

Abstract
Retinitis pigmentosa (RP), a common group of human retinopathic diseases, is characterized by late-onset night blindness, loss of peripheral vision, and diminished or absent electroretinogram (ERG) responses. Mutations in the photoreceptor-specific gene RP1 account for 5–10% of cases of autosomal dominant RP. We generated a mouse model of the RP1 form of RP by targeted disruption of the mouse ortholog (Rp1) of human RP1. In Rp1−/− mice, the number of rod photoreceptors decreased progressively over a period of 1 year, whereas that of cone photoreceptors did not change for at least 10 months. Light and electron microscopic analysis revealed that outer segments of Rp1−/− rods and cones were morphologically abnormal and became progressively shorter in length. Before photoreceptor cell death, rhodopsin was mislocalized in inner segments and cell bodies of Rp1−/− rods. Rod ERG amplitudes of Rp1−/− mice were significantly smaller than those of Rp1+/+ mice over a period of 12 months, whereas those of Rp1+/− mice were intermediate. The decreases in cone ERG amplitudes were slower and less severe than those in rods. These findings demonstrate that Rp1 is required for normal morphogenesis of photoreceptor outer segments and also may play a role in rhodopsin transport to the outer segments. The phenotype of Rp1 mutant mice resembles the human RP1 disease. Thus, these mice provide a useful model for studies of RP1 function, disease pathology, and therapeutic interventions.
Retinitis pigmentosa (RP) is a common inherited retinopathy that affects ≈1 in 3,500 persons worldwide (1). Clinical findings in RP include progressive loss of night and peripheral vision that usually culminates in severe visual impairment or blindness. The disease is characterized by an abnormal or absent response on electroretinography (ERG) and is associated with retinal atrophy, deposition of pigment, and attenuation of retinal vessels. RP is heterogeneous clinically and genetically (2).
We identified a gene, designated RP1, that is mutated in families with the RP1 form of autosomal dominant RP (3–8). The patients who are heterozygous for the RP1 mutations have very similar classic type 2 autosomal dominant RP phenotypes with relatively late onset of night blindness (usually by the third decade of life). However, within the same family, there is extensive variation in the age at which clinical disease is detected (7, 9). Moreover, in some families such as the UCLA-RP01, two members who are homozygous for an RP1 mutation have substantially more severe retinal degeneration than other family members who are heterozygous for the mutation (9). The human RP1 gene encodes a protein of 2,156 aa, the function of which is currently unknown. However, its N terminus shares significant homology with that of human doublecortin (DCX), a mutant form of which is involved in cerebral cortical abnormalities (10, 11). This region of DCX is known to interact with microtubules (12, 13).
To understand the function of the RP1 protein in the retina and the mechanism of retinopathy in RP1 disease, we cloned and characterized the mouse ortholog (Rp1) of the human RP1 gene. We have shown previously that Rp1 is specific to photoreceptors; in mice, its expression begins during the first postnatal week and persists through adulthood (3–5). Recently we showed that Rp1 is localized in the connecting cilia of both rod and cone photoreceptors (14). Here we report that a targeted disruption of Rp1 in mice results in progressive degeneration of photoreceptors, disorganization of photoreceptor outer segments (OSs), and reduced ERG signal. Furthermore, we demonstrate that rhodopsin (Rho) is mislocalized in Rp1−/− photoreceptors. Our results provide in vivo evidence of the function of the Rp1 protein. The phenotype of our Rp1 targeted mutant mice resembles that of RP1 patients; therefore, these mice provide a useful model of RP1 disease.
Materials and Methods
Generation of Rp1 Mutant Mice.
To generate Rp1 knockout mice, we replaced a 2.5-kb genomic fragment including exons 2 and 3 of the Rp1 gene with a 1.6-kb DNA fragment containing the neomycin gene. A 2.4-kb EcoRI-PstI fragment upstream of exon 2 and a 5.2-kb BglII-EcoRV fragment downstream of exon 3 were placed in the NTK Scrambler vector (Stratagene) flanking a 1.6-kb PGK-neo-pA fragment. AB 2.2 embryonic stem cells (Lexicon/Stratagene) derived from the 129/SvEv strain were electroporated with linearized targeting vector. DNA from embryonic stem cell lines was digested with BamHI and analyzed by Southern blot (Fig. 1). Four independently targeted cell lines were selected and microinjected into the C57BL/6 blastocysts to generate chimeras. Three chimeras from the C3 cell line underwent germline transmission. To genotype subsequent F2 and F3 generations of mice, we developed a semiquantitative PCR assay (23 cycles) with a pair of primers from the deleted region of Rp1 (5′-cttccatctgtggcatacag-3′ and 5′-cacagcatcaccaggctgg-3′; GenBank accession no. AF291754) and a pair of control primers from chromosome 13 (5′-ccgagtcttaagtcaggggtt-3′ and 5′-ttccttatccacagcagcct-3′). All animals used in this report were genotyped by PCR and confirmed by Southern analysis.
Figure 1.
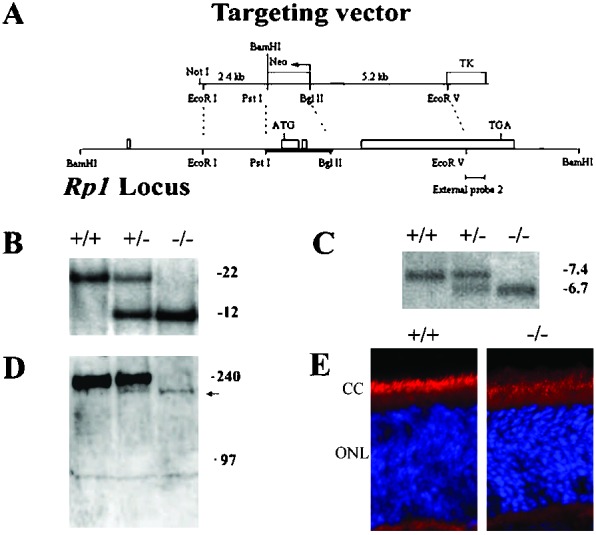
Generation of Rp1 mutant mice. (A) Targeted disruption of Rp1. The four exons are represented by rectangles. The targeting vector contains Neo- and thymidine kinase (TK)-selectable markers and deletes exons 2 and 3 in the Rp1 locus by homologous recombination. (B) Genomic Southern analysis of Rp1 mutant mice. After BamHI digestion of genomic DNA, external probe 2 detected a 22-kb band in the wild-type allele and a 12-kb band in the targeted allele. (C) Northern analysis of retinas of Rp1 mutant mice at postnatal day (P)14. An ≈270-bp fragment corresponding to the 5′ end of exon 4 was used as probe. A 7.4-kb band from the wild-type allele and a 6.7-kb band from the targeted allele were detected. (D) Western analysis of retinal homogenates from Rp1 mutant mice using a C-terminal Rp1 antibody. Each lane contains 150 μg of homogenates of the retinas of four mice of the same genotype and age (P14). A 240-kDa band seen in Rp1+/+ and Rp1+/− mice is absent in Rp1−/− mice; an ≈210-kDa band (arrow) is detected in Rp1−/− and Rp1+/− mice. (E) Immunofluorescent staining on frozen sections of Rp1+/+ and Rp1−/− retinas at P14 using a C-terminal Rp1 antibody (red) and 4′,6-diamidino-2-phenylindole (blue; ref. 14). CC, connecting cilia.
Light and Transmission Electron Microscopic Analysis of Retinal Sections.
All animals analyzed histologically were continuously maintained in a 12-hr light/dark cycle and were killed 8–12 h after the onset of the light phase. Anesthetized mice were perfused with 0.1 M PBS and then with 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) by intracardiac injection. The eyes were removed, left in the same fixative overnight at 4°C, and then embedded in epoxy medium. From each animal we obtained sections of four quadrants around the optic nerve and four sets of slides corresponding to the nasal peripheral-posterior optic nerve, temporal peripheral-posterior optic nerve, superior peripheral-posterior optic nerve, and inferior peripheral-posterior optic nerve regions. Sections of 0.5 μm thickness were stained with toluidine blue for light microscopy. Sections 60–80-nm thick were stained for transmission electron microscopy (TEM) with uranyl acetate in methanol and then with Reynolds lead citrate.
Measurements of Outer Nuclear Layer (ONL) Thickness and OS Length.
All images of slides were analyzed by the program BIOQUANTNOVA (R & M Biometrics, Nashville). The length of the OS and thickness of the ONL were measured at five consecutive points in a 100-μm segment located 300–400 μm from the optic nerve. The ONL thickness was measured from the base of the nuclei to the outer limiting membrane at a 90° angle. The OS length was measured from the base of the OS to the inner side of the retinal pigment epithelium. Sections in which columns of rod nuclei were apparent were used to ensure that sections were not oblique. Measurements from the four quadrants (see above) were averaged; the SD was less than 10% of the average value for each mouse.
Northern and Western Blot Analyses.
After removal of the lens, total RNA was extracted from the mouse eyes with TRIZOL (GIBCO/BRL), and 10 μg of total RNA per lane was analyzed. A PhosphorImager 445 SI was used for quantification of band intensity. Extracts from mouse retinas containing 50–150 μg of protein (14) were separated in 3–8% NuPage Tris-acetate polyacrylamide gel (NOVEX, San Diego) containing SDS. After transfer, the poly(vinylidene difluoride) membrane (Immobilon) was treated with primary antibodies, horseradish peroxidase-conjugated secondary antibody (Amersham Pharmacia), and SuperSignal (Pierce). The intensity of the bands was quantified by using a densitometer (MultiImage light cabinet).
Immunohistochemistry.
Eight to twelve hours after the onset of the light phase, mice were perfused with 0.1 M PBS and then with 4% paraformaldehyde solution in 0.1 M phosphate buffer (pH 7.3) by intracardiac injection; eyes were postfixed for 4 h and embedded in paraffin. Slides were deparaffinized and stained with primary antibodies. Reactions were visualized by treatment with a peroxidase-conjugated secondary antibody and then with 3,3′-diaminobenzidine reagents. Slides were observed under a light microscope (Olympus BX60). For terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) assays, paraffin sections were stained by using the ApopTag kit (Oncor). For double labeling to identify Rho and apoptosis, sections first were stained with ApopTag kit reagents and then were blocked in PBS containing 5% BSA for 1 h, treated with anti-Rho antibody (Leinco Technologies, St. Louis) overnight, washed, mounted and analyzed. Immunofluorescent staining on frozen sections was performed by using procedures described in ref. 14.
Staining of Retina Whole Mounts.
Eyes were removed, fixed in 4% paraformaldehyde in 0.1 M PBS for 4 h, and preserved overnight in 30% sucrose in 0.1 M PBS. Each eye cup was cut along the ora serrata. The cornea and lens were removed, and the retina was teased out. The retina was frozen and thawed three times, treated with peanut agglutinin conjugated to FITC (Biomedia, Foster City, CA), and washed. Small cuts were made in the retina to facilitate flat mounting. Images of the retinas were acquired by using a confocal laser scanning microscope (Leica TCS NT SP).
ERG and Fundus Photography.
Protocols for both rod and cone ERG recordings were described previously (15). Briefly, after 2 h of dark adaptation, mice were anesthetized by i.p. injection of 15 μg/g Ketamine and 7 μg/g xylazine. ERGs were recorded from the corneal surface of one eye after pupil dilation (1% atropine sulfate) using a gold loop corneal electrode together with a mouth reference and tail ground electrode. Response signals were amplified (CP511 AC amplifier, Grass Instruments, Quincy, MA), digitized (PCI-1200, National Instruments, Austin, TX), and computer-analyzed with custom software. Fundus photography was performed by using standard methodology (16). Briefly, after full dilation of the pupils, a small animal fundus camera (Kowa, Torrance, CA) and an indirect viewing lens were used, and posterior pole retinal photographs were obtained.
Results
Generation of Rp1−/− Mice.
We identified the mouse ortholog (Rp1) of RP1 by screening a mouse bacterial artificial chromosome library (Research Genetics, Huntsville, AL; catalog no. 96050) with human RP1 cDNA probes and determined the genomic sequence of 16,735 bp containing the entire Rp1 gene (GenBank accession no. AF291754). By comparing the genomic and cDNA sequences (GenBank accession no. AF155141), we found that the exon-intron structure of mouse Rp1 is identical to that of human RP1 (Fig. 1A). Similar to human RP1, there are three conserved putative CRX-binding motifs in the 5′ regulatory region (upstream of exon 1) of the Rp1 gene (data not shown; ref. 17).
To create a mouse model of RP1, we designed a targeting construct that deleted exons 2 and 3 of the Rp1 gene (Fig. 1A). This targeted disruption ensured the removal of the start codon (ATG) in exon 2 and the conserved Dcx functional domain in exons 2 and 3. Genomic Southern blot analysis of the mice confirmed the deletion in the Rp1 gene (Fig. 1B). The crosses between Rp1+/− mice yielded offspring with an ≈1:2:1 ratio of the Rp1+/+, Rp1+/−, and Rp1−/− genotypes; therefore, there were no deaths among the Rp1−/− embryos. Furthermore, Rp1−/− mice showed no gross developmental or behavioral abnormality.
Northern blot analyses of mouse retinas at ages P14, 1 month, and 3 months showed that a shorter mRNA was transcribed stably from the targeted allele (Fig. 1C). The difference in size between the normal Rp1 mRNA (≈7.4 kb) and the targeted Rp1 mRNA (≈6.7 kb) corresponded to the combined size of exons 2 and 3. We amplified mRNA from Rp1 mutant retinas by reverse transcription–PCR with primers from exon 1 and exon 4; sequence analysis of the products showed that the targeted deletion of exons 2 and 3 of the Rp1 gene resulted in an abnormal splicing between exon 1 and exon 4 (data not shown).
To confirm the ablation of the Rp1 protein in Rp1−/− mice, we performed Western blot analysis with two independent polyclonal antibodies to the middle portion and the C terminus (encoded by exon 4) of the Rp1 protein (Fig. 1D; ref. 14). Both Rp1+/+ and Rp1+/− mice expressed the expected ≈240-kDa Rp1 protein, but Rp1+/− mice expressed ≈50% less of this protein. No Rp1 protein with a mass of 240 kDa was detected in Rp1−/− retinas in either assay. However, by using the antibody against the C terminus on overloaded gels (80–150 μg of retinal extract per lane), we did detect a band of ≈210 kDa in both Rp1+/− and Rp1−/− retinas. The intensity of this 210-kDa band in the Rp1−/− retina is <5% of that of the 240-kDa band in the Rp1+/+ retina; that in the Rp1+/− retina is approximately half in the Rp1−/− retina. Furthermore, we performed immunofluorescent staining on Rp1 mutant retinas (14) and both Rp1 antibodies stained connecting cilia of the Rp1−/− retina (Fig. 1E). Noticeably, the intensity of staining in Rp1−/− retina is much fainter than that in the Rp1+/+ retina, consistent with results of Western analysis. Although further biochemical and genetic characterization is needed, it is very likely that a small amount (<5%) of truncated Rp1 protein lacking the conserved Dcx domain was made in Rp1 mutant retinas using alternative translation initiation sites in exon 4.
Progressive Degeneration of Photoreceptors.
We examined the retinal morphology of the F2 and F3 offspring of Rp1 mutant mice at ages P7 to 16 months. With the exception of the ONL and OS of Rp1−/− retinas, the gross retinal structure was normal in mice of all three genotypes (Fig. 2; measurements of inner nuclear layers not shown).
Figure 2.
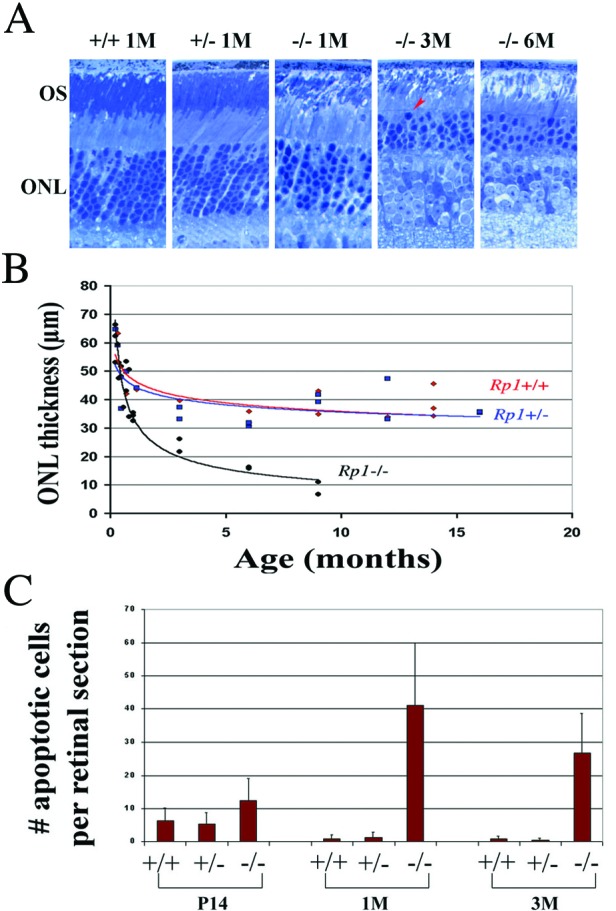
(A) Light micrographs of epoxy-embedded sections of the central retinas of Rp1 mutant mice at ages 1 (1M), 3 (3M), and 6 (6M) months. THe arrowhead labels an apparent pyknotic nucleus. (B) ONL thickness according to age in Rp1 mutant mice. Each point represents average measurements from one animal. (C) The number of apoptotic cells in ONL of retinal sections from the three genotypes at various ages. Each section was made either frontally or transversely near the optic nerve head. TUNEL-positive cells were counted from the ora serrata through the optic nerve; more than three evenly spaced sections were counted in each animal. The mean values for each animal were calculated. Group mean values and SDs (bars) are shown.
The thickness of the ONL did not differ significantly between Rp1+/+ and Rp1+/− mice at any of the ages examined (Fig. 2 A and B). Although ONL thickness gradually diminished in Rp1+/+ and Rp1+/− retinas, the magnitude and rate of decrease were similar to those observed during the normal aging process in other wild-type strains (18, 19). Up to age P21, Rp1−/− retinas had the same number of photoreceptors as did Rp1+/+ and Rp1+/− retinas (data not shown). Thereafter, however, the ONL thickness of Rp1−/− retinas showed a significant decrease over time; it had decreased to ≈50% of that in wild-type retinas by age 3 months, and only approximately one row of photoreceptors remained by age 9 months (Fig. 2B).
We assessed the loss of cones by staining with peanut agglutinin in whole-mount retinas at ages 1 and 10 months (Fig. 3A; ref. 20). Even at 10 months of age, the number of cones in Rp1−/− retinas did not differ significantly from the number in Rp1+/+ and Rp1+/− retinas (Fig. 3B). Similarly, when we stained retinal paraffin sections from 6-month-old mice with cone-specific antibodies (blue opsin, green opsin, and cone transducin γ subunit), the number of cones was similar in mice of all three genotypes (data not shown). We also counted cones in 0.5-μm epoxy-embedded sections by using criteria established by Carter-Dawson and La Vail (21) and found similar numbers of cones in mice of the three genotypes at ages up to 9 months (data not shown). These experiments demonstrated that cones did not degenerate significantly in Rp1−/− mice before age 10 months.
Figure 3.
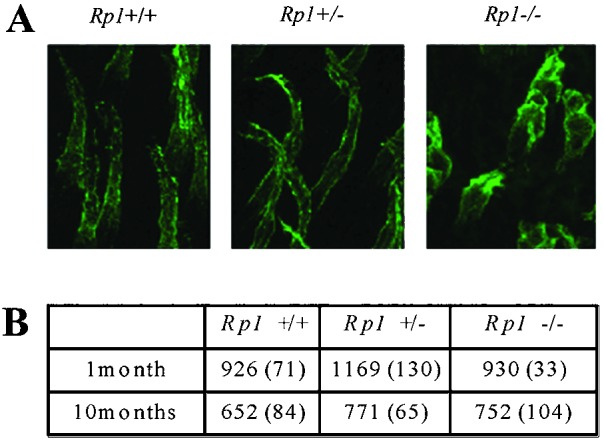
(A) Confocal microscopic images of cone OS and inner segment in Rp1 mutant retinas at 10 months of age. Whole-mount retinas were stained with peanut agglutinin, which binds specifically to cone OSs and inner segments. Serial transverse photomicrographs of retinas from each genotype were taken and superimposed. (B) The number of cones in the retinas of the three genotypes of Rp1 mutant mice at 1 and 10 months of age. From each retina, four different 250 × 250-μm areas (one from the periphery, one from the center, and two between the center and periphery) were selected, and the number of stained cones was counted. The mean values and SDs (in parentheses) are shown.
We further determined the nature of photoreceptor degeneration by TUNEL assays. We found significantly more apoptotic photoreceptors in Rp1−/− mice than in Rp1+/+ and Rp1+/− mice at ages 1 and 3 months, whereas at age P14, cell death did not differ significantly from that in Rp1+/+ and Rp1+/− mice (Fig. 2C). These findings were substantiated by the observation of pyknotic nuclei in Rp1−/− retinas at ages 1–6 months in 0.5-μm epoxy-embedded sections and in 60–80-nm TEM sections (Fig. 2A; TEM not shown). In addition, we examined expression of glial fibrillary acidic protein (GFAP) in the retinas of these mice and found that GFAP staining was more extensive in Rp1−/− retinas than in Rp1+/+ and Rp1+/− retinas (data not shown). Increased GFAP staining indicates the reactive response of Müller cells that are activated by the photoreceptor degeneration (22). These observations are consistent with our measurements of ONL thickness in Rp1 mutant mice and demonstrate that disruption of Rp1 caused apoptotic photoreceptor cell death.
Malformation and Disorganization of Photoreceptor OSs.
We observed not only the loss of photoreceptor nuclei in Rp1−/− animals but also abnormalities of the length and structure of the photoreceptor OS. The length of the OS was consistent among the three genotypes until P25 (Fig. 4A Inset). After that age, however, the OS length in Rp1−/− retinas progressively decreased until only a negligible amount remained at age 9 months (Fig. 4A).
Figure 4.
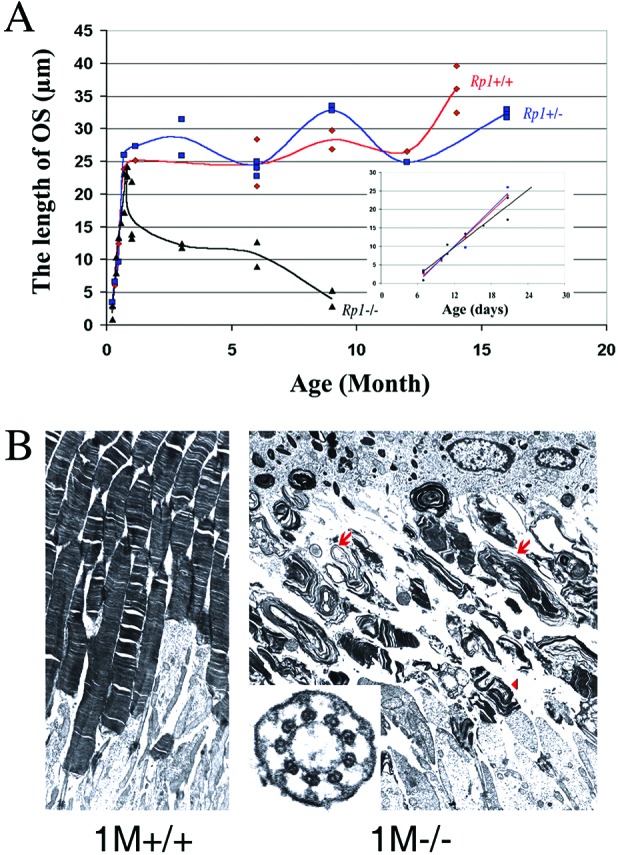
(A) Length of the OS (in μm) of retinas of Rp1 mutant mice at different ages. Each point represents average measurements from one animal. (Inset) Illustrates measurements at ages from P7 to P25. (B) TEM of the ultrastructure (×6,000) of OS in the Rp1+/+ (1M+/+) and Rp1−/− (1M−/−) retinas at 1 month. Cross section (×50,000) of connecting cilia of Rp1−/− retina appears normal (Inset). Disoriented discs (arrowhead), whorls, and disk membranes (arrows) of abnormal sizes were found in Rp1−/− retina, and the OS layer became shortened with age.
The fine structure of photoreceptors was characterized further by TEM. Shortened and disorganized OS were observed in Rp1−/− mice at all ages examined (Fig. 4B). The OS layer in Rp1−/− photoreceptors contained discs and whorls of various sizes that lacked the correct orientation; most of these discs were larger than normal discs, as are those of heterozygous Prph2+/− mice (23). Disorganization of OSs was noted first in Rp1−/− retinas at P7, the age at which OSs first form, and it progressed until age 9 months, at which time very few discs or whorls remained (data not shown). Whorls in Rp1−/− retinas contained peripherin (Prph2), as seen by immunocytochemistry (data not shown); they also contained Rho, identified by immunohistochemistry. In whole-mount preparations stained with peanut agglutinin (Fig. 3A), cone OSs in Rp1−/− retinas appeared disorganized, because their diameter and length varied. It is also possible that cone OSs in Rp1−/− retinas degenerated and the peanut agglutinin staining arose from remaining cone inner segment. Thus, the OSs of both rods and cones were abnormally organized and dysplastic in Rp1−/− animals. In contrast, the connecting cilia appeared to develop normally in Rp1−/− mice. In >48 longitudinal and >45 cross sections, the connecting cilia contained axonemal microtubules with the normal “9 + 0” structure and intact basal bodies at all ages examined (Fig. 4B Inset).
Mislocalization of Rho in Photoreceptors.
We used immunohistochemistry to determine whether photoreceptor proteins were distributed abnormally in Rp1 mutant mice. The distribution of blue and green cone opsin, Prph2, Rom1, transducin γ subunit of cones, and cGMP phosphodiesterase was comparable in the retinas of Rp1−/−, Rp1+/−, and Rp1+/+ littermates at ages P14, 1 month, and 3 months (data not shown). However, in Rp1−/− animals at age P14 and above, many cell bodies or inner segments of photoreceptors were reactive with anti-Rho antibody (Fig. 5) in the absence of extensive photoreceptor cell death. The pattern of Rho mislocalization was more appreciable at ages 1, 3, and 6 months (data not shown).
Figure 5.
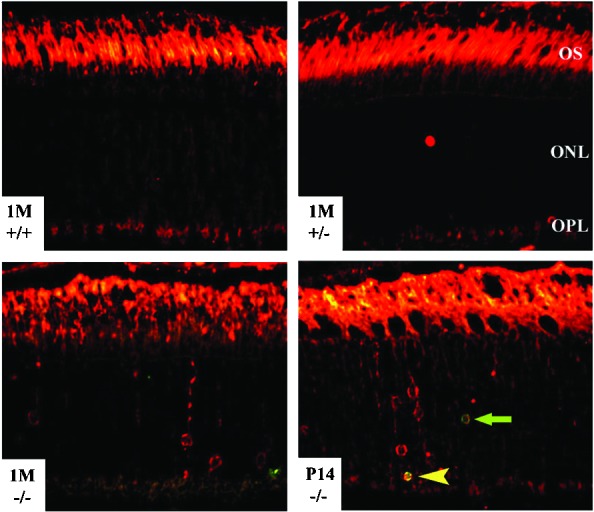
Mislocalization of Rho in Rp1−/− photoreceptors. Paraffin-embedded retinal sections were double-labeled for apoptosis (green) and Rho (red). Strong Rho labeling was observed in OSs of the retinas of all three genotypes. The green arrow indicates an apoptotic nucleus, and the yellow arrowhead indicates a double-labeled nucleus. Staining in the outer plexiform layer (OPL) is present in sections from all three genotypes, probably because of autofluorescence in the paraffin sections. It remains possible that Rho also is mislocalized in synaptic termini of photoreceptors in the outer plexiform layer of Rp1−/− retinas (22).
To confirm that the mislocalization of Rho was not the result of photoreceptor degeneration, we double-labeled Rp1 mutant retinas at ages P14 and 1 month to detect Rho and apoptosis. Most of the cell bodies that were labeled with anti-Rho antibody were not positive for the TUNEL assay used to detect apoptosis. The number of apoptotic cells was much smaller than the number of Rho-positive cells (Fig. 5), and only ≈10% of the apoptotic photoreceptor cells were positive also for Rho at P14 and 1 month (data not shown). These results demonstrate that Rho mislocalization occurred earlier than photoreceptor cell death and therefore was unlikely the result of photoreceptor degeneration.
Physiological Responses of Retinas.
To assess the relationship between the morphologic findings and the functional status of retinas in Rp1 mutant mice, we performed blinded ERG analyses of the mice (10 in each genotype) at 2, 6, and 12 months of age (Fig. 6). In Rp1−/− mice, ERGs showed a slow progressive dysfunction of rods and cones that clearly was present at 2 months of age and continued throughout the first year of life. ERG responses for both rods and cones were severely reduced by 12 months. ERG signals from the rod and cone systems diminished at a similar rate in Rp1−/− mice over a period of 1 year, although the rate was slightly slower in cones than in rods. Interestingly, the ERG amplitude values of Rp1+/+ and Rp1+/− mice were significantly different at all time points measured (P < 0.0001, Student's t test for each time point). These results suggest that Rp1+/− mice experience some retinal dysfunction despite their normal-appearing retinal morphology.
Figure 6.
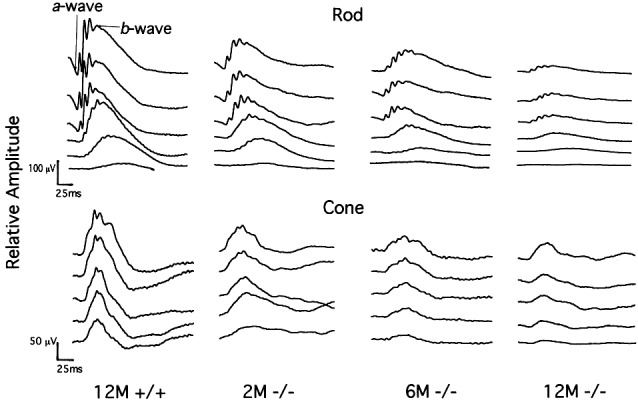
Representative rod and cone ERG recordings of Rp1+/+ and Rp1−/− mice at ages 2 (2M), 6 (6M) and 12 (12M) months. Serial recordings from rods show responses to flashes of short-wavelength (Wratten 47A; λmax = 470 nm) light increasing in intensity over a 3.0-log unit range (in 0.6-log unit steps), up to the maximum intensity (top traces, 0.668 cd⋅s/m2). Cone-dominated responses were stimulated by a series of white flashes of increasing intensity (in 0.3-log unit steps) on a rod-saturating background (32 cd/m2) up to the maximum intensity (8.46 cd⋅s/m2).
Finally, we examined the fundi of these animals at 11 months of age. The Rp1+/+ and Rp1+/− fundi had a normal appearance (data not shown) with minimal granularity to their retinal pigment epithelial layer and no evidence of retinal atrophy or vessel attenuation. The Rp1−/− fundi clearly were abnormal (data not shown), uniformly showing a pigmented granularity to their subretinal pigment epithelium and diffused multiple small areas of atrophy. The optic nerves showed pallor, and the retinal vessels were about one-half their normal diameter.
Discussion
Despite the importance of the RP1 gene (RP1 mutations account for 5–10% of cases of autosomal dominant RP; ref. 7), the function of RP1 in photoreceptors and the pathology of RP1 disease remain unknown. Our characterization of the Rp1 mutant mice provides in vivo evidence of the functional role of RP1. Our mice serve as a good animal model of RP1 and can be used for functional studies of Rp1 in retina.
Function of the RP1 Protein.
Rp1 is unlikely to be a transcription factor involved in photoreceptor development.
The targeted disruption of Rp1 did not change the mRNA levels of several outer segment-specific genes such as Rho, Prph2, Rom1, and Abcr (24) at P14 before the onset of extensive cell death (data not shown). These observations suggest that although Rp1 protein contains several putative nuclear localization domains (3–5), it is unlikely to function as a transcription factor in photoreceptor development as do Crx and Nrl (24, 25).
Rp1 may be involved in the transport of proteins from inner segments to OSs in photoreceptors.
Similar to Rpgr−/−, Myo7aSh1, Kinesin II−/−, and Tulp1−/− mice (22, 26–28) but unlike other mutant mice with Rho mislocalization (29), Rp1−/− mice had OSs and had little photoreceptor cell death when Rho mislocalization occurred. Furthermore, the RP1 protein has been localized recently to connecting cilia of both rods and cones (14), and the RP1 protein contains at its N terminus a DCX homologous domain that is known to bind and stabilize microtubules (12, 13). We thus hypothesize that mislocalization of Rho in Rp1−/− is caused by aberrant protein transport. Because the ultrastructure of connecting cilia in our Rp1−/− mice appeared normal, Rho mislocalization in these animals may be caused by aberrant motor activity rather than by structural defects in the ciliary microtubules. Because we did not observe such ectopic distribution in six other OS proteins, it is likely that transporting defects in Rp1−/− photoreceptors are specific to Rho in rods. However, because 85–90% of OS protein is Rho (30), we cannot rule out the possibility that other, less abundant proteins were mislocalized in Rp1−/− photoreceptors.
Rp1 is required for the morphogenesis of OSs of photoreceptors.
The earliest signs of abnormalities in Rp1−/− retinas that we can detect are disks with abnormal morphology and aberrant size at P7 just after the OS had begun to develop. The fact that the expression of Rp1 began at ≈P5 supports the notion that Rp1 is required for normal morphogenesis of OS. Interestingly, among many mutant mice with OS defects, the morphology of Rp1−/− disks appeared, in many aspects, similar to that seen in Prph2+/− mice (Fig. 4B; ref. 23). Thus, Rp1 may be involved in disk formation by genetically interacting with Prph2 and Rom1 (31). Alternatively, Rho mislocalization in Rp1−/− mice could reduce the transport of other OS proteins and thereby cause abnormal OS disk formation. We cannot exclude the possibility that Rp1 is involved in transporting as-yet-unidentified specific OS proteins that are involved directly in disk morphogenesis. Conversely, it remains possible that Rho mislocalization in Rp1−/− mice is the result of OS malformation, which eventually leads to photoreceptor cell death.
Distinguished from other rod-only retinal degenerative diseases, both cone ERGs and OSs are abnormal in Rp1−/− mice at young ages; these findings thus confirm the importance of Rp1 in cone OS morphogenesis and function and are consistent with our previous finding that Rp1 is present in both rod and cone cilia (14).
A Mouse Model of RP1.
To date, no retinas from patients with RP1 have been studied histologically. Detailed analysis of RP1 disease expression revealed a resemblance to our Rp1 mutant phenotypes (7, 8). Interestingly, the only two patients described who were homozygous for an RP1 mutation (4) had an earlier disease onset (ages 6–8 years) and much more severe symptoms than most patients who were heterozygous for the same mutation (ages 30s and 40s). Examination of Rp1+/− mice at older ages potentially could reveal a slow, progressive morphological abnormality. The phenotype of Rp1−/− mice resembles that of human RP1 heterozygous patients, but with more severe and faster progression, it may be more similar to that of human RP1 homozygous patients.
Acknowledgments
We thank D. Davis, S. Fraze, T. Quinn, J. Treadaway, and X. Li for technical assistance; J. Blanks, M. Fitzerald, D. Johnson, and J. Morgan for advice; and T. Curran for support. We also thank M. Applebury (blue and green cone opsin), L. Donoso (S-antigen), R. McInnes (Rom1), W. Moghrabi and G. Travis (Prph2), R. Molday (Prph2, Rom1, and Abcr), and T. Wensel and X. Zhang (RGS9) for generously providing antibodies. This work was supported in part by National Institutes of Health Cancer Center Support CORE Grant CA21765 and National Institutes of Health Grants EY12950, EY07142, EY12910, and EY08285, the American Lebanese Syrian Associated Charities, March of Dimes Birth Defects Foundation Research Grant 5-FY98-0725, and grants from the Foundation Fighting Blindness, Research to Prevent Blindness, the George Gund Foundation, the William Stamps Farish Fund, the M. D. Anderson Foundation, the Rosanne H. Silbermann Foundation, and the John S. Dunn Research Foundation.
Abbreviations
- RP
retinitis pigmentosa
- ERG
electroretinogram/electroretinography
- DCX
human doublecortin
- OS
outer segment
- Rho
rhodopsin
- P
postnatal day
- TEM
transmission electron microscopy
- ONL
outer nuclear layer
- TUNEL
terminal deoxynucleotidyltransferase-mediated UTP end labeling
- Prph2
peripherin
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF291754).
References
- 1.Humphries P, Kenna P, Farrar G J. Science. 1992;256:804–808. doi: 10.1126/science.1589761. [DOI] [PubMed] [Google Scholar]
- 2.Blackshaw S, Fraioli R E, Furukawa T, Cepko C L. Cell. 2001;107:579–589. doi: 10.1016/s0092-8674(01)00574-8. [DOI] [PubMed] [Google Scholar]
- 3.Pierce E A, Quinn T, Meehan T, McGee T L, Berson E L, Dryja T P. Nat Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan L S, Heckenlively J R, Bowne S J, Zuo J, Hide W A, Gal A, Denton M, Inglehearn C F, Blanton S H, Daiger S P. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guillonneau X, Piriev N I, Danciger M, Kozak C A, Cideciyan A V, Jacobson S G, Farber D B. Hum Mol Genet. 1999;8:1541–1546. doi: 10.1093/hmg/8.8.1541. [DOI] [PubMed] [Google Scholar]
- 6.Bowne S J, Daiger S P, Hims M M, Sohocki M M, Malone K A, McKie A B, Heckenlively J R, Birch D G, Inglehearn C F, Bhattacharya S S, Bird A, Sullivan L S. Hum Mol Genet. 1999;8:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson S G, Cideciyan A V, Iannaccone A, Weleber R G, Fishman G A, Maguire A M, Affatigato L M, Bennett J, Pierce E A, Danciger M, Farber D B, Stone E M. Invest Ophthalmol Visual Sci. 2000;41:1898–1908. [PubMed] [Google Scholar]
- 8.Berson E L, Grimsby J L, Adams S M, McGee T L, Sweklo E, Pierce E A, Sandberg M A, Dryja T P. Invest Ophthalmol Visual Sci. 2001;42:2217–2224. [PubMed] [Google Scholar]
- 9.Blanton S H, Heckenlively J R, Cottingham A W, Friedman J, Sadler L A, Wagner M, Friedman L H, Daiger S P. Genomics. 1991;11:857–869. doi: 10.1016/0888-7543(91)90008-3. [DOI] [PubMed] [Google Scholar]
- 10.des Portes V, Pinard J M, Billuart P, Vinet M C, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, et al. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 11.Gleeson J G, Allen K M, Fox J W, Lamperti E D, Berkovic S, Scheffer I, Cooper E C, Dobyns W B, Minnerath S R, Ross M E, Walsh C A. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 12.Gleeson J G, Lin P T, Flanagan L A, Walsh C A. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 13.Horesh D, Sapir T, Francis F, Wolf S G, Caspi M, Elbaum M, Chelly J, Reiner O. Hum Mol Genet. 1999;8:1599–1610. doi: 10.1093/hmg/8.9.1599. [DOI] [PubMed] [Google Scholar]
- 14.Liu Q, Daiger S P, Farber D B, Heckenlively J R, Sullivan L S, Zuo J, Milam A H, Pierce E A. Invest Ophthalmol Visual Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- 15.Nusinowitz S, Ridder W H, III, Heckenlively J R. In: Systematic Evaluation of the Mouse Eye: Anatomy, Pathology, and Biomethods. Smith R S, John S W M, Nishina P, Sundberg J P, editors. New York: CRC; 2002. pp. 320–344. [Google Scholar]
- 16.Hawes N L, Smith R S, Chang B, Davisson M, Heckenlively J R, John S W. Mol Vis. 1999;5:22. [PubMed] [Google Scholar]
- 17.Livesey F J, Furukawa T, Steffen M A, Church G M, Cepko C L. Curr Biol. 2000;10:301–310. doi: 10.1016/s0960-9822(00)00379-1. [DOI] [PubMed] [Google Scholar]
- 18.Sanyal S, De Ruiter A, Hawkins R K. J Comp Neurol. 1980;194:193–207. doi: 10.1002/cne.901940110. [DOI] [PubMed] [Google Scholar]
- 19.Clarke G, Goldberg A F, Vidgen D, Collins L, Ploder L, Schwarz L, Molday L L, Rossant J, Szel A, Molday R S, Birch D G, McInnes R R. Nat Genet. 2000;25:67–73. doi: 10.1038/75621. [DOI] [PubMed] [Google Scholar]
- 20.Blanks J C, Johnson L V. Invest Ophthalmol Visual Sci. 1984;25:546–557. [PubMed] [Google Scholar]
- 21.Carter-Dawson L D, LaVail M M. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 22.Hong D H, Pawlyk B S, Shang J, Sandberg M A, Berson E L, Li T. Proc Natl Acad Sci USA. 2000;97:3649–3654. doi: 10.1073/pnas.060037497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawkins R K, Jansen H G, Sanyal S. Exp Eye Res. 1985;41:701–720. doi: 10.1016/0014-4835(85)90179-4. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa T, Morrow E M, Li T, Davis F C, Cepko C L. Nat Genet. 1999;23:466–470. doi: 10.1038/70591. [DOI] [PubMed] [Google Scholar]
- 25.Kumar R, Chen S, Scheurer D, Wang Q L, Duh E, Sung C H, Rehemtulla A, Swaroop A, Adler R, Zack D J. J Biol Chem. 1996;271:29612–29618. doi: 10.1074/jbc.271.47.29612. [DOI] [PubMed] [Google Scholar]
- 26.Hagstrom S A, Adamian M, Scimeca M, Pawlyk B S, Yue G, Li T. Invest Ophthalmol Visual Sci. 2001;42:1955–1962. [PubMed] [Google Scholar]
- 27.Liu X, Udovichenko I P, Brown S D, Steel K P, Williams D S. J Neurosci. 1999;19:6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marszalek J R, Liu X, Roberts E A, Chui D, Marth J D, Williams D S, Goldstein L S. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 29.Sung C H, Tai A W. Int Rev Cytol. 2000;195:215–267. doi: 10.1016/s0074-7696(08)62706-0. [DOI] [PubMed] [Google Scholar]
- 30.Hall M O, Bok D, Bacharach A D. Science. 1968;161:787–789. doi: 10.1126/science.161.3843.787. [DOI] [PubMed] [Google Scholar]
- 31.Kedzierski W, Nusinowitz S, Birch D, Clarke G, McInnes R R, Bok D, Travis G H. Proc Natl Acad Sci USA. 2001;98:7718–7723. doi: 10.1073/pnas.141124198. [DOI] [PMC free article] [PubMed] [Google Scholar]