


Abstract
Anti-CRISPR (Acr) proteins are frequently co-encoded with the anti-CRISPR associated (Aca) proteins, which act as repressors for regulating Acr expression within acr-aca operons. We previously identified three aca genes (aca11–13) from Streptococcus mobile genetic elements, but their regulatory mechanisms remained unclear. Here, we showed that Aca11 and Aca13 mediate bidirectional regulation in acr-aca operons through recognition of their inverted repeat (IR) sequences within the acr promoters. Based on the bioinformatics search using Aca13 with its IR sequences, we discovered a novel type II-A Acr (named AcrIIA35). AcrIIA35 exhibits a potent inhibitory activity against St1Cas9 by interfering with DNA recognition of Cas9 in bacterial and human cells. We also developed a novel Aca-driven protein–protein interaction detection (APID) system by integrating Aca-tagged target proteins with fluorescently labeled IR-DNA probes. The APID system enables efficient detection of protein–protein interaction using proteins or crude cell lysates. Utilizing the APID system, we have further elucidated the mechanism of AcrIIA24, which can interact with the HNH nuclease domain of St3Cas9 to inhibit the DNA cleavage activity of Cas9. Collectively, our work expands the understanding of Aca functions to modulate Acrs and expands the potential for Aca-based applications in CRISPR technologies.
Graphical Abstract
Graphical Abstract.

Introduction
The CRISPR–Cas system is an adaptive immune system evolved by prokaryotes to target and degrade foreign genetic material, such as bacteriophage DNA or plasmids [1, 2]. This system is composed primarily of CRISPR arrays and associated Cas proteins. Following an initial infection, short fragments of invading DNA (protospacers) are integrated into the CRISPR array to form an immunological memory. In subsequent infections, the CRISPR array is transcribed into CRISPR RNA (crRNA), which assembles with Cas nucleases to recognize and cleave matching foreign DNA or RNA [3, 4]. Based on the composition and mechanism of Cas proteins, CRISPR–Cas systems are classified into two major classes: Class 1 (types I, III, and IV) and Class 2 (types II, V, and VI) [5]. Class 1 systems function through multi-subunit complexes, whereas Class 2 systems rely on single-subunit Cas proteins for their activity [6].
To counteract the CRISPR–Cas immune system, bacteriophages and plasmids have evolved anti-CRISPR (Acr) proteins, which enable the development of more controllable CRlSPR–Cas tools [7–9, 10]. To date, over a hundred Acr families have been identified, which can inhibit type I, II, III, V, and VI CRISPR–Cas systems [11]. Acrs inhibit CRISPR–Cas activity through various mechanisms, including preventing crRNA loading onto the Cas protein (e.g. AcrIIA17), blocking double-stranded DNA binding (e.g. AcrIIA4, AcrIIA27, AcrIIA31, and AcrIIA32), and suppressing nuclease cleavage activity (e.g. AcrIIA24 and AcrIIC1) [12–15, 16]. Although Acr proteins play a critical role in the arms race between phages and their hosts, the identification of Acr proteins remains challenging due to their high sequence diversity and lack of conserved structural domains [17–19]. Therefore, the classical homology-based approaches typically failed to recognize de novo Acr proteins.
Since acr genes are often co-encoded with conserved anti-CRISPR associated (aca) genes within acr-aca operons, aca genes serve as efficient markers for the identification of acr genes [20, 21]. Aca proteins, which are typically located downstream of acr genes, act as autorepressors to regulate the expression of Acrs [22]. To date, 13 distinct Aca families have been reported, and Aca1–10 have been characterized in detail [23–25]. These Aca proteins always contain a helix–turn–helix (HTH) DNA-binding motif, which can recognize inverted repeat (IR) sequences upstream of acr genes to regulate Acr expression [23–25, 26]. Previously, we identified three aca genes (aca11–13) from Streptococcus mobile genetic elements, which were proven to be effective markers for the identification of Acr proteins (AcrIIA24–34) [12, 27]. Structural predictions revealed that Aca11 and Aca12 contain HTH domains, whereas Aca13 lacks this canonical DNA-binding motif. Overall, their intrinsic regulatory functions as Aca proteins require further clarification.
In this work, we first elucidated the distinct regulatory mechanisms of Aca11 and Aca13. Differing from previously characterized Aca proteins, we showed that both Aca11 and Aca13 mediate bidirectional regulation in acr-aca operons through recognition of their IR sequences within the acr promoters. Notably, through a bioinformatics search that utilized Aca13 along with its IR sequences, we identified a novel type II-A anti-CRISPR protein, which we have named AcrIIA35. Furthermore, by combining the unique properties of Aca11 and Aca13, we developed a novel system named APID (Aca-driven protein–protein interaction detection) for efficient detection of Aca-based protein interactions using proteins or crude cell lysates. Collectively, this study advances our understanding of Aca-mediated regulation in phage defense systems and expands the potential for Aca-based applications in CRISPR technologies.
Materials and methods
Microbes
Escherichia coli strain TOP10 (Sangon Biotech) was employed for plasmid amplification and interference assays; E. coli strain DH5α (Tsingke) was used for plasmid-based fluorescence reporter assays; and strain T7 Express (Biomed) was utilized for protein expression. Unless otherwise indicated, all E. coli strains were cultured at 37°C in lysogeny broth (LB) medium with appropriate antibiotics (when required): ampicillin (50 μg/ml), kanamycin (50 μg/ml), or chloramphenicol (25 μg/ml).
Cell lines
HEK293T cells were cultured in DMEM (Gibco) medium supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) at 37°C in an incubator with 5% CO2.
Bioinformatics analysis
BLASTp program was used to identify homologs of Aca11 (accession: WP_009730540.1) and Aca13 (accession: RHB86839.1) in the non-redundant protein database (Supplementary Table S1). Candidate Acr proteins were identified from neighboring genes using the guilt-by-association method, based on our previous studies [12, 27]. HTH DNA-binding domains were predicted using HHpred (MPI Bioinformatics Toolkit) [28]. Genomic context analysis of diverse acr-aca operons (Fig. 1A and Supplementary Fig. S1) was based on the anti-CRISPR database (https://tinyurl.com/anti-CRISPR) [11]. Multiple sequence alignments of acr-aca promoters were performed using Jalview (https://www.jalview.org/) [29].
Figure 1.

The multifunctional regulatory roles of Aca11 and Aca13 in acr-aca operons. (A) Genomic organization of representative acr-aca operons (see Supplementary Fig. S1 for complete schematic). Acr genes are annotated with subtypes and numbers (e.g. IF1 denotes AcrIF1). (B) Schematic view of acr-aca11 locus and engineered plasmid constructs for assessing Aca11-mediated regulation of acr-aca11 promoters (Aca11-s1 and -s2) in E. coli. Acr genes are shown with numbers corresponding to AcrIIA numbers. HTH and AP2 DNA-binding motifs were identified using HHpred (see the “Materials and methods” section). AmpR, ampicillin resistance; KanR, kanamycin resistance. (C) Representative colony pictures of E. coli expressing mCherry under the control of acr-aca11 promoters (Aca11-s1 and -s2) in the presence or absence of Aca11 protein. Bright field (BF) and mCherry fluorescence channels are displayed on the left side of each row. (D) Quantitative analysis of mCherry fluorescence driven by acr-aca11 promoters. Bar graphs depict the median mCherry fluorescence values, normalized to the corresponding promoter controls without Aca11. Error bars represent mean ± SEM from three biological replicates. Statistical significance (***P < .001) was determined by unpaired t-test. (E) Schematic view of the genomic context of acr-aca13 locus, including acr genes, aca13 gene, and other neighboring genes (annotated based on NCBI database information). Schematic of the plasmids designed for the assay to measure regulation of the acr-aca13 promoters (Aca13-s1 and -s2) by AcrIIA32 (Aca13 fused in the C-terminal portion of AcrIIA32) in E. coli. (F) Domain architectures of AcrIIA32 protein, an Acr-Aca fusion protein. The protein comprises an N-terminal anti-CRISPR domain (A32NTD) and a C-terminal Aca13 domain (A32CTD). (G) Representative colony pictures of E. coli expressing mCherry under acr-aca13 promoters (Aca13-s1 and -s2) in the presence or absence of AcrIIA32, A32NTD, or A32CTD proteins. BF and mCherry fluorescence channels are displayed on the left side of each row. (H) Bar graphs show the median mCherry fluorescence values (normalized to promoter controls without AcrIIA32) under different AcrIIA32 variants. Error bars represent mean ± SEM from three biological replicates. Statistical significance was determined by unpaired t-test (***P< .001; n.s., not significant).
Structural predictions of all Acr proteins, Acr–Cas9 RNP (ribonucleoprotein) complexes, and Aca–DNA complexes were performed using AlphaFold 3 on the AlphaFold Server [30]. Protein structures were visualized using PyMol program.
Plasmid-based fluorescence reporter assays
Plasmids (pB001–pB019 and pB027–pB056) used in the plasmid-based fluorescence reporter system were designed based on our previous reports [12, 27] and listed in Supplementary Table S4. DNA sequences encoding Aca proteins were synthesized by Biomed or Tsingke and ligated into the pBAD24 vectors (colE1 replicon, ampicillin resistance) under the control of the arabinose-inducible araBAD promoter. Next, the mCherry transcriptional reporter plasmids were derived from pRSFduet-1 (RSF replicon, kanamycin resistance), with an mCherry reporter gene under the control of acr-aca promoters tested in the study.
The mCherry reporter plasmids were transformed into E. coli strain DH5α (Tsingke) with either the Aca-expressing pBAD24 vector (+Aca) or an empty control vector (−Aca) using the CaCl2 heat-shock procedure according to the manufacturer’s manual. Escherichia coli cells were recovered for 1 h in LB medium with 0.2% arabinose (Yeasen) and then plated on LB agar with antibiotics (50 μg/ml ampicillin and 50 μg/ml kanamycin) and inducers (0.2% arabinose) for 24–32 h at 37°C. The mCherry images of E. coli were taken on a Typhoon FLA 9500 (GE) under Cy5 condition, and the mCherry intensity was quantified using Image Lab software (Bio-Rad). Experiments were conducted in triplicates.
Plasmid interference assays using PICI system
Plasmids (pB020–pB026 and pB057–pB063) used in the plasmid interference with CRISPR interference (PICI) system were designed based on our previous reports [27] and listed in Supplementary Table S4. The exogenous CRISPR–Cas9 expression systems derived from pACYC184 plasmid (p15A origin, chloramphenicol resistance), while co-existing exogenous anti-CRISPR expression systems based on pBAD24 plasmid (colE1 origin, ampicillin resistance). The pT plasmid was derived from pRSFduet-1 (RSF origin, kanamycin resistance), with an mCherry reporter gene targeted by Cas9 plasmid with matching spacer.
Plasmids were transformed into E. coli using the CaCl2 heat-shock procedure as described previously [12, 27]. Colonies were imaged using a Tanon 3500 gel scanner and counted via ImageJ software. The inhibitory activity of anti-CRISPR can be measured by calculating the ratio of colony forming units between E. coli transformed with Cas9 plasmid with matching spacer and that of the mismatching spacer. In addition, the mCherry images of E. coli were taken on a Typhoon FLA 9500 (GE) under Cy5 condition, and the mCherry intensity was quantified using Image Lab software (Bio-Rad).
Protein expression and purification
DNA sequences encoding Aca and Acr proteins were ligated into the pET28a vector and transformed into E. coli (T7 Express, Biomed) for protein expression (pC003–pC015, Supplementary Table S4). The procedures of protein expression and purification were conducted according to our previous reports with slight modifications [12, 27]. Briefly, E. coli cells were grown in LB medium with kanamycin (50 μg/ml) for 16 h at 18°C supplemented with 1 mM IPTG (Yeasen) for induction of protein expression. Then, E. coli cells were collected and resuspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 10 mM imidazole, 1 mM dithiothreitol (DTT), and 500 mM NaCl) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and lysozyme. After sonication and centrifugation, a part of supernatant was filtered through a 0.22-μm syringe filter (Millipore) and used for APID system analysis. For protein purification, the supernatant was incubated with Ni-NTA agarose (QIAGEN), washed with buffer containing 20 mM imidazole, and eluted with 500 mM imidazole. The purified proteins were buffer-exchanged into Storage Buffer (20 mM HEPES–NaOH, pH 7.5, 5% (v/v) glycerol, 300 mM NaCl and 1 mM DTT) using an Amicon Ultra centrifugal filter (Millipore). The purified proteins were analyzed on 4%–20% SDS–PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) gel (ACE Biotechnology). In addition, AcrIIA4, AcrIIA31, and AcrIIA32 proteins were purified in our previous studies [12, 31].
In vitro DNA cleavage assays
The in vitro DNA cleavage assay was performed according to our previous studies [12, 27]. All sgRNAs were prepared from the purified PCR products using in vitro T7 transcription kit (Invitrogen) according to the manufacturer’s manual. SpyCas9 protein was purchased from IDT, while St1Cas9 and St3Cas9 proteins were purified in our pervious study [12]. The substrate DNA was generated by plasmid pC002 linearized through NotI restriction endonuclease (NEB) (Supplementary Fig. S3C and Supplementary Table S4). Cas9 protein (1 μM) was pre-incubated with sgRNA (500 nM) for 10 min at 37°C in a total volume of 10 μl with NEBuffer 3.1. Subsequently, excess Acr (20 μM) was added and incubated at room temperature for 20 min. Then, target DNA (300 ng) was added and incubated for 10 min at 37°C. The reaction was terminated by adding Proteinase K (PK) and DNA loading dye. The reaction products were analyzed on 1% agarose/1× TAE (Tris–acetate–EDTA) gels and imaged using a Tanon 3500 gel scanner.
T7 endonuclease 1 assay
The target sequence at the DYRK1A locus and the PCR primers used for T7 endonuclease 1 (T7E1) analysis are listed in Supplementary Table S2. HEK293T cells were cultured in 24-well plates and transfected with 1 μg of Cas9 plasmid, 0.5 μg of sgRNA plasmid, and 0.5 μg of Acr plasmid per well. Transfections were performed using Lipofectamine LTX reagent (Invitrogen) following the manufacturer’s instructions. After 72 h of transfection, genomic DNA from cells was extracted and amplified for PCR using Q5 High-Fidelity Polymerase (NEB). The PCR products were mixed with NEBuffer 2, denatured, annealed, and then treated with T7E1 (NEB) for 15 min at 37°C. The digestion products were analyzed on a 3% agarose/1× TAE gel, and bands were quantified using ImageJ software. Editing efficiency was calculated using the following formula: indel (%) = 100 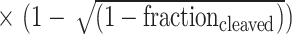 .
.
Next-generation sequencing
Gene editing efficiencies were assessed by next-generation sequencing (NGS) as described in our previous study [12]. Briefly, NGS assays were conducted using a two-step PCR-based approach. First, genomic DNA was extracted from cells and amplified via first-step PCR using Q5 High-Fidelity Polymerase (NEB). Primer sequences containing 5′ Illumina sequencing adaptors are provided in Supplementary Table S2. Subsequently, purified PCR products served as templates for second-step PCR using barcoded primers, followed by PE150 sequencing on an Illumina MiSeq platform (Tsingke Biotechnology). Genome editing efficiency was calculated from the sequencing data using CRISPResso2 [32].
Electrophoretic mobility shift assay of sgRNA
For RNA electrophoretic mobility shift assay (EMSA), St1Cas9 protein (2 μM) was incubated in Binding Buffer (20 mM Tris–HCl, pH 7.6, 150 mM KCl, 5 mM EDTA, 5 mM MgCl2, 1 mM DTT, 5% (v/v) glycerol, 50 μg/ml heparin, 0.01% Tween 20, 100 μg/ml BSA). The sgRNA (400 nM) and Acr protein (5 μM) were added in different order and incubated for 10 min at 37°C. Samples were analyzed on 6% Tris–borate–EDTA (TBE) (Biorigin), stained with SYBR Gold (Invitrogen), and imaged using the Typhoon 7000 (GE) system.
DNA EMSAs
For Figs 2E and 3C–E, DNA EMSAs were conducted with 100 nM fluorescently labeled substrate DNA (annealed oligonucleotides, Supplementary Fig. S4) in Binding Buffer (20 mM Tris–HCl, pH 7.6, 150 mM KCl, 5 mM EDTA, 5 mM MgCl2, 1 mM DTT, 5% (v/v) glycerol, 50 μg/ml heparin, 0.01% Tween 20, 100 μg/ml BSA). Aca proteins with titrations (100, 200, 400, and 800 nM) were added and incubated at room temperature for 20 min. Samples were analyzed on 6% TBE and imaged using the Typhoon 7000 (GE) system under Cy5 condition.
Figure 2.

Aca11 recognizes conserved IRs within the acr-aca11 promoters. (A) Nucleotide sequence alignment of acr-aca11 promoters (Aca11-s1 and -s2), spanning 80 bp upstream of the acr start codon. Predicted promoter elements, including the −10 and −35 regions, are highlighted by black boxes. IRs are denoted by orange boxes with arrowheads indicating their orientation. (B) Wild-type (WT) and scrambled IR (scIR) sequences in Aca11-s1 promoter context. Sequences of nucleotide substitutions in scIR mutant are shown in red. (C) Representative colony pictures of E. coli expressing mCherry under the control of Aca11-s1 (WT or scIR) promoters with or without Aca11 co-expression. BF and mCherry fluorescence channels are displayed on the left side of each row. (D) Bar graphs depict the median mCherry fluorescence values, normalized to the corresponding promoter controls without Aca11. Error bars represent mean ± SEM from three biological replicates. Statistical significance was determined by unpaired t-test (***P< .001; n.s., not significant). (E) EMSAs of the Aca11–DNA interactions with increasing concentrations (100, 200, 400, and 800 nM) of Aca11 indicated by black triangles. The assays were analyzed on the non-denaturing gel with Cy5-labeled WT/scIR probes (see Supplementary Fig. S4A). The gels are representative of three independent replicates. (F) The sequence of 24-bp Aca11-IR DNA used for molecular modeling. The core motif regions are highlighted. (G) AlphaFold 3-predicted structure of dimeric Aca11 in complex with 24-bp DNA [shown in panel (F)]. (H) Surface view of Aca11–IR complex with electrostatic potential (blue, positively charged; red, negatively charged). Aca11 exhibits a positively charged surface docked by IR DNA. (I) Spatial arrangement of key residues Y18, Q28, and R39 potentially engaged in base-specific contacts with palindromic motifs. (J) Representative colony pictures of E. coli expressing mCherry under the control of Aca11-s2 promoter in the presence of diverse Aca11 mutants. (K) Bar graphs depict the median mCherry fluorescence values of diverse Aca11 mutants, normalized to no Aca11 control. Error bars represent mean ± SEM from three biological replicates. Statistical significance was determined by unpaired t-test (***P< .001).
Figure 3.

Aca13 domain in C-terminal of AcrIIA32 acting on conserved IRs within acr-aca13 promoters. (A) Nucleotide sequence alignment of Aca13-s1 and Aca13-s2 promoters (80 bp upstream of the acr start codon). Two predicted palindromic IRs (IR1: green box; IR2: orange box) are shown with arrowheads indicating their orientation. (B) Sequences of the WT IR and mutated IR (scIR1 and scIR2) of Aca13-s1 promoter. scIR sequences are shown in red. EMSAs of the Cy5-labeled WT (C), scIR1 (D), and scIR2 (E) DNA probe of Aca13-s1 with increasing concentrations (100, 200, 400, and 800 nM) of AcrIIA32, A32NTD, or A32CTD proteins indicated by black triangles. The assays were analyzed on the non-denaturing gel with three independent replicates. (F) The sequence of 18-bp Aca13-IR DNA used for structural modeling. The core motif regions are highlighted. (G) AlphaFold 3-predicted structure of dimeric A32CTD in complex with 18-bp DNA [shown in panel (F)]. (H) Surface view of A32CTD-IR complex with electrostatic potential (blue, positively charged; red, negatively charged). A32CTD exhibits a positively charged surface docked by IR DNA. (I) Spatial arrangement of key residues K92, W99, R107, and S111 of A32CTD potentially engaged in base-specific contacts with palindromic motifs. (J) Representative colony pictures of E. coli expressing mCherry under the control of Aca13-s2 promoter in the presence of diverse A32CTD mutants. (K) Bar graphs depict the median mCherry fluorescence values of diverse A32CTD mutants, normalized to no A32CTD control. Error bars represent mean ± SEM from three biological replicates. Statistical significance was determined by unpaired t-test (***P< .001).
For Fig. 4G and Supplementary Fig. S8D, the DNA EMSA assays were performed as previously described [27]. Briefly, St1Cas9 protein (2 μM) was pre-incubated with sgRNA (400 nM) in Binding Buffer at 37°C for 10 min. Subsequently, Acr proteins with various titrations (4, 8, and 16 μM) or fluorescently labeled substrate DNA (100 nM, Cy5-labeled target strand) were added to the mix in different order at room temperature for 20 min. Samples were analyzed by biphasic polyacrylamide (the upper half of the gel is 6% and the lower half of the gel is 12%)/0.5× TBE gel electrophoresis and imaged using the Cy5 channel on the Typhoon 7000 (GE) system.
Figure 4.

A novel type II-A Acr (AcrIIA35) inhibiting St1Cas9 is identified by Aca13 with its IR sequences. (A) Schematic view of the genomic context of representative Aca13 loci from diverse Streptococcus phages and prophages. Candidate acr genes (shown in orange) and other neighboring genes (shown in gray and annotated by NCBI website or HHpred) are searched by aca13 (shown in blue). Acr genes are shown in red with numbers corresponding to AcrIIA numbers. acr-aca promoters (s11–s18) are highlighted by boxes and core sequences are shown with the −10 and −35 elements (green shading) and identified IRs (orange boxes) in the right panel. (B) Schematic view of the PICI system to characterize novel anti-CRISPR proteins. (C) Bar graphs show the calculated transformation efficiency of Acrs and Acr candidates against St1Cas9 in E. coli as colony forming units per 25 ng of each plasmid DNA (left panel in C). Bar graphs show the median mCherry fluorescence value of Acrs, which were normalized to each Acr with mismatching spacer control (right panel in C). Error bars represent the mean ± SEM with three biological replicates. Sign “#” represents “below detection limit of this assay”. *P < .05, **P < .01, and ***P < .001, determined by unpaired t-test. n.s., not significant. (D) DNA cleavage assays targeting linearized plasmid DNA by St1Cas9 in the presence or absence of Acrs. Open and closed arrowheads indicate intact substrate and cleavage products, respectively. Representative data from triplicate experiments are shown. Representative gel images (E) and quantification of gene editing efficiencies (F) of T7E1 assay to manifest the inhibitory activities of Acrs against St1Cas9 in human cells. Error bars represent the mean ± SEM with three biological replicates. *P < .05, **P < .01, and ***P < .001, determined by unpaired t-test. n.s., not significant. (G) DNA EMSA assays were conducted to analyze the effect of AcrIIA35 on DNA binding of St1Cas9 RNP, when added prior to or after the addition of target DNA. Assays were conducted with St1Cas9 RNP (400 nM) and Acr titrations (4, 8, and 16 μM). The assays were analyzed on the non-denaturing gel with target DNA labeled by Cy5. The gels are representative of three independent replicates. (H) RNA EMSA assays were conducted to analyze the effect of AcrIIA35 on St1Cas9 binding to sgRNA, when Acrs were added prior to or after the addition of sgRNA. The gels are representative of three independent replicates.
DNA EMSAs with APID system
For Supplementary Fig. S9, DNA EMSAs were conducted with fluorescently labeled substrate DNA (100 nM, Supplementary Fig. S4) in Binding Buffer. Cell lysates of Aca proteins with titrations (described in figure legend) were added and incubated at room temperature for 20 min. Samples were analyzed on 6% TBE (Biorigin), and imaged using the Typhoon 7000 (GE) system under Cy5 or FAM condition.
For Fig. 5D and E, DNA EMSAs were conducted by incubating St1Cas9 protein (4 μM) with or without sgRNA (1.6 μM) in Binding Buffer at 37°C for 10 min. AcrIIA35-Aca13 fusion proteins (400 nM) or their cell lysates (0.03 μl of supernatant) were added and incubated at room temperature for 20 min. Then, fluorescently labeled substrate DNA (40 nM) were added and incubated at room temperature for another 20 min. Samples were analyzed by biphasic polyacrylamide (the upper half of the gel is 6% and the lower half of the gel is 12%)/0.5× TBE gel electrophoresis and imaged using the Cy5 channel on the Typhoon 7000 (GE) system.
Figure 5.

Establishment of Aca-based EMSAs for protein–protein interaction detection. (A) Schematic view of the APID system based on EMSAs in this study. The target protein is conjugated with Aca13, which can specifically bind to fluorescently labeled Aca13 probe. Free and protein-bound probe can be separated in the non-denaturing polyacrylamide gel after electrophoresis. (B) Schematic view of AcrIIA35-Aca13 constructs. A glycine- and serine-rich linker (32aa) connects the Acr and Aca terminus. (C) Experimental workflow of AcrIIA35-Aca13 hybrid expression and purification from E. coli. Supernatant of cell lysis and proteins of AcrIIA35-Aca13 were used for subsequent EMSAs in panels (D) and (E). DNA EMSA assays of APID system were conducted with AcrIIA35-Aca13 protein (D) or AcrIIA35-Aca13 supernatant of cell lysis (E). Lanes 4 and 8 contain the purified AcrIIA35-Aca13 proteins as controls. Hollow arrowheads indicate shifted protein–DNA complex of AcrIIA35-Aca13 with Aca13 probe. AcrIIA35 can only bind to sgRNA-bound St1Cas9, leading to a DNA supershift. The gels are representative of three independent replicates. (F) Summary of the inhibitory mechanism identified in this study. For AcrIIA35, it binds solely to sgRNA-bound Cas9 to block the binding of Cas9 to target DNA. In contrast, AcrIIA24 abrogates the DNA cleavage activity of Cas9.
For Fig. 6D and E, DNA EMSAs were conducted with cell lysates of Aca proteins combining FAM-labeled Aca11 probe and Cy5-labeled Aca13 probe. Briefly, cell lysates of St3_HNH-n-Aca11 hybrid (0.25 μl of supernatant) were incubated with Aca13 hybrids (0.015625 μl of supernatant for each hybrid) in Binding Buffer at room temperature for 20 min. Then, fluorescently labeled substrate DNA (100 nM) were added and incubated at room temperature for another 20 min. Samples were analyzed on 6% TBE (Biorigin), and imaged using the Typhoon 7000 (GE) system under Cy5 or FAM condition.
Figure 6.

Detection of AcrIIA24 binding to the HNH domain of St3Cas9 using the optimized APID system. (A) Schematic view of the optimized APID (APID-v2) system. This system is constructed using Aca11 and Aca13 proteins along with doubly fluorescent-labeled probes. Supershifted DNA band indicates the co-localization of protein–protein interaction complexes. (B) AlphaFold 3-predicted structure of St3Cas9–gRNA–DNA–AcrIIA24 complex. AcrIIA24 comprises two α-helices and five β-sheets and bounds to the HNH domain of St3Cas9. The loop (E32–D38) between β1 and β2 sheets represents key residues engaged in interaction. (C) Schematic view of Aca-tagged protein constructs. Acr subtypes and numbers are indicated. A, AcrIIA; C, AcrIIC. (D) DNA EMSA assays were conducted with supernatant of cell lysis from E. coli expressing Acr-Aca13 and St3_HNH-Aca11 fusion proteins, in the presence of FAM-labeled Aca11 probe. The gels are representative of three independent replicates. (E) DNA EMSA assays of APID-v2 system were performed using the supernatant of cell lysis from E. coli expressing Acr-Aca13 and St3_HNH-Aca11 hybrids. Both FAM-labeled Aca11 probe and Cy5-labeled Aca13 probe were used in the assay. Asterisk indicates the supershifted DNA band, which demonstrates the co-localization of Aca11 and Aca13 probes bound by AcrIIA24-St3_HNH interaction complexes. (F) Bar graphs show the calculated transformation efficiency of AcrIIA24 variants against St3Cas9 in E. coli as colony forming units per 25 ng of each plasmid DNA. Error bars represent the mean ± SEM with three biological replicates. Sign “#” represents “below detection limit of this assay.” *P < .05, **P < .01, and ***P < .001, determined by unpaired t-test. n.s., not significant. (G) Schematic view of the inhibition mechanism of AcrIIA24. AcrIIA24 prevents Cas9 from DNA cleavage by binding to the HNH domain of Cas9.
Results
The multifunctional regulatory roles of Aca11 and Aca13 in their acr-aca operons
Aca genes are typically positioned downstream of acr genes, functioning as autorepressors to regulate Acr expression [23]. Consistent with previous findings, our genomic context analysis of aca1–13 genes revealed that acr genes are almost invariably located upstream of aca genes (Supplementary Fig. S1). Notably, Aca11 and Aca13 displayed a unique genomic architecture, with multiple acr genes also clustered in downstream positions. This is a distinct spatial arrangement compared to other aca family members (Fig. 1A and Supplementary Fig. S1). This observation prompted us to investigate whether Aca11 and Aca13 were able to regulate acr genes positioned both upstream and downstream.
We first analyzed the genomic context of the acr-aca11 locus within prophage region of Streptococcus sp. F0441, where AcrIIA25 and AcrIIA3 flank aca11 in upstream and downstream positions, respectively (Fig. 1B). We then established a plasmid-based fluorescence reporter system by individual insertion of AcrIIA25 and AcrIIA3 promoters (Aca11-s1 and Aca11-s2, respectively) into an mCherry transcriptional reporter plasmid, given that Aca proteins always repress Acr expression by binding the promoter region of acr genes (Fig. 1B and Supplementary Fig. S2A). Aca11 was co-expressed from a second plasmid under control of the arabinose-inducible araBAD promoter (Fig. 1B). In the absence of Aca11, we observed high mCherry fluorescence value of E. coli colonies for both reporter constructs. However, upon Aca11 induction, mCherry fluorescence decreased by ∼60% in colonies harboring the Aca11-s1 promoter construct and by 90% in those with the Aca11-s2 promoter construct (Fig. 1C and D). These results suggest that Aca11 can repress acr promoters located both upstream and downstream of aca gene.
To assess the repression activity of Aca13, we analyzed the genomic context of the acr-aca13 locus within prophage region of Streptococcus uberis NZ01 (Fig. 1E). We also constructed fluorescence reporter plasmids individually containing the upstream AcrIIA3 promoter (Aca13-s1) and the downstream AcrIIA27 promoter (Aca13-s2) (Fig. 1E and Supplementary Fig. S2B). Interestingly, AcrIIA32 is a fusion protein that comprises an N-terminal Acr domain (named A32NTD) and a C-terminal Aca13 domain (named A32CTD) (Fig. 1F). To explore their functions, we separated AcrIIA32 into its N-terminal domain (NTD) and C-terminal domain (CTD) and tested their regulatory activities using the fluorescence reporter system. The results showed that E. coli exhibited robust mCherry expression in the absence of AcrIIA32, whereas only full-length AcrIIA32 significantly suppressed mCherry transcription for both reporter constructs, reducing fluorescence intensity by ∼90% (Fig. 1G and H). Neither A32NTD (Acr domain) nor A32CTD (Aca domain) affected the Aca13-s1 promoter, leading to high mCherry fluorescence value in E. coli. Notably, A32CTD (Aca domain) exhibited robust repressive activity on the Aca13-s2 promoter, leading to a marked reduction in mCherry fluorescence (Fig. 1G and H). Intriguingly, the A32NTD (Acr domain) was found to mediate weak repression of the Aca13-s2 promoter. In addition, we investigated the anti-CRISPR activity of A32NTD (Acr domain) and A32CTD (Aca domain) against SpyCas9 and St3Cas9, based on plasmid interference and in vitro DNA cleavage assays (Supplementary Fig. S3). The data showed that A32NTD (Acr domain) displayed anti-CRISPR activity, while A32CTD (Aca domain) displayed negligible CRISPR–Cas inhibitory activity under in vitro and in vivo conditions (Supplementary Fig. S3A and D). These results indicate that AcrIIA32 is a bifunctional protein with both Acr and Aca13 functionality. Specifically, A32NTD exerts anti-CRISPR function, while A32CTD predominantly serves to regulate adjacent acr genes.
Aca11 represses acr-aca11 promoters through recognizing conserved IRs in its operons
As DNA-binding transcriptional regulators, Aca proteins typically contain an HTH domain and repress Acr expression by binding to IR sequences within acr promoter regions [23, 24]. Since Aca11 represses both upstream and downstream acr promoters, we hypothesized that Aca11 binds to the promoter regions of both Aca11-s1 (associated with AcrIIA25) and Aca11-s2 (associated with AcrIIA3). We then performed nucleotide sequence alignment of the Aca11-s1 and Aca11-s2 promoters (Fig. 2A). We identified the conserved IR sequences located between the −35 and −10 elements in both promoters, suggesting these motifs serve as putative Aca11 binding sites. To verify this hypothesis, we employed our fluorescence reporter system, comparing mCherry transcription driven by either the WT Aca11-s1 promoter or a mutant variant with scIR sequences (Fig. 2B). In the absence of Aca11, we observed high mCherry fluorescence value of E. coli colonies for both reporter constructs. However, upon Aca11 induction, mCherry fluorescence decreased by ∼90% in colonies harboring the WT Aca11-s1 promoter construct, while those with the scIR mutant showed no significant reduction (Fig. 2C and D). These results indicate that Aca11-mediated repression depends on its interaction with conserved IR sequences within acr-aca11 promoters.
To determine whether Aca11 directly interacts with IR sequences, we conducted EMSAs using purified Aca11 protein and Cy5-labeled DNA probes corresponding to the WT promoter and the scIR mutant (Supplementary Figs S3B and S4A). A clear dose-dependent band shift was observed upon Aca11 binding to the WT promoter probe, while no shifted complexes formed with the scIR probe at any tested protein concentration (Fig. 2E). These findings demonstrated the sequence-specific binding of Aca11 to the IR motif.
Previous studies have demonstrated that Aca proteins typically function in a dimeric form to bind IRs [26, 33]. We then utilized AlphaFold 3 to predict the structure of a dimeric Aca11 complex bound to IR DNA (Fig. 2F and G). In the DNA-bound Aca11 prediction model, IR DNA tightly localized on the positively charged surface of the Aca11 dimer, with each monomer binding to one half of the IR sequence (Fig. 2G and H). AlphaFold 3-predicted structure reveals that Aca11 consists of multiple α-helices with helices α2 and α3 likely forming an HTH domain, where Y18, Q28, and R39 might be involved in specific contacts with the IR sequence (Fig. 2I). To validate these predictions, we introduced alanine substitutions at these positions and assessed repression activity using our plasmid-based fluorescence reporter system. Remarkably, Q28A and R39A mutations nearly abolished Aca11-mediated transcriptional repression, while the Y18A mutant retained partial functionality compared to WT Aca11 (Fig. 2J and K). Our results demonstrate that Q28 and R39 are essential for the interaction between Aca11 and DNA.
Aca13 domain of AcrIIA32 binds to conserved IRs within acr-aca13 promoters
AcrIIA32 consists of an N-terminal Acr domain (A32NTD) and a C-terminal Aca13 domain (A32CTD), and its anti-CRISPR mechanism of AcrIIA32 has been comprehensively elucidated in previous studies [12, 16]. Thus, we mainly focused on investigating its regulatory function as an anti-CRISPR transcriptional repressor. Similar to our approach with Aca11, we performed nucleotide sequence alignment of the Aca13-s1 (associated with AcrIIA3) and Aca13-s2 (associated with AcrIIA27) promoters. This analysis revealed two conserved IR regions (Aca13-IR1 and Aca13-IR2) located between the −35 and −10 promoter elements (Fig. 3A). To determine whether Aca13 directly interacts with these IR motifs, we conducted EMSAs using purified AcrIIA32, A32NTD, and A32CTD proteins along with Cy5-labeled DNA probes corresponding to WT, scIR1, or scIR2 sequences (Fig. 3B and Supplementary Figs S3B and S4B). Both AcrIIA32 and A32CTD exhibited dose-dependent binding to WT and scIR1 sequences, while A32NTD showed no detectable binding activity (Fig. 3C and D). Notably, scIR2 mutations completely abolished AcrIIA32 and A32CTD binding at all tested concentrations (Fig. 3E). These results demonstrate that the C-terminal Aca13 domain (A32CTD) can specifically interact with Aca13-IR2 motif.
Subsequently, we used AlphaFold 3 to predict the structure of a dimeric A32CTD (Aca13) complex bound to Aca13-IR2 DNA (Fig. 3F and G). Similar to Aca11, the IR DNA tightly localize on the positively charged surface of the A32CTD dimer, with each monomer engaging one half of the IR sequence (Fig. 3G and H). Intriguingly, Aca13 contains a coiled α-helix interacting with IR sequence in our predicted DNA-bound Aca13 model, a feature distinct from the canonical HTH motif observed in other Aca proteins (Fig. 3G). Further analysis identified four critical residues (K92, W99, R107, and S111) within the DNA-binding interface (Fig. 3I). Alanine substitutions at these positions completely abrogated A32CTD-mediated repression in our mCherry reporter system using the Aca13-s2 promoter (Figs 1G and H, and 3J and K), confirming their essential role in DNA recognition.
Previous experiments showed that A32NTD deletion eliminates AcrIIA32’s repression of the Aca13-s1 promoter (Fig. 1G and H), suggesting a functional synergy existing between the distinct domains of AcrIIA32. The structural models of full-length AcrIIA32 in its bound state with Aca13-IR2 DNA, as predicted by AlphaFold 3, revealed structural congruence between the predicted A32NTD and its cryo-EM structure (PDB: 8YE6) (Supplementary Fig. S5A and B) [16]. AlphaFold 3-predicted structure reveals that residues R104 and E105 in the Aca domain might interact dynamically with loop1 (M1–S11) and loop2 (T36–T46) of the Acr domain (Supplementary Fig. S5C). We then generated multiple mutants of AcrIIA32, by introducing alanine substitutions at R104 and E105, truncating the N-terminus of AcrIIA32 (delete M1–S11), and replacing residues T36–T46 with a GSGSGS linker. The results showed that R104A and R104A/E105A mutations abolished regulatory activity of AcrIIA32 on Aca13-s1 promoter, but have no effect on Acr activity of AcrIIA32 against St3Cas9 (Supplementary Fig. S5D–G). Deletion of loop2 specifically impaired the anti-CRISPR activity of AcrIIA32, while deletion of loop1 disrupted both regulatory and anti-CRISPR activities of AcrIIA32 (Supplementary Fig. S5D–G). These data suggest that Acr domain may enhance the regulatory activity of Aca domain, which represent the mechanism of functional synergy in anti-CRISPR systems.
Subsequently, we investigated the tolerance of Aca11 and Aca13 proteins for mismatches within their IR sequences using a plasmid-based fluorescence reporter system in E. coli (Supplementary Fig. S6A and E). E. coli cells were transformed with plasmids co-expressing Aca proteins and an mCherry reporter. The reporter’s expression was controlled by the acr-aca promoter harboring various indicated mutants of the corresponding IR DNA sequences (Supplementary Fig. S6B and F). The results showed that both Aca11 and Aca13 (A32CTD) strongly repressed mCherry expression upon recognition of WT IR sequences in acr-aca promoter regions. In contrast, scIR mutants, containing substitutions of all nucleotides within the IR sequences, completely abolished the regulatory activity of Aca11 and Aca13 proteins (Supplementary Fig. S6C and G). In addition, single nucleotide substitutions in the core motif of IR sequences altered the regulatory activity of Aca proteins on their acr-aca promoters (Supplementary Fig. S6C and G). Interestingly, we observed that full-length AcrIIA32 displayed greater tolerance to mismatches in the IR sequence compared to Aca13, further verifying that the Acr domain (A32NTD) enhances the regulatory activity of the Aca13 domain (A32CTD) (Supplementary Fig. S6C and D).
A novel type II-A Acr identified by Aca13 with its IR sequences
To investigate whether other homologs of Aca11 and Aca13 function as multifunctional regulators, we examined the genomic context of Aca11 and Aca13 loci with similar acr-aca spatial arrangement from diverse Streptococcus phages and prophages (Supplementary Fig. S7A). We identified conserved IR sequences located between the −35 and −10 promoter elements in the homologous loci of both Aca11 and Aca13. Results from fluorescence reporter system demonstrate that these Aca11 and Aca13 homologs can repress the expression of their operons through recognition of IR sequences in acr promoter regions (Supplementary Fig. S7B–E). Notably, we observed several unknown genes (Cand1, Cand2, and Cand3) under the control of acr-aca13 promoters, strongly indicating that these genes are acr genes (Fig. 4A and Supplementary Table S1). Subsequently, Acr candidates were tested against Streptococcus Cas9 systems (SpyCas9, St1Cas9, and St3Cas9) using our established PICI system in E. coli (Fig. 4B) [27]. The result showed that only one Acr candidate (Cand2) can potently inhibit St1Cas9 in E. coli, and its inhibitory effect is similar to that of AcrIIA31 (Fig. 4C and Supplementary Fig. S8A). Following standard nomenclature, we designated Cand2 as AcrIIA35. In addition, we observed a relatively high mCherry fluorescence value of E. coli colonies, which transformed St1Cas9 with a matching spacer in the presence of AcrIIA35 (Fig. 4C). Our results suggest that AcrIIA35 inhibit St1Cas9 by blocking Cas9 binding to target DNA or at an upstream stage (interfering with crRNA loading).
To further investigate the inhibitory activity of AcrIIA35, we purified AcrIIA35 and performed DNA cleavage assays in vitro (Fig. 4D and Supplementary Fig. S8B). We found that AcrIIA35 potently inhibited St1Cas9 DNA cleavage activity, regardless of whether it was added prior to or after the formation of the St1Cas9–sgRNA RNP complex (Fig. 4D). These data suggest that AcrIIA35 mainly act on Cas9 RNP to impact the downstream function of Cas9 RNP. To examine whether AcrIIA35 can inhibit St1Cas9 in human cells, human endogenous DYRK1A loci were targeted by St1Cas9 in HEK293T cells and analyzed using T7E1 and NGS (Supplementary Table S2). These results showed that AcrIIA35 can efficiently inhibit St1Cas9-mediated genome editing in human cells, with inhibitory efficacy comparable to that of AcrIIA31 (Fig. 4E and F, and Supplementary Fig. S8E).
Given that AcrIIA35 effectively inhibits St1Cas9 in human cells, we further explored its mechanism using EMSAs. DNA EMSAs were conducted with a fluorescently labeled substrate DNA (Cy5-labeled target strand) probe (Supplementary Fig. S8C). The EMSA results showed that AcrIIA35 can abrogate DNA binding of St1Cas9 RNP, only when added prior to the addition of target DNA (Fig. 4G and Supplementary Fig. S8D). Additionally, RNA EMSAs were performed to determine whether AcrIIA35 can prevent the formation of Cas9–sgRNA RNP complex. Our results showed that AcrIIA35 had no impact on the formation of St1Cas9–sgRNA RNP, regardless of whether it was added prior to or after the addition of sgRNA (Fig. 4H).
Establishment of Aca-based EMSAs for protein–protein interaction detection
We have demonstrated that AcrIIA35 inhibits St1Cas9 by blocking DNA target recognition rather than interfering with sgRNA loading (Fig. 4G and H). However, it remains unclear whether AcrIIA35 interacts with apo-St1Cas9 or exclusively targets the mature RNP complex. To further investigate the molecular mechanism of AcrIIA35, we developed an APID system based on EMSA. The APID system employs Aca13-tagged target protein and fluorescent DNA probe containing conserved IRs specifically recognized by Aca13 (Fig. 5A). This system enables efficient detection of protein–protein interactions based on migration differences. If an interaction occurs between the untagged protein and the Aca13-tagged target protein, a larger complex will be formed causing supershift of DNA.
Subsequently, we fused Aca13 (A32CTD) to the C- or N-terminus of AcrIIA35 using a glycine and serine-rich linker to generate A35-c-Aca13 and A35-n-Aca13, respectively (Fig. 5B and Supplementary Table S3). DNA EMSAs using the APID system were performed with these fusion proteins, which were expressed in E. coli and purified through standard methods (Fig. 5C). The results showed that both A35-c-Aca13 and A35-n-Aca13 bind to the Aca13 probe, causing DNA band shifts (Fig. 5D). Furthermore, these fusion proteins can only trap the St1Cas9 in the presence of gRNA to cause DNA band supershifts, indicating that AcrIIA35 binds to and inhibits St1Cas9 exclusively after RNP complex formation (Fig. 5D). Notably, clearer supershifted DNA bands were observed with A35-c-Aca13 compared to A35-n-Aca13, suggesting enhanced stability of the St1Cas9–gRNA complex with the C-terminal fusion. Given the natural C-terminal localization of Aca13 (A32CTD) in AcrIIA32, we speculate that mimicking native domain arrangements (e.g. A35-c-Aca13) enhances the stability of fusion motifs.
To further simplify the APID workflow, we tested the feasibility of using crude cell lysates as alternatives to purified proteins for DNA EMSAs. First, we evaluated the DNA-binding ability of Aca proteins in crude lysates. The results showed that Aca proteins in lysates retained their IR sequence recognition specificity, displaying concentration-dependent effect comparable to that of purified proteins (Supplementary Fig. S9A and B). Next, we performed DNA EMSA using cell lysates containing AcrIIA35-Aca13, optimizing lysate dilution to match the conditions used for purified proteins (Supplementary Fig. S9C). The electrophoretic migration patterns observed with crude lysates aligned with those from purified protein experiments (Fig. 5D and E), suggesting that the APID system can detect Aca-based protein–protein interactions with feasibility of using crude cell lysates as alternatives. Through APID system analysis, we have clarified the inhibitory mechanism of AcrIIA35. It binds to St1Cas9 after sgRNA loading and blocks the interaction between RNPs and target DNA (Fig. 5F). Given that AcrIIA35 exhibits specific inhibition of St1Cas9, we speculate that AcrIIA35 may bind to a unique site on St1Cas9 to exert its function. These results enhance our knowledge of Acr protein function and the utility of the APID system in exploring protein interactions.
Detection of AcrIIA24 binding to the HNH domain of St3Cas9 using the optimized APID system
To improve the detection efficiency of the APID system, we developed the APID-v2 system by integrating two Aca proteins (Aca11 and Aca13) with their corresponding DNA probes labeled by different fluorescent dyes (Fig. 6A). Our prior work demonstrated that AcrIIA24 inhibits St3Cas9 activity by preventing the Cas9 RNP complex from cleaving target DNA, exhibiting a distinct inhibitory strategy compared to AcrIIA35 (Fig. 5F) [12]. However, the precise mechanism of AcrIIA24 remained unknown. The structure of the St3Cas9–gRNA–DNA–AcrIIA24 complex, as predicted by AlphaFold 3, revealed that AcrIIA24 consists of two α-helices and five β-strands and specifically interacts with the HNH domain of St3Cas9 (Fig. 6B). Notably, a loop region between the β1 and β2 strands (residues E32–D38) was predicted to play a crucial role in the interaction with the St3Cas9 HNH domain (Fig. 6B).
To validate this AlphaFold 3-predicted structural model, we utilized the APID-v2 system to examine the interaction between AcrIIA24 and the HNH domain of St3Cas9. We fused Aca11 to the N-terminus of the St3Cas9 HNH domain to generate St3_HNH-n-Aca11 hybrid. Additionally, we constructed the A24-c-Aca13 fusion protein by fusing Aca13 (A32CTD) to the C-terminus of AcrIIA24 (Fig. 6C). We also constructed a deletion mutant of AcrIIA24 based on A24-c-Aca13 to generate A24-(Δ32–38)-c-Aca13 hybrid, by replacing residues E32–D38 with a GSGSGS linker. AcrIIC1-c-Aca13 served as a negative control due to its known inability to inhibit St3Cas9. We also evaluated the DNA-binding ability of these Aca fusion proteins in crude lysates. The results showed that these fusion proteins in lysates retained their specificity in recognizing the IR sequences, and they exhibited concentration-dependent effect (Supplementary Fig. S9D and E).
Next, we performed DNA EMSAs using APID system by cell lysates of these fusion proteins with only the FAM-labeled Aca11 probe. The results showed that only St3_HNH-n-Aca11 hybrid can bind to the Aca11 probe to cause DNA band shifts, while Aca13 fused Acrs cannot (Fig. 6D). Furthermore, a supershifted DNA band was observed in the presence of St3_HNH-n-Aca11, only when A24-c-Aca13 instead of A24-(Δ32–38)-c-Aca13 was added in the reaction (Fig. 6D). These data indicate that AcrIIA24 binds to the St3Cas9 HNH domain through its E32–D38 loop region.
Moreover, we conducted DNA EMSAs using the APID-v2 system, with cell lysates of these fusion proteins combing both FAM-labeled Aca11 and Cy5-labeled Aca13 probes. We observed the supershifted DNA band displaying co-localization of Aca11 and Aca13 probes, only when St3_HNH-n-Aca11 and A24-c-Aca13 were included in the reaction (Fig. 6E). In contrast, neither A24-(Δ32–38) nor AcrIIC1 can interact with St3Cas9 HNH domain to produce any co-localized signal. Plasmid interference assays further validated complete loss of AcrIIA24 inhibitory activity against St3Cas9 in E. coli upon E32–D38 deletion (Fig. 6F). These findings demonstrated that AcrIIA24 directly interacts with the St3Cas9 HNH domain, and its loop region (E32–D38) are key sites for both the interaction with St3Cas9 and the inhibitory activity of AcrIIA24. In summary, AcrIIA24 binds to the HNH domain to specifically inhibit DNA cleavage activity of St3Cas9 (Fig. 6G). Furthermore, we show that our APID system provides a robust and efficient platform for protein–protein interaction analysis.
Discussion
Aca proteins are regulatory elements linked to anti-CRISPR systems, typically positioned downstream of Acr operons to prevent excessive Acr expression that could harm the host [24]. Conventionally, Aca proteins regulate Acr expression through negative feedback, minimizing the fitness cost to the host during the later stages of phage infection [22, 23]. In this study, we elucidated the distinct regulatory mechanisms of Aca11 and Aca13, which mediate bidirectional regulation of acr-aca operons by recognizing their IR sequences within acr promoters. We proposed that this bidirectional regulatory mechanism provides phages with a significant advantage over the conventional unidirectional control of a single set of acr genes observed in other acr-aca operons [25]. This mechanism allows phages to swiftly initiate high-level Acr transcription from two robust promoters. Subsequently, it attenuates Acr expression via a single Aca protein. This mechanism manages to establish an optimized finely tuned equilibrium between immune evasion and energy metabolism. As a result, this form of regulation can enhance phage survival against CRISPR–Cas defenses. Our findings reveal a novel regulatory paradigm in anti-CRISPR systems. They not only distinguish Aca11 and Aca13 from previously characterized Aca-associated operons, but also provide fresh insights into the intrinsic interplay between phages and bacterial immune systems.
The identification of Aca proteins has mainly relied on the presence of an HTH domain [7, 21, 23, 34]. However, Aca13 lacks this canonical DNA-binding motif, leading to its initial oversight in early Aca study [34]. In this study, we demonstrated that Aca13 acts as a typical Aca protein, playing a crucial role in the transcriptional regulation of Acrs by binding to specific IR sequences within acr-aca13 promoters. This finding challenges the conventional criteria for Aca identification and suggests that Aca proteins may exhibit a broader range of structural features and regulatory mechanisms than previously recognized. Furthermore, our functional characterization of AcrIIA32 revealed that its Acr domain (A32NTD) displays anti-CRISPR activity, whereas its Aca13 domain (A32CTD) primarily regulates Acr expression. Interestingly, we found the Acr domain (A32NTD) enhances the regulatory activity of Aca13 domain (A32CTD), unveiling a previously unrecognized mechanism of functional domain synergy in anti-CRISPR systems. This study provides new insights into the regulatory landscape of anti-CRISPR systems and identifies structural domains that perform dual roles in CRISPR-mediated immunity.
Currently, the identification of Acr proteins remains challenging due to their remarkable sequence diversity and the absence of conserved structural domains [18, 19]. Since aca genes within acr-aca operons tend to be more conserved, they serve as effective markers for identifying adjacent acr genes through the “guilt-by-association” method [12, 20, 21]. Here, we present an enhanced approach based on “guilt-by-association”, combing Aca proteins along with identification of its IR sequences. This refinement enables us to consider genes regulated by Aca proteins as promising candidates for potential Acr proteins. By utilizing this improved strategy, we screened three candidate genes (Cand1, Cand2, and Cand3) against Streptococcus Cas9 systems (SpyCas9, St1Cas9, and St3Cas9) and successfully identified a novel type II-A Acr, AcrIIA35. Our findings demonstrate the effectiveness of this bioinformatics strategy for Acr discovery. Furthermore, although Cand1 and Cand3 were found not to inhibit SpyCas9, St1Cas9, and St3Cas9, we hypothesize that they might still function as Acr proteins capable of inhibiting other CRISPR–Cas systems, which requires further validation.
To elucidate the molecular mechanism of AcrIIA35 and AcrIIA24, we developed an APID system based on EMSA. The APID system employs Aca-tagged target proteins and fluorescent DNA probes containing conserved IRs that are specifically recognized by Aca proteins. Through APID analysis, we uncovered the inhibitory mechanism of AcrIIA35 and AcrIIA24 against Cas9. Specifically, AcrIIA35 binds to St1Cas9 after sgRNA loading and prevents RNP complex from interacting with target DNA, whereas AcrIIA24 binds to the HNH domain to specifically inhibit DNA cleavage activity of St3Cas9. The APID system enables efficient detection of Aca-mediated protein–protein interactions using either purified proteins or crude cell lysates, significantly simplifying experimental workflows.
In summary, our work provides novel insights into anti-CRISPR regulatory strategies, revealing unique mechanisms that distinctly differentiate Aca11 and Aca13 from other previously characterized Aca proteins. Through development of a bioinformatics strategy combining Aca protein and IR sequence analysis, we discovered a new anti-CRISPR protein, AcrIIA35. Furthermore, we established the APID system as a robust platform for protein interaction studies, providing a versatile tool for functional proteomics in CRISPR–Cas research [35].
Supplementary Material
Acknowledgements
We thank Jianhui Li for technical support in protein purification. We are grateful to Guohong Li for the help with E. coli imaging.
Author contributions: G.S.: Conceptualization, Methodology, Investigation, Formal analysis, Writing – original draft, Writing – review & editing; J.L.: Conceptualization, Methodology, Investigation, Formal analysis, Writing – original draft; J.H.: Investigation; X.G.: Investigation; F.Z.: Investigation; C.T.: Investigation; Y.T.: Supervision, Formal analysis, Writing – review & editing.
Contributor Information
Guoxu Song, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Jiahui Li, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Jun Han, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Xing Gao, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Chunhong Tian, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Fei Zhang, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yong Tian, State Key Laboratory of Epigenetic Regulation and Intervention, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
None declared.
Funding
This work is supported by grants from the National Key R&D Program of China (2022YFF0710700, 2024YFF0728700, 2021YFF0702800, 2020YFA0803501), the National Natural Science Foundation of China (32270567, 32070533), and Biological Resource Program of Chinese Academy of Sciences (KFJ-BRP-005). Funding to pay the Open Access publication charges for this article was provided by National Key R&D Program of China.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
References
- 1. Mayo-Munoz D, Pinilla-Redondo R, Birkholz N et al. A host of armor: prokaryotic immune strategies against mobile genetic elements. Cell Rep. 2023; 42:112672. 10.1016/j.celrep.2023.112672. [DOI] [PubMed] [Google Scholar]
- 2. Garneau JE, Dupuis ME, Villion M et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010; 468:67–71. 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 3. Wang JY, Doudna JA CRISPR technology: a decade of genome editing is only the beginning. Science. 2023; 379:eadd8643. 10.1126/science.add8643. [DOI] [PubMed] [Google Scholar]
- 4. Wang JY, Pausch P, Doudna JA Structural biology of CRISPR–Cas immunity and genome editing enzymes. Nat Rev Microbiol. 2022; 20:641–56. 10.1038/s41579-022-00739-4. [DOI] [PubMed] [Google Scholar]
- 5. Makarova KS, Wolf YI, Iranzo J et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020; 18:67–83. 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koonin EV, Gootenberg JS, Abudayyeh OO Discovery of diverse CRISPR–Cas systems and expansion of the genome engineering toolbox. Biochemistry. 2023; 62:3465–87. 10.1021/acs.biochem.3c00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bondy-Denomy J, Pawluk A, Maxwell KL et al. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–32. 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marino ND, Pinilla-Redondo R, Csorgo B et al. Anti-CRISPR protein applications: natural brakes for CRISPR–Cas technologies. Nat Methods. 2020; 17:471–9. 10.1038/s41592-020-0771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bubeck F, Hoffmann MD, Harteveld Z et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR–Cas9. Nat Methods. 2018; 15:924–7. 10.1038/s41592-018-0178-9. [DOI] [PubMed] [Google Scholar]
- 10. Aschenbrenner S, Kallenberger SM, Hoffmann MD et al. Coupling Cas9 to artificial inhibitory domains enhances CRISPR–Cas9 target specificity. Sci Adv. 2020; 6:eaay0187. 10.1126/sciadv.aay0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bondy-Denomy J, Davidson AR, Doudna JA et al. A unified resource for tracking anti-CRISPR names. The CRISPR J. 2018; 1:304–5. 10.1089/crispr.2018.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song G, Zhang F, Tian C et al. Discovery of potent and versatile CRISPR–Cas9 inhibitors engineered for chemically controllable genome editing. Nucleic Acids Res. 2022; 50:2836–53. 10.1093/nar/gkac099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rauch BJ, Silvis MR, Hultquist JF et al. Inhibition of CRISPR–Cas9 with bacteriophage proteins. Cell. 2017; 168:150–8. 10.1016/j.cell.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harrington LB, Doxzen KW, Ma E et al. A broad-spectrum inhibitor of CRISPR–Cas9. Cell. 2017; 170:1224–33. 10.1016/j.cell.2017.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Li X, Ma Y et al. Inhibition mechanisms of CRISPR–Cas9 by AcrIIA17 and AcrIIA18. Nucleic Acids Res. 2022; 50:512–21. 10.1093/nar/gkab1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng J, Zhu Y, Huang T et al. Inhibition mechanisms of CRISPR–Cas9 by AcrIIA25.1 and AcrIIA32. Sci China Life Sci. 2024; 67:1781–91. 10.1007/s11427-024-2607-8. [DOI] [PubMed] [Google Scholar]
- 17. Zhang F, Song G, Tian Y Anti-CRISPRs: the natural inhibitors for CRISPR–Cas systems. Anim Models Exp Med. 2019; 2:69–75. 10.1002/ame2.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wiegand T, Karambelkar S, Bondy-Denomy J et al. Structures and strategies of anti-CRISPR-mediated immune suppression. Annu Rev Microbiol. 2020; 74:21–37. 10.1146/annurev-micro-020518-120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davidson AR, Lu WT, Stanley SY et al. Anti-CRISPRs: protein inhibitors of CRISPR–Cas systems. Annu Rev Biochem. 2020; 89:309–32. 10.1146/annurev-biochem-011420-111224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pawluk A, Amrani N, Zhang Y et al. Naturally occurring off-switches for CRISPR–Cas9. Cell. 2016; 167:1829–38. 10.1016/j.cell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pinilla-Redondo R, Shehreen S, Marino ND et al. Discovery of multiple anti-CRISPRs highlights anti-defense gene clustering in mobile genetic elements. Nat Commun. 2020; 11:5652. 10.1038/s41467-020-19415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Birkholz N, Kamata K, Feussner M et al. Phage anti-CRISPR control by an RNA- and DNA-binding helix–turn–helix protein. Nature. 2024; 631:670–7. 10.1038/s41586-024-07644-1. [DOI] [PubMed] [Google Scholar]
- 23. Stanley SY, Borges AL, Chen KH et al. Anti-CRISPR-associated proteins are crucial repressors of anti-CRISPR transcription. Cell. 2019; 178:1452–64. 10.1016/j.cell.2019.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Birkholz N, Fagerlund RD, Smith LM et al. The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res. 2019; 47:9658–65. 10.1093/nar/gkz721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shehreen S, Birkholz N, Fineran PC et al. Widespread repression of anti-CRISPR production by anti-CRISPR-associated proteins. Nucleic Acids Res. 2022; 50:8615–25. 10.1093/nar/gkac674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee SY, Birkholz N, Fineran PC et al. Molecular basis of anti-CRISPR operon repression by Aca10. Nucleic Acids Res. 2022; 50:8919–28. 10.1093/nar/gkac656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song G, Tian C, Li J et al. Rapid characterization of anti-CRISPR proteins and optogenetically engineered variants using a versatile plasmid interference system. Nucleic Acids Res. 2023; 51:12381–96. 10.1093/nar/gkad995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zimmermann L, Stephens A, Nam SZ et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018; 430:2237–43. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 29. Waterhouse AM, Procter JB, Martin DM et al. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–91. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abramson J, Adler J, Dunger J et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024; 630:493–500. 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song G, Zhang F, Zhang X et al. AcrIIA5 inhibits a broad range of Cas9 orthologs by preventing DNA target cleavage. Cell Rep. 2019; 29:2579–89. 10.1016/j.celrep.2019.10.078. [DOI] [PubMed] [Google Scholar]
- 32. Clement K, Rees H, Canver MC et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol. 2019; 37:224–6. 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Y, Zhang L, Guo M et al. Structural basis for anti-CRISPR repression mediated by bacterial operon proteins Aca1 and Aca2. J Biol Chem. 2021; 297:101357. 10.1016/j.jbc.2021.101357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang B, Zheng J, Yin Y AcaFinder: genome mining for anti-CRISPR-associated genes. mSystems. 2022; 7:e0081722. 10.1128/msystems.00817-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Song G, Ma Y, Gao X et al. CRISPR/Cas9-mediated genetic correction reverses spinocerebellar ataxia 3 disease-associated phenotypes in differentiated cerebellar neurons. Life Med. 2022; 1:27–44. 10.1093/lifemedi/lnac020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.