K., Loh K.P., Howard L.M. Thrombosis in a bleeding disorder: case of thromboembolism in factor VII deficiency. Clin. Case Rep. 2017;5:277–279. doi: 10.1002/ccr3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaczmarek R., Herzog R.W. Treatment-induced hemophilic thrombosis? Mol. Ther. 2022;30:505–506. doi: 10.1016/j.ymthe.2022.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu S., Razon L., Ritchie O., Sihn C.-R., Handyside B., Berguig G., Woloszynek J., Zhang L., Batty P., Lillicrap D., et al. Application of in-vitro-cultured primary hepatocytes to evaluate species translatability and AAV transduction mechanisms of action. Mol. Ther. Methods Clin. Dev. 2022;26:61–71. doi: 10.1016/j.omtm.2022.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gray J.T., Zolotukhin S. Design and construction of functional AAV vectors. Methods Mol. Biol. 2011;807:25–46. doi: 10.1007/978-1-61779-370-7_2. [DOI] [PubMed] [Google Scholar]
- 56.Verhaagen J., Hobo B., Ehlert E.M.E., Eggers R., Korecka J.A., Hoyng S.A., Attwell C.L., Harvey A.R., Mason M.R.J. Small Scale Production of Recombinant Adeno-Associated Viral Vectors for Gene Delivery to the Nervous System. Methods Mol. Biol. 2018;1715:3–17. doi: 10.1007/978-1-4939-7522-8_1. [DOI] [PubMed] [Google Scholar]
- 57.Grimm D., Kay M.A., Kleinschmidt J.A. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 2003;7:839–850. doi: 10.1016/s1525-0016(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 58.Hermens W.T., ter Brake O., Dijkhuizen P.A., Sonnemans M.A., Grimm D., Kleinschmidt J.A., Verhaagen J. Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum. Gene Ther. 1999;10:1885–1891. doi: 10.1089/10430349950017563. [DOI] [PubMed] [Google Scholar]
- 59.Hartholt R.B., Wroblewska A., Herczenik E., Peyron I., Ten Brinke A., Rispens T., Nolte M.A., Slot E., Claassens J.W., Nimmerjahn F., et al. Enhanced uptake of blood coagulation factor VIII containing immune complexes by antigen presenting cells. J. Thromb. Haemost. 2017;15:329–340. doi: 10.1111/jth.13570. [DOI] [PubMed] [Google Scholar]
- 60.Herczenik E., van Haren S.D., Wroblewska A., Kaijen P., van den Biggelaar M., Meijer A.B., Martinez-Pomares L., ten Brinke A., Voorberg J. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J. Allergy Clin. Immunol. 2012;129:501-9–509.e1-5. doi: 10.1016/j.jaci.2011.08.029. [DOI] [PubMed] [Google Scholar]
- 61.Roet K.C.D., Eggers R., Verhaagen J. Noninvasive bioluminescence imaging of olfactory ensheathing glia and schwann cells following transplantation into the lesioned rat spinal cord. Cell Transplant. 2012;21:1853–1865. doi: 10.3727/096368911X627471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, J. Voorberg, upon reasonable request.