

Abstract
Posttranscriptional processing of mRNA is an integral component of the gene expression program. By using DNA microarrays, we precisely measured the decay of each yeast mRNA, after thermal inactivation of a temperature-sensitive RNA polymerase II. The half-lives varied widely, ranging from ∼3 min to more than 90 min. We found no simple correlation between mRNA half-lives and ORF size, codon bias, ribosome density, or abundance. However, the decay rates of mRNAs encoding groups of proteins that act together in stoichiometric complexes were generally closely matched, and other evidence pointed to a more general relationship between physiological function and mRNA turnover rates. The results provide strong evidence that precise control of the decay of each mRNA is a fundamental feature of the gene expression program in yeast.
Athough initiation of transcription is well studied and its importance in regulation is clear, we know much less about the specificity, precision and regulatory role of mRNA decay. The abundance of each mRNA in the cell is determined not only by the rate at which it is produced, but also by its rate of degradation. mRNA decay rates determine how quickly each message can adapt to a new steady-state level after a change in transcription rate, and dynamic control of the decay of specific transcripts can have an important role in their regulation (1). The decay rates of specific mRNAs can vary by 100-fold or more (2, 3) and are affected by a wide variety of stimuli and cellular signals, including specific hormones (2, 4), iron (5, 6), cell cycle progression (7, 8), cell differentiation (9, 10), and viral infection (11). Characterizing this stage in the natural history of each mRNA is an important step toward understanding the logic and molecular mechanisms underlying the regulation of the gene expression program of a genome.
With its ease of biochemical and genetic manipulation, yeast makes an excellent model for studying eukaryotic mRNA turnover. Simple and reliable procedures have been developed to measure the decay rates of individual mRNAs in yeast, including global or specific transcriptional shut-off assays (12–14) and in vivo kinetic labeling (15). However, the global profile of yeast mRNA turnover has not been systematically and quantitatively determined. In this report, we present the results of a genomewide determination of mRNA decay rates, coupling the global transcriptional shut-off assay with DNA microarray analysis.
Materials and Methods
Yeast Strain.
Saccharomyces cerevisiae strain Y262 (MATa ura3–52 his4–939am rpb1-1), carrying a temperature-sensitive mutation in RNA polymerase II (13), was used in this study.
Determination of mRNA Decay by Transcriptional Shut-Off Assay.
Y262 was grown in 500 ml of yeast extract/peptone/dextrose (YPD) medium at 24°C to OD600 ∼0.5. The temperature of the culture was abruptly shifted to 37°C by adding an equal volume of YPD medium that had been prewarmed to 49°C. Aliquots of the culture (100 ml) were removed at 0, 5, 10, 15, 20, 30, 40, 50, and 60 min after the temperature shift. Cells were rapidly harvested on a nitrocellulose filter (Whatman no. 141109) followed by immediate freezing in liquid N2. Total RNA was prepared from cells harvested at each time point by hot phenol extraction (16).
Microarray Analysis.
Yeast DNA microarrays were produced and hybridized as described (17–19) with modifications outlined below. For total RNA sample (15–75 μg) at each time point, a fluorescently labeled cDNA probe was prepared by reverse transcription in the presence of Cy5-dUTP, using a random primer (http://www-genome.stanford.edu/turnover). Yeast genomic DNA (200 ng) was digested with DpnII (New England Biolabs) into 0.3–2-kb fragments, labeled with Cy3-dUTP by using reverse transcriptase (http://www-genome.stanford.edu/turnover), and used as an internal standard for each hybridization. Microarrays were scanned with an Axon Instruments (Foster City, CA) scanner, and the data were collected with GENEPIX PRO 3.0 software (Axon Instruments). Only spots for which the signal-to-background ratio was greater than 1.2 were selected for analysis. A background corresponding to ∼0.1 transcripts per cell, determined from results of control experiments, was subtracted from the Cy5/Cy3 fluorescence ratio measured for each mRNA, and the resulting values for each mRNA were then plotted as a function of time after the temperature shift.
For normalization between time points, an internal standard was prepared with a pool of in vitro-transcribed Bacillus subtilis RNAs. PCR products representing five B. subtilis DNAs (ATCC nos. 87482, 87483, 87484, 87485, and 87486) were printed onto the yeast DNA microarrays (∼125 spots on each array in different locations). RNA transcripts of each of these B. subtilis DNAs were prepared in vitro and pooled at a final concentration ∼4 ng/μl of each B. subtilis mRNA; a detailed protocol is presented at http://www-genome.stanford.edu/turnover. This internal standard mixture was added to each total RNA sample analyzed in the decay time course, at a final concentration 400 pg of standard RNA mix per 15 μg of total RNA, and to the genomic DNA sample, at a final concentration 400 pg per 200 ng of genomic DNA, before labeling and hybridization. The internal standard gives us a way to normalize the results from each array to a constant level of total RNA, based on the assumption that the total amount of RNA remains approximately constant even though the mRNAs are decaying. We believe this assumption is reasonable because mRNAs account for less than 5% of cellular RNAs.
mRNA Decay Profile Analysis.
The imagedisplay program was developed and used to automate the display of mRNA decay profiles. The program can be downloaded from http://www-genome.stanford.edu/turnover.
A nonlinear least squares model was fit to determine the decay rate constant (k) and half-life (t1/2) of each mRNA. The decay rate constant, k, is the value that minimized Σi = 1,n[y(ti) − exp(−k⋅ti)]2, where y(t) is the mRNA abundance at time t and the summation is taken over all observations for the particular mRNA. The half-life is t1/2 = ln2/k. The goodness of fit of the decay model for each gene was assessed with the F statistic (20), based on the null hypothesis that the data fit a first-order decay model.
A bootstrap method was used to calculate confidence intervals for both t1/2 and k (21). We assumed that residuals between and within time points were independent. We also assumed that residuals in triplicate measurements within a time point were equally distributed, but residuals between time points were not. For each bootstrap iteration, new time courses were simulated by sampling the residuals within each time point with replacement and adding the resampled residuals to the fitted value at their respective time points. Confidence intervals were calculated only for mRNAs for which there were replicates for at least 4 time points. This procedure yielded bootstrap decay rate constants k(b) for b = 1, … , 1,000. By taking the 2.5% and 97.5% quartiles values of k(b) as lower and upper confidence limits, respectively, we formed a 95% bootstrap confidence interval for k. By setting t1/2(b) = ln2/k(b), we similarly formed a 95% bootstrap confidence interval for t1/2.
Statistical Analysis of mRNA Decay in Stoichiometric Protein Complexes.
The concordance of the decay rate constants of transcripts encoding components of stoichiometric protein complexes was assessed by comparison to size-matched sets of decay rate constants for randomly selected transcripts. Because the SDs of sets of decay rate constants were not independent of the means, a normalized SD (sd*) was calculated by a nonparametric statistical method so that they were functionally independent from the means of the decay rates. This transformation is accomplished by first plotting the SDs of decay rate constants for random groups of genes as a function of their means. Then a natural cubic spline was fit to the scatter plot (22). The SDs were then divided by the fitted value of the spline at their corresponding means. To measure the concordance of the decay rates for a complex of size N, we calculated the normalized SD sdn of its decay rates. We also calculated the normalized SDs sd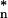 of 104 randomly selected groups of N mRNAs. The P value of the test was calculated as the proportion of sd
of 104 randomly selected groups of N mRNAs. The P value of the test was calculated as the proportion of sd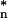 less than or equal to sdn.
less than or equal to sdn.
An additional statistic was used to evaluate the concordance of subunit decay rates for the 33 heterodimeric complexes. Let ki,1 and ki,2 be the two decay rate constants for the ith heterodimeric complex. The statistic we used was Σ(ki,1 − ki,2)2. This statistic was compared with 104 sets of random pairings of the 66 mRNAs.
Results
Whole-Genome Determination of mRNA Half-Lives.
As a first step toward determining the mechanism and role of mRNA decay in the yeast gene expression program, we measured mRNA decay on a genomic scale, using DNA microarrays coupled with global transcriptional shut-off assay (Fig. 1A).
Figure 1.

Whole-genome determination of mRNA half-lives. (A) Schematic of the DNA microarray procedure for determining genomewide mRNA half-lives. Total RNA was isolated at specified intervals after inactivation of RNA polymerase II. Fluorescently labeled cDNA probes were prepared from each RNA sample by reverse transcription in the presence of Cy5-dUTP. Yeast genomic DNA was similarly labeled with Cy3-dUTP to provide an internal hybridization standard for every gene. For each time point, Cy5-labeled probe was mixed with the Cy3-labeled genomic DNA standard and the mixtures were hybridized to DNA microarrays. (B) Examples of mRNA decay profiles determined by quantitative microarray analysis. The different symbols represent data from three independent time courses; the solid lines represent the nonlinear least squares fit to an exponential decay model; the dashed lines represent the upper and lower limits of the 95% confidence interval for the decay curves, determined by the bootstrapping procedure. The calculated half-life and 95% confidence interval for the decay of each transcript are listed. (C) Northern analysis of the decay of mRNAs encoding PGK1 and RPS6B (quantified with a PhosphorImager).
Three independent time courses were analyzed. Triplicate decay curves, each defined by 9 time points after the temperature shift, were determined for each mRNA. By using a custom imagedisplay program, a set of 4,687 genes with at least 2 independent time courses was selected for further analysis. The triplicate decay curves of each mRNA were then fitted to an exponential decay model with a nonlinear least squares method. This model, although a simplification (23), provided a good fit to the data and allowed simple parameters (the decay rate constant, k, and the half-life, t1/2) to be derived for each mRNA for use in the subsequent analysis. Fig. 1B shows examples of the decay profile for individual mRNAs, and similar graphs of the complete set of data are available at http://www-genome.stanford.edu/turnover. A bootstrap method was also used to determine the SD and the 95% confidence interval of the decay rate constant (k) and half-life (t1/2) for each mRNA. For 3,735 genes (80%), the half-lives varied by less than ±15% in independent measurements. Independent Northern analysis of two individual mRNA species confirmed that the decay rates measured by microarrays were consistent with the decay of the full-length transcripts (Fig. 1C). Furthermore, with few exceptions, the mRNA half-lives determined with DNA microarrays were in reasonable agreement with those of 34 mRNAs previously determined by Northern analysis [correlation coefficient (Cor. Coeff.) = 0.74] (http://www-genome.stanford.edu/turnover). The exceptions are mainly mRNAs with short half-lives (via Northern analysis). The fitting protocol used for analysis of all of the data assumed a zero endpoint to maximize consistency in the data treatment. This assumption, however, introduces additional error in the individual fits, especially for the short-lived mRNAs, and errors for these mRNAs are further exacerbated by the choice of time points, with the first occurring at 5 min (20). We cannot make extensive comparisons between our data and previous global decay data obtained from a single time point (45 min) (24), because no error analysis is possible from a single measurement and because most of the mRNA half-lives are shorter than 45 min.
The half-lives of the 4,687 mRNAs analyzed varied widely, ranging from ∼3 min to more than 90 min, with a mean of 23 min and median of 20 min (Fig. 2A). No simple correlation was found between the decay rates of mRNAs and their abundance (Cor. Coeff. = 0.06), the size of the ORF (Cor. Coeff. = −0.01), codon adaptation index (Cor. Coeff. = 0.04), or the density of ribosomes bound to the mRNA (Cor. Coeff. = 0.08) (Y. Arava, D.H., and P.O.B., unpublished data) (http://www-genome.stanford.edu/turnover).
Figure 2.

Comparison between overall mRNA decay rates and poly(A)+ mRNA decay rates. (A) Distribution of half-lives of mRNA overall decay (blue) and poly(A)+ mRNA decay (red). (B) Scatter plot of half-lives of mRNA overall decay and poly(A)+ mRNA decay for the 4,661 mRNAs. Cor. Coeff. = 0.50. The pink dashed line indicates a slope of 1. The green solid line is the best least-squares linear fit of the data, with a slope of 0.41 and y intercept of 1 min (in log scale).
Determination of Poly(A) Shortening Rates.
Three pathways for mRNA degradation have been identified in yeast (23). Degradation can be initiated by poly(A) shortening or arrest of translation at a premature nonsense codon. Poly(A) shortening has been proposed to be an initial and rate-limiting step in the decay of many eukaryotic mRNAs (25, 26).
To examine the global relationship between poly(A) shortening and mRNA turnover, we made a separate series of measurements of the fate of each mRNA, using an anchored oligo(dT) primer (5′-T20VN-3′), rather than random primers, in the cDNA probe synthesis. This approach allowed us to track specifically the mRNAs that retained poly(A) tails of sufficient length to allow priming. The poly(A)+ mRNA decay half-lives, as defined in our assay, were distributed within a narrower range and were significantly shorter (peak at 10–15 min) than the overall mRNA decay half-lives (Fig. 2A). Moreover, the rate at which most individual mRNAs disappeared from the poly(A)+ population was faster than their overall decay rates (Fig. 2B). A small number of mRNAs (512 of 4,661) has shorter apparent half-lives for overall decay than poly(A) shortening. Of these, 445 (87%) have half-lives for poly(A) shortening that are less than their overall decay half-lives plus 1 SD, suggesting that the small number of exceptions can be accounted for by experimental uncertainty. A monotonic and roughly linear relationship was observed between the poly(A)+ mRNA decay rates and the overall decay rates (Fig. 2B), consistent with a current model for the major decay pathway in yeast, in which poly(A) shortening precedes the decay of the entire transcript, and the downstream events in the decay program depend on poly(A) shortening (23).
Precision in mRNA Decay.
How are the decay rates of individual mRNAs related to the function of the proteins they encode, and how precisely specified is the decay of each mRNA? To address these questions, we needed a model that makes a strong, specific prediction for the decay rates of specific transcripts, based on the functions of the products. In general, we could not hope to predict, with any precision, what the decay rate of a specific mRNA ought to be based on what we know of its function. Nevertheless, a specific functional relationship between genes, the empirical evidence for which is relatively easy to assess, provides a basis for a robust, quantitative prediction for hundreds of mRNAs: We expect the transcripts of genes whose protein products invariably work together in the cell as components of heteromultimeric complexes with defined stoichiometry to be regulated in a very similar pattern. Indeed, genomewide studies have demonstrated a striking covariation in the abundance of transcripts that encode subunits of the same stoichiometric complexes (17, 27, 28). We therefore focused the questions of specificity and precision by asking whether, and with what precision, the decay rates of mRNAs that encode protein subunits of stoichiometric complexes are matched to one another.
The nucleosome core is composed of four histone subunits, in equimolar proportions, assembled into an octamer (29). The 4 distinguishable histone mRNAs have closely matched, rapid decay rates, with t1/2 = 7 ± 2 min (Fig. 3A). The 20S proteasome core is a stoichiometric complex of 14 different protein subunits (30). We were able to obtain good measurements of the decay of 13 of the 14 corresponding mRNAs; all decayed at closely matched rates, with t1/2 = 13 ± 3 min (Fig. 3A). The ribosome, the largest stoichiometric protein complex, is composed of 32 different proteins in the small subunit and 46 different proteins in the large subunit. The ribosomal proteins are encoded by 137 different mRNAs (there are 59 duplicated genes) (31). The 131 ribosomal protein mRNAs analyzed in this study had remarkably similar half-lives, with t1/2 = 22 ± 6 min (Fig. 3A). The last illustrative example is the trehalose phosphate synthase complex, which plays an important role in carbohydrate metabolism and stress responses (32, 33). The mRNAs encoding the four distinct subunits of this stoichiometric complex exhibited uniformly slow decay rates, with t1/2 = 105 ± 12 min (Fig. 3A).
Figure 3.

mRNA encoding the subunits of stoichiometric protein complexes exhibit coordinated decay. (A) Examples of coordinated decay of transcripts for four stoichiometric protein complexes. The number of unique components in each complex is indicated in parentheses; decay curves for 40 randomly selected mRNAs, excluding five outliers (see Fig. 2D), encoding unique ribo some subunits are shown. (B) Clustering of the decay half-lives of mRNAs encoding subunits of protein complexes. The number of unique components in each complex is indicated in parentheses. Red open circles, half-lives of individual mRNAs within each complex; thick black bar, the mean half-life for each complex; error bars indicate ±1 SD. The complexes are sorted along the vertical axis (top to bottom) in the order of increasing mean half-lives. (C) Statistical test for the coordinated decay of subunits of stoichiometric protein complexes with N ≥ 2 components. A P value for the clustering of decay rates of transcripts for each physical complex of size of N was calculated. The probability of obtaining a smaller P value from random sampling (104 times) of N samples from 4,482 unique mRNA half-lives was then determined and summarized in the histogram. The dashed line represents the uniform distribution expected for the null hypothesis in which there is no coordination of decay rates. (D) A small set of mRNAs encoding ribosomal proteins has anomalously fast decay rates. Blue curve, average decay curve of 131 RP mRNAs; error bars indicate ±1 SD; green curves, decay curves of five individual mRNAs—RPS4A, RPS4B, RPL3, RPS27A, and RPS28B (average of triplicate measurements)—with very short half-lives (t1/2 < 10 min).
As illustrated by these examples, the transcripts encoding subunits of a stoichiometric complex appeared generally to be programmed to decay at rates matched with remarkable precision. To evaluate the generality of this observation, we identified a set of 95 heteromultimeric protein complexes that satisfied the following criteria. (i) They are documented as protein complexes by both the MIPS database (http://www.mips.biochem.mpg.de/proj/yeast) and the YPD database (http://www.proteome.com/databases); in either case, physical interactions detected solely by yeast two-hybrid analysis were not considered. (ii) We had technically adequate measurements of decay rates for the transcripts encoding at least two of the subunits. A similar coordination in mRNA decay was observed for many of these protein complexes (Fig. 3B).
To test the significance of the apparent coordination, we used a permutation method to model the distribution of subunit decay rates under the null hypothesis: For a protein complex containing N different subunits, the null distribution of the mRNA decay rates was modeled by randomly sampling sets of N mRNA decay rates from our set of 4,482 unique measurements.‖ We could then ask whether the decay rates of transcripts encoding members of a bona fide protein complex of size N were more closely matched than would be expected for a random set of N transcripts (see Materials and Methods). The results are summarized in Fig. 3C. The observed coordination in decay rates was highly significant. For example, of the 95 protein complexes of size N ≥ 2 that were suitable for this analysis, in 57 (60%) the concordance in the decay rates of the subunit transcripts was at a level that would be expected to occur in fewer than 20% of random complexes of the corresponding size (http://www-genome.stanford.edu/turnover). Of the remaining 38 complexes, 16 are heterodimeric. Because the significance of the similarity in any single pair of decay rates is difficult to evaluate, we evaluated the concordance of subunit decay rates in the 33 heterodimeric complexes as a group. The variance of the two decay rate constants for any given heterodimeric complex was compared with 104 random pairings of the mRNA pool that contained all of the components of the heterodimeric complexes. The observed concordance in the decay rates was highly significant (P < 10−4). We conclude that the decay rates of most of these transcripts, and presumably of yeast mRNAs in general, are programmed with remarkable precision, to satisfy requirements related to the function of the product they encode.
The discordance in mRNA decay rates observed for some of the complexes we analyzed may reflect imprecision in our measurements, imprecision in the regulation of decay, or a significant feature of the corresponding regulatory program. A closer look at some of the outliers suggests that the mRNAs with apparently anomalous decay rates may have a physiological function. For example, there were a few noticeable exceptions of the otherwise precisely coordinated decay of ribosomal protein (RP) transcripts (Fig. 3D). Among the 131 RP mRNAs analyzed in this study, 5 mRNAs (those encoding RPS4A, RPS4B, RPL3, RPS27A, and RPS28A) had aberrantly short half-lives (t1/2 < 10 min). Perhaps significantly, although more than 70% of RP transcripts have at least one intron and more than 90% are regulated by the transcription factor Rap1 (34), four of these five unstable transcripts (except RPS28A) lack introns and are not Rap1 targets. Although the altered RP gene transcript levels in amplified strains suggest that in general RP genes are not individually autoregulated at this level (35, 36), RPS4A and RPS4B have been reported to use autoregulation to regulate turnover of their transcripts (37, 38). The RPL3 gene product is one of the earliest ribosomal proteins to assemble in the nascent large subunit during ribosome biosynthesis (39). Levels of the RPL3 transcript, uniquely among ribosomal protein mRNAs, are subject to regulation by the Rrb1 protein, which has been shown to interact physically in vivo with the RPL3 protein, suggesting a specialized autoregulatory mechanism (40). The anomalous decay rates we observed for five specific ribosomal mRNAs may therefore point to specialized regulatory programs and distinct functions.
Physiological Coherence in mRNA Decay.
Relationships between mRNA decay rates and more broadly defined functional groupings provided additional evidence of the physiological logic of mRNA decay. For example, transcripts encoding the enzymes that participate in the central systems of energy metabolism, including glycolysis/gluconeogenesis, the tricarboxylic acid cycle, and the glyoxylate cycle, are characteristically among those that live the longest (Fig. 4A). In contrast, virtually all of the transcripts encoding the proteins of the mating pheromone signal transduction pathway turn over relatively rapidly (Fig. 4B). As shown in Fig. 4C, mRNAs encoding translation initiation factors and termination factors turn over rapidly, whereas transcripts encoding translation elongation factors lived 2–4-fold longer. One general relationship between the stability of an mRNA and the physiological function of its product seems to be that mRNAs involved in central metabolic functions are generally relatively long-lived, whereas those involved in regulatory systems turn over relatively rapidly. Such a relationship has been suggested based on studies of a number of individual mRNAs (12) and is strongly supported by our genomewide analysis of mRNA turnover.
Figure 4.

Coherence of mRNA turnover in physiological systems. The range of half-lives, from 0 to more than 45 min, was continuously color-coded with a green-yellow-red gradient (green = shortest half-lives, red = longest half-lives). For protein complexes with multiple subunits, smaller blocks were individually color-coded to represent the mRNA half-lives for each subunit. White boxes represent transcripts for which we did not obtain an adequate measurement of decay. TCA, tricarboxylic acid. mRNA turnover in (A) central energy metabolism systems (modified from ref. 17); (B) the pheromone signal transduction pathway (modified from ref. 43); and (C) translation factors.
In summary, our results provide strong evidence for functional coordination of mRNA decay. Nevertheless, we cannot determine whether there is greater precision in the coordination of decay for mRNAs encoding proteins that form stoichiometric complexes than other mRNAs encoding proteins that function in the same biological process. Such analysis is hindered by the limitations in defining the functional interrelationships of gene products.
Discussion
By systematically measuring the decay of mRNAs from more than 4,000 yeast genes, we found that this key step in gene expression is specified for individual genes with considerable precision. The programmed decay of each mRNA, like initiation of transcription, seems to be intimately related to the physiological role of the encoded product. The mRNA levels can be adjusted more rapidly for mRNAs with short half-lives. Furthermore, theoretical studies have shown that coordinated regulation of mRNA turnover, transcription, translation, and protein turnover can provide precision, speed, and flexibility in biological regulation beyond what would be possible with any subset of these regulatory mechanisms (1). Indeed, steroid hormones affect the stability of the target mRNAs as well as the transcription of the target genes (2, 4). In erythroid differentiation, induction of globin gene transcription is coupled with destabilization of a large number of cellular mRNAs (9), permitting a rapid redirection of cellular resources to the production of hemoglobin. There is evidence to suggest that the stability of mRNAs and the proteins they encode are often correlated, although the systems that mediate their degradation are distinct (1).
In this study, mRNA decay was measured under only a single, nonphysiological condition. Indeed, features of the decay program observed in this experiment may reflect a stress response.** We think it unlikely that the mRNA decay rate is a static property of individual mRNAs, indifferent to changing physiological demands. It will be of great interest to determine whether and how mRNA stability is dynamically regulated in response to changing conditions.
What molecular mechanisms account for the specificity and precision of mRNA decay? The decay rates must be specified by sequence or structural features that dictate their susceptibility to the degradation machinery, either directly or indirectly. These mRNA-specific features could include sequences or secondary structures recognized by transacting factors (e.g., endo- and exonucleases, RNA binding regulatory proteins) and more general features of the mRNA sequence and structure. Previous studies have suggested that the decay rates of individual yeast mRNAs may be correlated with the frequency of rare codons (12) or inversely related to the length of the protein coding sequence (42). We were unable to detect any significant correlation between mRNA half-lives and codon usage or ORF lengths. Nor was there any detectable correlation between the decay rate of individual mRNA species and their abundance or translation rates. This systematic picture of mRNA decay provides a rich resource for further in-depth investigations of the molecular codes and mechanisms that regulate the turnover of individual mRNAs.
Acknowledgments
We thank Dr. Richard Young (Massachusetts Institute of Technology) for providing yeast strain Y262 and Yoav Arava for permission to cite unpublished results. This work was supported by National Institutes of Health Grant HG00983 (to P.O.B.) and by the Howard Hughes Medical Institute (P.O.B.). P.O.B. is an Associate Investigator of the Howard Hughes Medical Institute. Y.W. was supported by National Institutes of Health Research Fellowship F32 GM20873-01.
Abbreviations
- Cor. Coeff.
correlation coefficient
- RP
ribosomal protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
The measured mRNA half-lives of transcripts that share more than 70% identity in nucleotide sequences were averaged and treated as a single mRNA species.
For 30 genes, most with known heat shock or stress-response functions, mRNA levels increased slightly instead of decreasing after temperature shift. This behavior presumably results from the low level of leakiness of the temperature-sensitive phenotype of the rpb1–1 mutant and the strong preference for transcription of stress-response genes under these conditions (41).
References
- 1.Hargrove J L, Schmidt F H. FASEB J. 1989;3:2360–2370. doi: 10.1096/fasebj.3.12.2676679. [DOI] [PubMed] [Google Scholar]
- 2.Ross J. Microbiol Rev. 1995;59:423–450. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabrera C V, Lee J J, Ellison J W, Britten R J, Davidson E H. J Mol Biol. 1984;174:85–111. doi: 10.1016/0022-2836(84)90366-8. [DOI] [PubMed] [Google Scholar]
- 4.Ross J. Trends Genet. 1996;12:171–175. doi: 10.1016/0168-9525(96)10016-0. [DOI] [PubMed] [Google Scholar]
- 5.Casey J L, Hentze M W, Koeller D M, Caughman S W, Rouault T A, Klausner R D, Harford J B. Science. 1988;240:924–928. doi: 10.1126/science.2452485. [DOI] [PubMed] [Google Scholar]
- 6.Thomson A M, Rogers J T, Leedman P J. Int J Biochem Cell Biol. 1999;31:1139–1152. doi: 10.1016/s1357-2725(99)00080-1. [DOI] [PubMed] [Google Scholar]
- 7.Heintz N, Sive H L, Roeder R G. Mol Cell Biol. 1983;3:539–550. doi: 10.1128/mcb.3.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris T D, Weber L A, Hickey E, Stein G S, Stein J L. Mol Cell Biol. 1991;11:544–553. doi: 10.1128/mcb.11.1.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krowczynska A, Yenofsky R, Brawerman G. J Mol Biol. 1985;181:231–239. doi: 10.1016/0022-2836(85)90087-7. [DOI] [PubMed] [Google Scholar]
- 10.Jack H M, Wabl M. EMBO J. 1988;7:1041–1046. doi: 10.1002/j.1460-2075.1988.tb02911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorenson C M, Hart P A, Ross J. Nucleic Acids Res. 1991;19:4459–4465. doi: 10.1093/nar/19.16.4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrick D, Parker R, Jacobson A. Mol Cell Biol. 1990;10:2269–2284. doi: 10.1128/mcb.10.5.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nonet M, Scafe C, Sexton J, Young R. Mol Cell Biol. 1987;7:1602–1611. doi: 10.1128/mcb.7.5.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decker C J, Parker R. Genes Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- 15.Kim C H, Warner J R. J Mol Biol. 1983;165:79–89. doi: 10.1016/s0022-2836(83)80243-5. [DOI] [PubMed] [Google Scholar]
- 16.Iyer V R, Eisen M B, Ross D T, Schuler G, Moore T, Lee J C, Trent J M, Staudt L M, Hudson J, Jr, Boguski M S, et al. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 17.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 18.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 19.Spellman P T, Sherlock G, Zhang M Q, Iyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neter J, Kutner M H, Nachtsheim C J, Wasserman W. Applied Linear Statistical Models. Chicago: Irwin; 1996. [Google Scholar]
- 21.Efron B, Tibshirani R J. An Introduction to the Bootstrap. New York: Chapman & Hall; 1993. [Google Scholar]
- 22.Hastie T, Tibshirani R. Generalized Additive Models. New York: Chapman & Hall; 1990. [DOI] [PubMed] [Google Scholar]
- 23.Beelman C A, Parker R. Cell. 1995;81:179–183. doi: 10.1016/0092-8674(95)90326-7. [DOI] [PubMed] [Google Scholar]
- 24.Holstege F C, Jennings E G, Wyrick J J, Lee T I, Hengartner C J, Green M R, Golub T R, Lander E S, Young R A. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 25.Shyu A B, Belasco J G, Greenberg M E. Genes Dev. 1991;5:221–231. doi: 10.1101/gad.5.2.221. [DOI] [PubMed] [Google Scholar]
- 26.Muhlrad D, Parker R. Genes Dev. 1992;6:2100–2111. doi: 10.1101/gad.6.11.2100. [DOI] [PubMed] [Google Scholar]
- 27.Brown P O, Botstein D. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 28.Marcotte E M, Pellegrini M, Thompson M J, Yeates T O, Eisenberg D. Nature (London) 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- 29.Fukuma M, Hiraoka Y, Sakurai H, Fukasawa T. Yeast. 1994;10:319–331. doi: 10.1002/yea.320100305. [DOI] [PubMed] [Google Scholar]
- 30.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik H D, Huber R. Nature (London) 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 31.Planta R J, Mager W H. Yeast. 1998;14:471–477. doi: 10.1002/(SICI)1097-0061(19980330)14:5<471::AID-YEA241>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 32.Reinders A, Burckert N, Hohmann S, Thevelein J M, Boller T, Wiemken A, De Virgilio C. Mol Microbiol. 1997;24:687–695. doi: 10.1046/j.1365-2958.1997.3861749.x. [DOI] [PubMed] [Google Scholar]
- 33.Thevelein J M, Hohmann S. Trends Biochem Sci. 1995;20:3–10. doi: 10.1016/s0968-0004(00)88938-0. [DOI] [PubMed] [Google Scholar]
- 34.Lieb J D, Liu X, Botstein D, Brown P O. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- 35.Hughes T R, Roberts C J, Dai H, Jones A R, Meyer M R, Slade D, Burchard J, Dow S, Ward T R, Kidd M J, et al. Nat Genet. 2000;25:333–337. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- 36.Gasch A P, Huang M, Metzner S, Botstein D, Elledge S J, Brown P O. Mol Biol Cell. 2001;12:2987–3003. doi: 10.1091/mbc.12.10.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Presutti C, Ciafre S A, Bozzoni I. EMBO J. 1991;10:2215–2221. doi: 10.1002/j.1460-2075.1991.tb07757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Presutti C, Villa T, Hall D, Pertica C, Bozzoni I. EMBO J. 1995;14:4022–4030. doi: 10.1002/j.1460-2075.1995.tb00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venema J, Tollervey D. Annu Rev Genet. 1999;33:261–311. doi: 10.1146/annurev.genet.33.1.261. [DOI] [PubMed] [Google Scholar]
- 40.Iouk T L, Aitchison J D, Maguire S, Wozniak R W. Mol Cell Biol. 2001;21:1260–1271. doi: 10.1128/MCB.21.4.1260-1271.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gasch A P, Spellman P T, Kao C M, Carmel-Harel O, Eisen M B, Storz G, Botstein D, Brown P O. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santiago T C, Purvis I J, Bettany A J, Brown A J. Nucleic Acids Res. 1986;14:8347–8360. doi: 10.1093/nar/14.21.8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrick K, Garvik B, Hartwell L H. Genetics. 1997;147:19–32. doi: 10.1093/genetics/147.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]