

Abstract
Organic semiconductors based on π-conjugated oligomers and polymers constitute the active elements in new generations of plastic (opto)electronic devices. The performance of these devices depends largely on the efficiency of the charge-transport processes; at the microscopic level, one of the major parameters governing the transport properties is the amplitude of the electronic transfer integrals between adjacent oligomer or polymer chains. Here, quantum-chemical calculations are performed on model systems to address the way transfer integrals between adjacent chains are affected by the nature and relative positions of the interacting units. Compounds under investigation include oligothienylenes, hexabenzocoronene, oligoacenes, and perylene. It is shown that the amplitude of the transfer integrals is extremely sensitive to the molecular packing. Interestingly, in contrast to conventional wisdom, specific arrangements can lead to electron mobilities that are larger than hole mobilities, which is, for instance, the case of perylene.
Organic π-conjugated materials offer remarkable potential as active elements in (opto)electronic devices that exploit their semiconducting properties, such as field-effect transistors (FETs; refs. 1–8), light-emitting diodes (LEDs; refs. 9–13), or photovoltaic and solar cells (14–17). Such devices are expected to be ultimately incorporated, for instance, into all-plastic integrated circuits for low-end and cheap electronics (7, 8, 18) and all-plastic light-emitting displays, where each pixel consists of an organic LED driven by an organic FET (19, 20). In all of these applications, the efficiency of charge transport within the organic layer(s) plays a key role. In light-emitting diodes, it is desirable that the injected holes and electrons have large and similar mobilities to prevent electroluminescence quenching that can occur when charges recombine close to a metallic interface (21); high-charge mobilities favor recombination processes in the bulk (where charges can be confined further by means of organic–organic interfaces; ref. 22). In solar cells, the charges created upon photoexcitation of the active material have to be transported efficiently to be collected at the metallic contacts and stored under the form of electrical energy. Another challenge is to develop materials displaying high electron and hole mobilities in field-effect architectures to design complex organic circuits.
The charge-transport properties critically depend on the degree of ordering of the chains in the solid state as well as on the density of chemical and/or structural defects (23–25). This dependence explains why, over the last decades, the experimental characterization of the transport properties in organic thin films or crystals has led to results that vary with sample quality. Recently, the synthesis of ultra-pure single crystals of organic semiconductors such as oligoacenes has allowed Batlogg and coworkers (28) to demonstrate remarkable features that in many instances had been thought to be restricted to inorganic materials; for example, (i) the appearance of very high hole and electron mobilities in the 104–105 cm2 V−1 s−1 range at low temperature (26–30); (ii) the observation of superconductivity (29); (iii) the observation of integer or fractional quantum-Hall effect (27); or (iv) the possibility of fabricating a solid-state injection laser. Because charge transport is intrinsic in high-purity compounds, a direct comparison between experimental data and the results of quantum-chemical calculations becomes possible (31, 32).
At the microscopic level, one of the key parameters for transport in organic conjugated materials is the interchain transfer integral t, that expresses the ease of transfer of a charge between two interacting chains. The transfer integral associated with a given electronic level is related to the energetic splitting of that level when going from an isolated chain to a system of interacting chains, as sketched in Fig. 1. In organic semiconductors, the splitting of the highest occupied molecular orbital (HOMO) lowest unoccupied molecular orbital (LUMO) level resulting from the interaction of adjacent chains along given directions yields the transfer integral to be used for the description of hole (electron) transport in these directions. For simple cosine-shaped bands, the higher the HOMO (LUMO) bandwidth, the higher the expected hole (electron) mobility.
Figure 1.

Illustration of the bonding–antibonding interactions between the HOMO/LUMO levels of two ethylene molecules in a cofacial configuration; we also illustrate the formation of the valence and conduction bands when a large number of stacked molecules interact.
It turns out that, at low temperature, the charge transport in a number of organic crystals and highly organized thin films can be described in a band-like regime similar to that in inorganic semiconductors (33). In that case, the total widths and shapes of the valence and conduction bands formed by the interaction of the HOMO and LUMO levels of the π-conjugated chains, respectively, determine the hole and electron mobilities. In a tight-binding model, the total bandwidth of a one-dimensional infinite stack is simply equal to four times the transfer integral between neighbors (34); by extension, the total width can be expressed for any molecular packing from the amplitude of the transfer integrals between the various interacting units. When temperature increases, the effective bandwidths are progressively reduced by phonon-scattering processes; this result translates into a decrease in charge mobilities leading to a high-temperature regime where the charge carriers get localized over single chains and transport operates by a thermally activated hopping mechanism (33, 35). At the microscopic level, the charge transport mechanism can then be described as involving an electron transfer from a charged oligomer to an adjacent neutral oligomer. In the context of semiclassical electron-transfer theory (36–38), the electron-transfer (hopping) rate, kET, can be described to a good approximation as
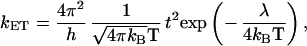 |
1 |
where T is the temperature, λ is the reorganization energy, t is the transfer integral, and h and kB are the Planck and Boltzmann constants. The transfer integrals reflect the strength of the interaction between the two oligomers; the reorganization energy term describes the strength of the electron-phonon (vibration) and can be reliably estimated as twice the relaxation energy of a polaron localized over a single unit. Eq. 1 shows that fast charge transfer processes within a hopping regime require large transfer integrals and a weak coupling of the charges to the (vibrations of the) conjugated backbones. We also note that the presence of t in Eq. 1 applies to situations where the charge transfer between the initial and final states involved in the hopping process can be described from the simple consideration of molecular orbital levels, i.e., within a one-electron picture.
Recent ab initio calculations describing hole transfer between two benzene molecules have shown that, when the intermolecular distance is reduced, the electronic couplings calculated at the one-electron level progressively deviate from those obtained by considering the full electronic wave functions of the initial and final states (39). However, the electronic couplings are underestimated only by a factor of 2–3 at the one-electron level in a cofacial dimer made of two benzene molecules separated by 3.5 Å, where the interactions are maximized because of the very small size of the interacting units. The discrepancies between the two theoretical approaches will be significantly smaller for the much bigger molecules involved in our study, thus making the one-electron approach a very efficient and simple way to estimate interchain transfer integrals.
Also, it is useful to recall that the carrier residency time, τ, on a given unit goes as
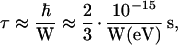 |
where W is the full effective bandwidth. This means, as a general rule of thumb, that an effective bandwidth on the order of about 0.1–0.2 eV or more is needed to ensure that the carrier residency time is smaller than the typical phonon/vibration times (then, the molecules do not have the time to geometrically relax and trap the charge). These vibrations introduce a loss of coherence among the interacting units, leading to effective bandwidths (transfer integrals) smaller than the total values provided by our calculations; our results have thus to be considered as upper limits.
Thus, the transfer integrals are seen to play a central role in the understanding of transport properties in both the band and hopping regimes. In this contribution, we seek to establish structure-transport properties relationships by analyzing how these integrals are affected at the molecular level by the nature, size, and relative positions of the interacting units. This analysis is done by carrying out semiempirical intermediate neglect of differential overlap (INDO) calculations on model systems. We first focus on oligothiophenes as prototypical compounds for organic semiconductors; the reason is that these oligomers as well as the parent polymer have well characterized transport properties and are considered as excellent candidates for the fabrication of high performance FETs (3, 5, 40, 41). The following sections are devoted to hexabenzocoronene, oligoacenes (from naphthalene to pentacene), and to perylene.
Theoretical Methodology.
As detailed in previous works, the first step in our approach consists of optimizing the geometry of an isolated oligomer molecule with the semiempirical Hartree-Fock Austin Model 1 (AM1) Hamiltonian, which has been parameterized to reproduce the ground-state geometry of organic molecules (42). Then, we use the semiempirical Hartree-Fock INDO method (as developed by Zerner and coworkers for spectroscopic purposes, refs. 43 and 44) to compute the electronic structure of isolated molecules and of supermolecular systems made of dimers or larger molecular clusters; the latter are built in various ways that will be described as we proceed (note that the intramolecular geometries are not reoptimized when performing the supermolecular dimer or cluster calculations). The choice of the INDO/S Hamiltonian is driven by the fact that INDO calculations have been shown to provide descriptions of the one-electron structure of isolated and interacting conjugated molecules in excellent agreement with corresponding experimental data (31, 32) and theoretical data obtained at the ab initio level (45).
The amplitudes of the calculated transfer integrals are rationalized by an analysis of (i) the shape of the frontier orbitals of the isolated units (visualized here by means of a home-made program (ZOA v.2.0, available at http://zoa.freeservers.com/download.htm) and (ii) the bonding–antibonding overlap pattern of the HOMO and LUMO wave functions in the clusters, which is determined by the relative positions of the interacting units.
We apply this methodology to various model systems to assess the influence on the transfer integral values of the following aspects: (i) we investigate the influence of intermolecular separation by considering a perfectly cofacial dimer of sexithienyl (6T) molecules and varying the distance between the molecular planes in the range 3.2–5.0 Å; (ii) setting the intermolecular distance in the sexithienyl dimer at 4.0 Å, we examine the impact of lateral displacements of one of the oligomers, along both the long and short molecular axes; (iii) the role of chain length is addressed by looking at dimers of oligothiophenes ranging in size from thiophene to octathiophene; (iv) finally, we evaluate the evolution of the transfer integral values as a function of the number of oligomers constituting a one-dimensional cofacial π-stack, i.e., as a function of cluster size. In the following sections, we apply our approach to clusters of hexabenzocoronene, oligoacene, and perylene molecules, either in the form of a cofacial π-stack or in the configuration found in the crystalline structure.
Results and Discussion
As an introduction to the issues discussed below, it is useful to consider the simple example of a dimer made of two perfectly superimposed ethylene molecules. In the isolated ethylene molecule, the HOMO level corresponds to the bonding situation where the lobes of the same sign of the two π-atomic orbitals interact (thus leading to the absence of node in the electronic wave function), whereas the LUMO level corresponds to the antibonding situation where lobes of opposite sign interact (thus introducing a node in the electronic wave function; see Fig. 1).
In a cofacial dimer, the interaction between the two molecules leads to a splitting of the HOMO level and a splitting of the LUMO level, as illustrated in Fig. 1. The HOMO splitting is very large, 0.539 eV (for an intermolecular distance of 4 Å); the reason is that the interaction between the HOMO wave functions of the two molecules is either fully bonding (which leads to the much stabilized HOMO−1 level of the dimer) or fully antibonding (leading to the much destabilized dimer HOMO level). For the LUMO level, the splitting is found to be much smaller, 0.148 eV, because either direct bonding interactions are compensated for by diagonal antibonding interactions in the dimer LUMO wave function or direct antibonding interactions are compensated for by diagonal bonding interactions in the LUMO+1 level, see Fig. 1.
Thus, in the case of perfectly cofacial configurations, one can state that, as a qualitative rule, the lower the number of nodes in the wave function of a given frontier level of an isolated chain, the larger the splitting of that level in a dimer (or larger clusters). Because in isolated π-conjugated chains, the LUMO wave function has usually one more node than the HOMO wave function, the LUMO splitting is expected to be smaller than the HOMO splitting. Qualitatively, for large clusters, this difference will translate into larger HOMO bandwidths, as will be discussed below. This feature is what has given rise to the conventional wisdom that in crystals or crystalline films of π-conjugated chains, the hole mobility is expected to be higher than the electron mobility.
Cofacial Configurations of Sexithienyl Clusters.
Influence of intermolecular separation.
We first consider perfectly cofacial dimers made of two sexithienyl molecules and examine the evolution of the electronic splittings of the HOMO and LUMO levels as a function of the distance between the molecular planes. Although fully cofacial configurations are rarely encountered in actual crystal structures, it is of interest to study such geometries because they provide a highly symmetric reference point and lead to the largest electronic splittings (here, for instance, 0.236 and 0.191 eV for the HOMO and LUMO levels, respectively, for an interchain distance of 4 Å).
The results are illustrated in Fig. 2. Consistent with our previous discussion, the HOMO splitting is calculated to be larger than the LUMO splitting whatever the interchain separation. The amplitudes of the electronic splittings are observed to decay exponentially when the interchain distance is increased; this simply translates the exponential decay in intermolecular overlap of the π-atomic orbitals when the two oligomers are pulled apart. An important result is that the electronic splittings vary by as much as a factor of three to four between 3.4 and 4.0 Å, a range that corresponds to the typical intermolecular distances found in organic conjugated crystals and films (46, 47).
Figure 2.

Evolution of the INDO-calculated electronic splittings of the HOMO and LUMO levels in a cofacial dimer made of two sexithienyl molecules as a function of the intermolecular separation.
Influence of relative displacements.
In many instances, cofacial packing involves the displacements of adjacent molecules along their long and/or short molecular axes. Fig. 3 describes the evolution of the HOMO and LUMO splittings in dimers where the top 6T molecule is translated along its main chain axis (while keeping the interchain distance fixed at 4 Å). The most interesting result is the appearance of strong oscillations in the values of the splitting, with a periodicity that is different for the HOMO and LUMO splittings; it is about twice as small for the HOMO as for the LUMO. The important consequence of this difference in oscillation period is that small translations can lead to situations where the electronic splitting is larger for the LUMO than for the HOMO and, hence, where electrons can be expected to be more mobile than holes. For instance, for a shift of about 1.5 Å, the reversal in the relative amplitude of the splittings is very significant: we calculate a LUMO splitting of 0.12 eV and a HOMO splitting of 0.02 eV.
Figure 3.

Evolution of the INDO-calculated electronic splittings of the HOMO and LUMO levels in a dimer formed by two sexithienyl molecules separated by 4.0 Å as a function of the degree of translation of one molecule along its main chain axis.
The calculated evolutions can be rationalized from the shape of the HOMO and LUMO orbitals of a single 6T molecule. In the HOMO level, the distribution of the positive and negative linear combination of atomic orbital (LCAO) coefficients shows a change in the sign of the wave function every half monomer unit (see Fig. 4). This pattern leads to extrema in the calculated electronic splittings for degrees of translation corresponding to multiples of half the monomer unit size. Large electronic splittings are dominated by full bonding or antibonding interactions between the π-atomic orbitals localized over the carbon–carbon double bonds within the thiophene rings. In contrast, the minima are calculated for geometries where the double bonds of one oligomer are superimposed over the center of the thiophene rings or the inter-ring bonds of the other chain; in such configurations, the global overlap (and hence the HOMO splitting) is considerably reduced by the compensation of bonding and antibonding interactions between the double bonds of one chain and the two adjacent double bonds of the other chain.
Figure 4.

Illustration of the LCAO bonding–antibonding pattern of the HOMO (Upper) and LUMO (Lower) levels in the sexithienyl molecule. The color and size of the circles are representative of the sign and amplitude of the LCAO coefficients, respectively; the dashed lines delimit half a monomer unit.
The evolution of the LUMO splitting also displays maxima and minima; however, for small degrees of translation, these do not reach values as low as in the situation observed with the HOMO level. This finding is explained once again by considering the shape of the LUMO orbital. In this case, there is no sign change along the translation axis (see Fig. 4). This pattern systematically leads to dominant bonding (antibonding) overlaps in the LUMO (LUMO+1) level of the dimer, and hence to significant electronic splittings. Maxima (minima) are observed when the thiophene rings of one chain overlap the thiophene rings (the inter-ring bonds) of the second chain; the oscillation period of the curve is, thus, twice as large as that calculated for the HOMO splitting.
Note that there occurs an overall decrease in the HOMO and LUMO splittings for increasing translational shifts. This evolution simply results from the progressive reduction in the overall extent of spatial overlap between the two oligomers.
We now turn to the impact of translating the top oligomer along its short axis. As expected, the HOMO splitting is found to decay progressively when the molecule is shifted by up to 3.5 Å, whereas the LUMO splitting exhibits two minima for the same lateral displacement. Again, these evolutions can be rationalized by looking at the wave functions. The HOMO wave function does not change sign along the short axis; thus, the translation preserves dominant bonding (antibonding) interactions in the HOMO−1 (HOMO) level of the dimer; these interactions attenuate as the translational shift increases. In contrast, the antibonding character over the double bonds found in the LUMO level leads to the appearance of a minimum in the course of the translation.
Influence of ring orientation.
In addition to the perfectly cofacial dimer situation where any two superimposed rings point to the same direction, we have also examined the dimer configuration where one oligomer is rotated by 180° along the long-chain axis. In this case, the molecular planes are still fully parallel, but superimposed rings point in opposite directions.
Taking an interchain distance of 4 Å, we calculate for the rotated geometry splittings of 0.078 and 0.142 eV for the HOMO and LUMO levels, respectively, to be compared with 0.236 and 0.190 eV for the cofacial dimer. The LUMO splitting has a similar order of magnitude in the two dimers because of the fact that the interactions between the β carbon atoms in the cofacial dimer are compensated for by the interaction between the sulfur atoms and the C–C single bonds in the rotated geometry. In contrast, the absence of electronic density on the sulfur atoms in the HOMO level prevents a similar compensation from occurring and leads to a significant reduction in splitting when going from cofacial to rotated geometry. Thus, this indicates another type of configuration for which electron mobilities can be favored over hole mobilities.
Influence of oligomer length.
To make the discussion easy, it is useful to refer back to the case of the ethylene dimer that we described at the beginning of this section and then to consider an extension of the size of the molecule to go to longer polyenes. We find that the HOMO splittings decrease with chain length and saturate; the LUMO splittings increase and tend to a saturated value only slightly lower than that for the HOMO splittings.
These evolutions can be rationalized by the fact that, as chain length increases, the HOMO level gains an increasing number of nodes (in the polyene with n double bonds, the HOMO has n − 1 nodes); this contributes to decreasing the HOMO splitting, because of the appearance of an increasing number of antibonding interactions between the top and bottom molecules of the dimer. The situation is opposite for the LUMO level, for which there appears, with chain length, an increasing number (n − 1) of bonds over which the LUMO wave function is bonding. At the limit of long chains, the relative difference in the number of nodes between the HOMO and LUMO levels becomes increasingly smaller, hence the HOMO/LUMO splitting differences also become smaller.
The evolutions of the HOMO and LUMO splittings for oligothienylene dimers with chain lengths going from thiophene to octathiophene are given in Fig. 5 (interchain distance fixed here at 3.5 Å). The results are qualitatively similar to those in the polyene dimers and have the same explanation.
Figure 5.

Evolution of the INDO-calculated HOMO and LUMO splittings in cofacial dimers formed by two oligothiophenes separated by 3.5 Å as a function of the number of thiophene rings in the chains.
Influence of cluster size.
We now discuss the evolution of the total HOMO and LUMO bandwidths formed by the interaction of the HOMO and LUMO levels in clusters containing from one to six sexithienyls stacked in a perfectly cofacial configuration, with interchain separations of 4 Å. The INDO-calculated splittings rapidly saturate with the size of the cluster. Most interestingly, the splittings follow a linear relationship when plotted as a function of cos[π/(n + 1)], with n the number of oligomers in the stack, see Fig. 6. Such a linear evolution is the one expected in the framework of tight-binding models, for which the total bandwidth for a one-dimensional stack is expressed as 4t × cos[π/(n + 1)]. The excellent linear match indicates that the splittings calculated for large clusters at the INDO level are dominated by interactions between adjacent neighbors. Thus, in this context, the total valence and conduction bandwidths of an infinite one-dimensional stack can be estimated from a simple dimer calculation (4t being then simply twice the total dimer splitting).
Figure 6.

Evolution of the INDO-calculated HOMO and LUMO splittings in cofacial stacks made of sexithienyl chains separated by 4.0 Å as a function of cos[π/(n + 1)], with n = the number of molecules in the stack.
Hexabenzocorene.
Hexabenzocoronene (HBC), C42H18, has recently received much attention because it displays a wide range of interesting structural and electrical properties (48–51). It is a discotic molecule made of 13 fused benzene rings and it possesses D6h symmetry; HBC molecules substituted by saturated chains can be arranged into one-dimensional stacks in liquid crystalline phases, for which remarkable room temperature mobilities approaching 1 cm2/Vs have been reported recently (52). The HBC crystal structure presents a columnar arrangement in which the molecules are tilted by 40° with respect to the column axis (47). Within a column, the distance between two molecular planes is 3.5 Å; this stacking is likely to induce strong interactions between the molecules and thus good transport properties along the columns; because the interactions between columns are very weak, we focus here on a single column.
The HOMO and LUMO levels of a single HBC molecule are doubly degenerate because of the D6h symmetry. For comparison purposes, we first consider a perfectly cofacial dimer (in this case, the degenerate levels split in the same way). For the distance of 3.5 Å between the molecular planes, the HOMO and LUMO splittings calculated for stacks of increasing size extrapolate to valence (HOMO) and conduction (LUMO) bandwidths that are extremely high: 1.93 and 1.13 eV, respectively.
We now turn to a dimer adopting the same configuration as in a column of the single crystal structure (47). With respect to the perfectly cofacial dimer, this configuration corresponds to sliding slightly the top molecule over the bottom one. In the displaced case, the degenerate HOMO levels lead to markedly different intermolecular interactions; the same is true for the degenerate LUMO levels. Considering the splittings for clusters of increasing size leads to valence and conduction bandwidths of 1.04 and 0.69 eV, respectively, as estimated by extrapolation from the evolution of the calculated bandwidths as a function of cos[π/(n + 1)]. Thus, going away from a perfectly cofacial stack leads to bandwidth reductions on the order of 40–45%. Despite this result, the valence and conduction bandwidths remain remarkably large; such a large valence bandwidth in the crystal is qualitatively consistent with the large one-dimensional mobilities reported for the discotic liquid crystal phases (52).
Oligoacenes.
The crystal structures of oligoacenes containing 2, 3, and 5 rings (i.e., naphthalene, anthracene, and pentacene) are characterized by a layered herringbone packing (53–55), as illustrated in Fig. 7 for pentacene. Interestingly, significant HOMO and LUMO splittings are calculated not only along the a axis but also along the diagonal axes (d1 and d2 in Fig. 7), although the molecular planes of adjacent molecules along the latter form an angle of ≈50° (55). Vanishingly small splittings are obtained from interactions among molecules located in adjacent layers (along c); this confirms that transport in oligoacene single crystals is mostly two-dimensional (26). The total widths of the valence and conduction bands can be expressed from the transfer integrals calculated along the a, d1, and d2 directions (31).
Figure 7.

Illustration of the herringbone packing of pentacene molecules (from ref. 55).
The results show that, for each compound, the total widths of the valence and conduction bands, and hence to a first approximation, the electron and hole mobilities, are similar. They reach values higher than 0.6 eV in pentacene; these are consistent with recent experimental estimates on the order of 0.5 eV deduced from mobility measurements (26). Interestingly, in these herringbone-packed systems, both the valence and conduction bandwidths increase with chain length. This result contrasts with the trends obtained when considering cofacial stacks where the valence bandwidth decreases with chain length. These results demonstrate the subtle interplay between crystal packing and electronic splittings and emphasize that the latter cannot be assessed simply at the mere view of the crystalline structure.
Also, it is most important to realize that a simple comparison of the overall valence and conduction bandwidths can only provide a qualitative picture of the respective hole and electron mobilities. To determine the latter on a more quantitative basis, it is required to evaluate the three-dimensional band structures of the crystals under investigation; the curvatures (effective masses) at the top of the valence band and at the bottom of the conduction band directly impact the hole and electron mobilities. The three-dimensional band structures and mobilities of oligoacenes need to be evaluated. [We note that preliminary results for pentacene indicate a stronger curvature at the top of the valence band than at the bottom of the conduction band, thus pointing to a lower effective mass and higher mobility for holes than electrons (Y. C. Chen, R. Silbey, D.A.d.S.F., J.P.C., J.C., J.L.B., unpublished work); this is in agreement with the experimental data at low temperature; ref. 26].
Perylene.
Perylene possesses a particular crystal structure in the sense that the molecules pack as displaced dimers that arrange themselves in a herringbone fashion (56). We have considered a perfect cofacial dimer of perylene as well as the dimer configuration present in the crystal structure. In the former case, the HOMO and LUMO splittings are both large (0.811 and 0.564 eV, respectively), even if, as expected for such a configuration, the LUMO splitting is lesser. In the crystal dimer configuration, the molecular planes remain parallel, but there occur displacements along both the long and short axes. These displacements result in a marked reduction of the LUMO splitting and, to an even larger extent, of the HOMO splitting; they become 0.188 and 0.067 eV, respectively, with the HOMO splitting nearly three times as small as the LUMO splitting. Thus, as described earlier, lateral displacements of one dimer partner over the other can lead to the reversal of the relative HOMO/LUMO splitting ratio. When taking into account the herringbone packing of the dimers (details of these calculations will be presented elsewhere), we found that the conduction band is 0.55 eV wide, and the valence band is 0.40 eV wide.
Field-effect mobility data have been recently reported by Batlogg and coworkers (57) for ultra-pure perylene crystals. At low temperature, the electron mobility is on the order of 102 cm2/Vs, whereas the hole mobility is about 25 times lower. At room temperature, the electron transport seems to remain band-like and large, ≈5 cm2/Vs; the hole transport switches to a hopping regime around 250 K. Batlogg and coworkers (58) concluded that their experimental results “overthrow the dogma that electron field-effect mobilities in organic semiconductors are much smaller than hole mobilities.” The theoretical results described in the present work are qualitatively consistent with the experimental data and provide an explanation of why this dogma has indeed no reason to be valid.
Conclusion
In this work, we have illustrated how the amplitude of the transfer integrals that govern charge transport in organic conjugated materials is highly sensitive to various aspects of molecular packing. In the fully π-conjugated systems investigated here, the largest splittings for the HOMO and LUMO levels are found consistently calculated for perfectly cofacial configurations; in this case, hole mobilities are expected to be higher than electron mobilities.
However, from a detailed examination of molecular orbital shapes and interactions, it is observed that specific arrangements can promote higher mobilities for electrons than for holes. This is, for instance, the case in perylene, which confirms that one can abandon the “dogma” according to which organic semiconductors necessarily have smaller electron mobilities than hole mobilities.
Thus, quantum-chemical calculations seem to be a useful tool to characterize the intrinsic transport properties of organic semiconductors and to design materials with enhanced electron and hole mobilities.
Acknowledgments
We thank B. Batlogg, J. H. Schön, and R. Silbey for stimulating discussions. The work at Arizona is partly supported by the Office of Naval Research, National Science Foundation Grant CHE-0078819, the Petroleum Research Fund, the Department of Energy, and the IBM Shared University Research program. The work in Mons is partly supported by Belgian Federal Government Grant PAI 4/11, the European Commission Training and Mobility of Researchers project LAMINATE, and Fonds National de la Recherche Scientifique (FNRS/FRFC). J.C. is an FNRS Research Associate; J.Ph.C. holds a scholarship from the Fonds pour la Formahon et la Recherche pour l'Industrie et l'Agriculture (FRIA). D.A.d.S.F. is supported by the Brazilian agency Comissao de Aperfeiçoamento de Pessoal de Nival Superior.
Abbreviations
- HOMO
highest occupied molecular orbital
- LUMO
lowest unoccupied molecular orbital
- INDO
intermediate neglect of differential overlap
References
- 1.Garnier F, Hajlaoui R, Yassar A, Srivastava P. Science. 1994;265:1684–1686. doi: 10.1126/science.265.5179.1684. [DOI] [PubMed] [Google Scholar]
- 2.Katz H E. J Mater Chem. 1997;7:369–376. [Google Scholar]
- 3.Horowitz G. Adv Mater. 1998;10:365–377. [Google Scholar]
- 4.Nelson S F, Lin Y Y, GunYdlach D J, Jackson T N. Appl Phys Lett. 1998;72:1854–1856. [Google Scholar]
- 5.Sirringhaus H, Brown P J, Friend R H, Nielsen M N, Bechgaard K, Langeveld-Voss B M W, Spiering A J H, Janssen R A J, Meijer E W, Herwig P, de Leeuw D M. Nature (London) 1999;401:685–688. [Google Scholar]
- 6.Katz H E, Lovinger A J, Johnson J, Kloc C, Siegrist T, Li W, Lin Y Y, Dodabalapur A. Nature (London) 2000;404:478–480. doi: 10.1038/35006603. [DOI] [PubMed] [Google Scholar]
- 7.Bao Z. Adv Mater. 2000;12:227–230. [Google Scholar]
- 8.Gelinck G H, Geuns T C T, de Leeuw D M. Appl Phys Lett. 2000;77:1487–1489. [Google Scholar]
- 9.Tang C W, Van Slyke S A. Appl Phys Lett. 1987;51:913–915. [Google Scholar]
- 10.Burroughes J H, Bradley D D C, Brown A R, Marks R N, Friend R H, Burn P L, Holmes A B. Nature (London) 1990;347:539–541. [Google Scholar]
- 11.Sheats J R, Antoniadis H, Hueschen M, Leonard W, Miller J, Moon R, Roitman D, Stocking A. Science. 1996;273:884–888. doi: 10.1126/science.273.5277.884. [DOI] [PubMed] [Google Scholar]
- 12.Friend R H, Burroughes J, Shimoda T. Physics World. 1999;June:35–40. [Google Scholar]
- 13.Friend R H, Gymer R W, Holmes A B, Burroughes J H, Marks R N, Taliani C, Bradley D D C, dos Santos D A, Brédas J L, Lögdlund M, Salaneck W R. Nature (London) 1999;397:121–128. [Google Scholar]
- 14.Sariciftci N S, Smilowitz L, Heeger A J, Wudl F. Science. 1992;258:1474–1476. doi: 10.1126/science.258.5087.1474. [DOI] [PubMed] [Google Scholar]
- 15.Halls J J M, Walsh C A, Greenham N C, Marseglia E A, Friend R H, Moratti S C, Holmes A B. Nature (London) 1995;376:498–500. [Google Scholar]
- 16.Yu G, Wang J, McElvain J, Heeger A J. Adv Mater. 1998;17:1431–1434. [Google Scholar]
- 17.Brabec C J, Sariciftci N S, Hummelen J C. Adv Funct Mater. 2001;11:15–26. [Google Scholar]
- 18.Sirringhaus H, Kawase T, Friend R H, Shimoda T. Science. 2000;290:2123–2126. doi: 10.1126/science.290.5499.2123. [DOI] [PubMed] [Google Scholar]
- 19.Dodabalapur A, Bao Z, Makhija A, Laquindaum J G, Raju V R, Feng Y, Katz H E, Rogers J. Appl Phys Lett. 1998;173:142–144. [Google Scholar]
- 20.Sirringhaus H, Tessler N, Friend R H. Science. 1998;280:1741–1744. doi: 10.1126/science.280.5370.1741. [DOI] [PubMed] [Google Scholar]
- 21.Schön J H, Dodabalapur A, Kloc C, Batlogg B. Science. 2000;290:963–965. doi: 10.1126/science.290.5493.963. [DOI] [PubMed] [Google Scholar]
- 22.Greenham N C, Moratti S C, Bradley D D C, Friend R H, Holmes A B. Nature (London) 1993;365:628–630. [Google Scholar]
- 23.Laquindanum J G, Katz H E, Lovinger A J, Dodabalapur A. Chem Mater. 1996;8:2542–2544. [Google Scholar]
- 24.Gundlach D J, Lin Y Y, Jackson T N, Nelson S F, Schlom D G. IEEE Electron Device Lett. 1997;18:87–89. [Google Scholar]
- 25.Fichou D. J Mat Chem. 2000;10:571–588. [Google Scholar]
- 26.Schön J H, Kloc C, Batlogg B. Science. 2000;288:2338–2340. doi: 10.1126/science.288.5475.2338. [DOI] [PubMed] [Google Scholar]
- 27.Schön J H, Berg S, Kloc C, Batlogg B. Science. 2000;287:1022–1023. doi: 10.1126/science.287.5455.1022. [DOI] [PubMed] [Google Scholar]
- 28.Schön J H, Kloc C, Dodabalapur A, Batlogg B. Science. 2000;289:599–601. doi: 10.1126/science.289.5479.599. [DOI] [PubMed] [Google Scholar]
- 29.Schön J H, Kloc C, Batlogg B. Science. 2000;406:702–704. doi: 10.1038/35021011. [DOI] [PubMed] [Google Scholar]
- 30.Schön J H. Synt Met. 2001;122:157–160. [Google Scholar]
- 31.Cornil J, Calbert J P, Brédas J L. J Am Chem Soc. 2001;123:1250–1251. doi: 10.1021/ja005700i. [DOI] [PubMed] [Google Scholar]
- 32.Cornil J, Beljonne D, Calbert J P, Brédas J L. Adv Mater. 2001;13:1053–1067. [Google Scholar]
- 33.Schön J H, Kloc C, Batlogg B. Phys Rev Lett. 2001;86:3843–3846. doi: 10.1103/PhysRevLett.86.3843. [DOI] [PubMed] [Google Scholar]
- 34.Haddon R C, Siegriest T, Fleming R M, Bridenbaugh P M, Laudise R A. J Mater Chem. 1995;5:1719–1724. [Google Scholar]
- 35.Horowitz G, Hajlaoui R, Bourguiga R, Hajlaoui M. Synt Met. 1999;101:401–404. [Google Scholar]
- 36.Marcus R A. Rev Mod Phys. 1993;65:599–610. [Google Scholar]
- 37.Sakanoue K, Motoda M, Sugimoto M, Sakaki S. J Phys Chem. 1999;103:5551–5556. [Google Scholar]
- 38.Malagoli M, Brédas J L. Chem Phys Lett. 2000;327:13–17. [Google Scholar]
- 39.Li X Y, Tang X S, He F C. Chem Phys. 1999;248:137–146. [Google Scholar]
- 40.Horowitz G, Hajlaoui M E. Adv Mater. 2000;12:1046–1050. [Google Scholar]
- 41.Torsi L, Dodabalapur A, Rothberg L J, Fung A W P, Katz H E. Phys Rev B Condens Matter. 1998;57:2271–2275. [Google Scholar]
- 42.Dewar M J S, Zoebisch E G, Healy E F, Stewart J J P. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 43.Ridley J, Zerner M C. Theor Chim Acta. 1973;32:111–134. [Google Scholar]
- 44.Zerner M C, Loew G H, Kichner R F, Mueller-Westerhoff U T. J Am Chem Soc. 1980;102:589–599. [Google Scholar]
- 45.Newton M D. Int J Quant Chem. 2000;77:255–263. [Google Scholar]
- 46.Li X C, Sirringhaus H, Garnier F, Holmes A B, Moratti S C, Feeder N, Clegg W, Teat S J, Friend R H. J Am Chem Soc. 1998;120:2206–2207. [Google Scholar]
- 47.Goddard R, Haenel M W, Herndon W C, Krüger C, Zander M. J Am Chem Soc. 1995;117:30–41. [Google Scholar]
- 48.Müller M, Kübel C, Müllen K. Chem Eur J. 1998;4:2099–2109. [Google Scholar]
- 49.Herwig P, Kayser C W, Müllen K, Spiess H W. Adv Mater. 1996;8:510–513. [Google Scholar]
- 50.van de Craats A M, Warman J M, Müllen K, Geerts Y, Brand J D. Adv Mater. 1998;10:36–38. [Google Scholar]
- 51.Keil M, Samori P, dos Santos D A, Kugler T, Stafström S, Brand J D, Müllen K, Brédas J L, Rabe J B, Salaneck W R. J Phys Chem B. 2000;104:3967–3975. [Google Scholar]
- 52.van de Craats A M, Warman J M, Fechtenkötter A, Brand J D, Harbison M A, Müllen K. Adv Mater. 1999;11:1469–1472. [Google Scholar]
- 53.Ponomarev V I, Filipenko O S, Atovmyan L O. Kristallografiya. 1976;21:392–394. [Google Scholar]
- 54.Brock C P, Dunitz J D. Acta Crystallogr B. 1990;46:795–806. [Google Scholar]
- 55.Holmes D, Kumaraswamy S, Matzger A J, Vollhardt K P C. Chem Eur J. 1999;5:3399–3412. [Google Scholar]
- 56.Camerman A, Trotter J. Proc R Soc London. 1964;279:129–146. [Google Scholar]
- 57.Schön J H, Kloc Ch, Batlogg B. Appl Phys Lett. 2000;77:3776–3778. [Google Scholar]