

Abstract
Mycobacterium tuberculosis, the causative agent of human tuberculosis, and Mycobacterium bovis each express two genes, glbN and glbO, encoding distantly related truncated hemoglobins (trHbs), trHbN and trHbO, respectively. Here we report that disruption of M. bovis bacillus Calmette–Guérin glbN caused a dramatic reduction in the NO-consuming activity of stationary phase cells, and that activity could be restored fully by complementing knockout cells with glbN. Aerobic respiration of knockout cells was inhibited markedly by NO in comparison to that of wild-type cells, indicating a protective function for trHbN. TyrB10, which is highly conserved in trHbs and interacts with the bound oxygen, was found essential for NO consumption. Titration of oxygenated trHbN (trHbN⋅O2) with NO resulted in stoichiometric oxidation of the protein with nitrate as the major product of the reaction. The second-order rate constant for the reaction between trHbN⋅O2 and NO at 23°C was 745 μM−1⋅s−1, demonstrating that trHbN detoxifies NO 20-fold more rapidly than myoglobin. These results establish a role for a trHb and demonstrate an NO-metabolizing activity in M. tuberculosis or M. bovis. trHbN thus might play an important role in persistence of mycobacterial infection by virtue of trHbN′s ability to detoxify NO.
Mycobacterium tuberculosis infects over 1.8 billion persons worldwide, causing 1.5 million deaths per year. Of the infected population, 90% harbor the bacterium but do not display symptoms (1). In most healthy individuals, the initial M. tuberculosis infection is contained by the immune system, which forces the bacteria to enter a latent state, often for several years, with possible reactivation later in life. Animal models suggest that the initial event in Mycobacterium infection involves entry and multiplication within inactivated macrophages. Within the macrophages the bacteria multiply in a specialized vacuolar compartment called the phagosome. After an initial rapid growth phase, infected macrophages are surrounded and walled off by newly recruited activated macrophages to form the characteristic caseous granuloma (2). Nitric oxide (NO) produced by the activated macrophages contributes strongly to induction and maintenance of bacilli dormancy (2–11). However, tuberculous infection is in dynamic balance that teeters for years in a competition between host immunity and M. tuberculosis growth, indicating that an endogenous mechanism for NO resistance operates in the tubercule bacillus. How M. tuberculosis copes with NO therefore is a key issue.
NO plays an important role in host defense against microbial pathogens by inhibiting or inactivating key enzymes such as the terminal respiratory oxidases (12–16) and the iron/sulfur protein aconitase (17, 18), which is a member of the citric acid cycle. NO also combines at near diffusion-limited rate with superoxide produced by respiring cells to form the highly oxidizing agent peroxynitrite (19, 20).
In response to the host defense, microorganisms have developed various resistance mechanisms by which toxic effects of NO can be evaded. One such mechanism is mediated by the two-domain flavohemoglobins (FHbs) that are found in certain bacteria and yeasts. FHb detoxifies NO in a rapid reaction of the heme-bound O2 with NO to form the innocuous nitrate and ferric FHb [Fe(II)O2 + NO → Fe(III) + NO3−] (21–26). This same function is fulfilled by animal myoglobin (Mb) and Hb (27–29).
M. tuberculosis does not possess an FHb gene (30) but does encode small Hbs, called truncated Hbs (trHbs). trHbs represent a class of small oxygen-binding hemeproteins widely distributed in bacteria, plants, and unicellular eukaryotes and forming a group separate from other Hbs including FHbs (31, 32). Three distantly related groups are distinguished within the trHb family, and some organisms express trHb encoded by genes in more than one group (31). Thus far, no functional roles have been described for trHbs, although recently M. tuberculosis trHbO has been shown to enhance oxygen uptake of Escherichia coli membrane fractions (33).
In M. tuberculosis and Mycobacterium bovis two genes, glbN and glbO, encode trHbs from group I (trHbN) and group II (trHbO), respectively (34, 35). Sequence analysis showed that M. tuberculosis trHbN and trHbO are identical to their M. bovis counterparts (31). Physiological studies performed with M. bovis bacillus Calmette–Guérin (BCG) have demonstrated that trHbO is expressed throughout the growth phase, but in contrast trHbN expression is enhanced greatly during the stationary phase (35). trHbN binds oxygen with great affinity (P50 = 0.013 mm Hg at 20°C) because of a fast combination (25 μM−1⋅s−1) and slow dissociation (0.2 s−1) of O2 (35). Resonance Raman studies in conjunction with site-directed mutagenesis suggested that trHbN heme iron coordination may be optimized for performing O2/NO chemistry (36). It has been proposed that in vivo trHbN, by retaining bound O2, may ensure a low but critical level of local O2 availability necessary to afford protection against NO, assuring survival of M. tuberculosis in the hypoxic environment of the granuloma (35, 36). Here we report that trHbN actively detoxifies NO and protects aerobic respiration from NO inhibition.
Material and Methods
Bacterial Growth.
M. bovis BCG (ATCC 35734) was grown in Middlebrook 7H9 (Becton Dickinson) medium supplemented with 0.6% glycerol/10% ADC (5% BSA/2% glucose/0.85% NaCl)/0.05% Tween-80 as described (35). E. coli XL1-Blue strain (Stratagene) was grown in Luria–Bertani medium supplemented with the appropriate antibiotic.
Construction of a glbN Mutant of M. bovis BCG.
Plasmid pMG-03 was used for the disruption of glbN and was constructed as follows. A 4.7-kb BamHI fragment containing the entire glbN and lprI genes flanked by 0.7 and 3.0 kb of upstream and downstream sequences, respectively, was subcloned into the BamHI site of pBluescript SK(−) (Stratagene) to give pMG-01 (ref. 35; Fig, 1A). The glbN gene was disrupted by cloning into the unique ApaI site a 1.2-kb cassette containing the aph gene for resistance to kanamycin to give pMG-02. Finally, the 5.9-kb BamHI fragment of pMG-02 was subcloned into the BamHI site of pPR27, a mycobacterial vector carrying a temperature-sensitive origin of replication and the sacB gene (37) to give pMG-03. Plasmid DNA containing the disrupted glbN gene was electroporated into M. bovis BCG cells, and transformants were selected on 7H10 plates containing 25 μg/ml of kanamycin at 30°C. Two independent kanamycin-resistant clones were grown in 7H9 medium to mid-log and plated on 7H10 plates supplemented with ADC/0.5% glycerol/25 μg/ml kanamycin/2% sucrose at 39°C. After 8 weeks, transformants were picked and analyzed by Southern hybridization.
Complementation of the glbN Mutant.
A 1.4-kb PCR fragment carrying glbN and lprI coding sequences as well as 125 and 208 bp of 5′- and 3′-flanking sequences was amplified from M. bovis BCG (ATCC 35734) genomic DNA by using the forward primer 5′-CAGGTACCCGACCAAGGATCTGTTTGC-3′ and reverse primer 5′-TAGGTACCGTTGCATCACCCATGTTGC-3′. Both primers contain a KpnI restriction site. The amplicon was digested with KpnI and subcloned into the vector pBluescript to give pMG-11. A second plasmid, pMG-12, containing a frame-shift mutation in the lprI gene, was constructed from pMG-11 by introducing an 8-bp BclI linker at the StuI of lprI. The tyrosine found at the position 33 (B10) in trHbN was changed to a phenylalanine by site-directed mutagenesis (QuikChange site-directed mutagenesis kit, Stratagene) into pMG-11 to give pMG-13. These three plasmids were sequenced on both strands using local sequencing facilities. The three 1.4-kb fragments containing the glbN-lprI genes then were subcloned into pMV206/hyg (38) bearing the hyg gene for resistance to hygromycin to give pMG-14, pMG-15, and pMG-16. These constructs were electroporated into ΔHbN cells, and transformants were isolated at 37°C on 7H10 plates containing 50 μg/ml hygromycin.
RNA Isolation and Reverse Transcription–PCR.
Total cellular RNA was isolated from M. bovis BCG (ATCC 35734) cells harvested 24 h after the beginning of the stationary phase by using an RNeasy kit (Qiagen, Chatsworth, CA). First-strand synthesis was performed with Omniscript reverse transcriptase (Qiagen) and a reverse primer (5′-AGCCGGTGGTCCAGACTG-3′) located within the coding sequence of lprI gene according to the manufacturer's protocol. PCR amplification was performed by using Taq DNA polymerase (GIBCO/BRL, Life Technologies, Burlington, ON, Canada) and a set of primers (forward, 5′-GGAAATCCTCGGCGTCAT-3′, and reverse, 5′-AGCCGGTGGTCCAGACTG-3′) located in glbN and lprI genes, respectively.
Preparation of NO-Saturated Solutions.
NO solutions were prepared by first deoxygenating water or a 50 mM phosphate buffer (pH 7.5) containing 50 μM EDTA for at least 30 min with N2 and then saturating them with NO. The final NO concentration (1.5–2.0 mM) was measured with an NO electrode (World Precision Instruments, Sarasota, FL). Diluted NO solutions were prepared in 50 mM phosphate buffer (pH 7.5) containing 50 μM EDTA inside an MBraun Labmaster 100 glovebox using gas-tight Sample-Luer Lock Hamilton syringes. Hamilton gas-tight syringes were used to withdraw and inject NO solutions.
Determination of NO Consumption and Respiration Rates.
NO and O2 consumption was measured in a thermostated 2-ml reaction chamber (World Precision Instruments) with ISO-NO and ISO2 electrodes (World Precision Instruments), respectively. The NO and O2 electrodes were calibrated according to the manufacturer's recommendations. Cells were harvested after 24 h in stationary phase, centrifuged at 3,000 × g at 4°C for 15 min and resuspended at either 1.0 or 4.0 OD580 nm (1 A580 ≅ 1 × 108 cells per ml) in PBS buffer containing 0.05% (vol/vol) Tween-80. NO consumption was measured after the addition of NO to the cell suspension (2 ml) at 37°C. We tested the effect of adding glucose to the cell suspension and found no difference in either NO or O2 consumption.
Titration of trHbN⋅O2 with NO.
trHbN was purified as described (35). trHbN⋅O2 samples were prepared by exposing to air the ligand-free ferrous trHbN prepared anaerobically. NO was added sequentially from a saturated stock solution (1.5–2.0 mM) with a gas-tight Hamilton syringe into a 1.5-ml quartz cuvette sealed with a rubber septum and containing 1.4 ml of 15 μM trHbN⋅O2 in 50 mM phosphate buffer (pH 7.5) containing 50 μM EDTA. Spectra were recorded at 23°C with a Cary 3E spectrophotometer until equilibrium was achieved.
Determination of Nitrite and Nitrate Formed in the Reaction of trHbN⋅O2 with NO.
All solutions were prepared by using ultra pure water (Fluka) containing less than 0.000001% nitrate). trHbN⋅O2 and oxygenated horse heart Mb (Mb⋅O2, Sigma–Aldrich) were prepared at a concentration of 25 μM as described above. NO was added at a final concentration of 25 μM from a saturated solution (1.5–2.0 mM). Protein samples were denatured by heat (100°C) for 5 min and centrifuged at 13,000 × g for 5 min. The supernatants were assayed for the nitrite and nitrate content by using the Griess reagent and vanadium(III) (39). We measured no nitrate and 0–0.2 μM nitrite in the saturated NO solutions.
Stopped-Flow Kinetic Analysis.
Kinetic studies were carried out at 23°C with an Applied Photophysics SX.18MV single-wavelength stopped-flow spectrophotometer equipped with a photodiode array detector. The optical path length was 1 cm. The dead time of the apparatus was 0.9 ms. Solutions of Mb⋅O2 and trHbN⋅O2 (1–5 μM) were prepared in 50 mM phosphate buffer (pH 7.5) containing 50 μM EDTA. Protein concentrations were determined by measuring the absorbance at 417 and 542 nm (ɛ417 = 128 mM−1⋅cm−1, ɛ542 = 13.9 mM−1⋅cm−1) for Mb⋅O2 and 416 and 545 nm (ɛ416 = 94.4 mM−1⋅cm−1, ɛ545 = 10.0 mM−1⋅cm−1) for trHbN⋅O2. Kinetic traces (averages of at least five single traces of three separate experiments) taken at 409 (horse heart Mb) and 404 nm (trHbN) were analyzed with the SX.18MV operating software.
Results
Organization of the glbN Locus.
Sequence analysis of the M. bovis and M. tuberculosis genomes revealed that the genomic organization of the glbN locus is conserved (Fig. 1A; refs. 34 and 35). It also indicated the presence of a second gene (lprI), the initiation codon of which was separated from the stop codon of glbN by only 58 nucleotides, suggesting that both genes were cotranscribed. To verify cotranscription of both genes, total RNA was isolated from M. bovis BCG cells harvested 24 h after the beginning of the stationary phase and used to perform reverse transcription–PCR with a pair of primers that allows coamplification of glbN and lprI. A single product of the expected size was detected after electrophoresis on agarose gel, indicating that both genes are cotranscribed in vivo (data not shown).
Figure 1.

Inactivation of glbN from M. bovis BCG (ATCC 35734). (A) Schematic representation of the glbN locus in M. bovis before and after the insertion of the kan resistance gene. The same organization is observed in M. tuberculosis. (B) Southern blot of BamHI-digested genomic DNA showing the glbN-containing fragment of WT BCG (lane 3, expected size of 4,600 bp) and the hybridizing fragment of two glbN mutants (lanes 1 and 2, expected size of 5,800 bp). The fragment size of the mutants is consistent with the insertion of the 1.2-kb kan resistance gene. (C) Western blot analysis of total proteins (10.0 μg) from WT BCG (lane 1) and a ΔHbN mutant (lane 2) harvested 24 h after entry into stationary phase. The detection was performed by using polyclonal antibodies raised against trHbN.
Gene Disruption of glbN in M. bovis BCG.
To inactivate glbN, a 1.2-kb aph gene (kanamycin resistance) was inserted at its unique ApaI site (Fig. 1A). The BamHI fragment containing disrupted glbN was cloned in the delivering vector pPR27 and used to transform M. bovis BCG cells (Fig. 1A). This plasmid allows a gene replacement strategy that uses the counterselective properties of the sacB gene and a mycobacterial thermosensitive (ts) origin of replication (37). The presence of the ts origin of replication in conjunction with sacB facilitates the selection of mutants. Kanamycin-resistant transformants that grew at 39°C on 7H10/kanamycin-containing 2% sucrose plates were conserved for Southern blot analysis. As expected, for allelic exchange mutants, clones were found that presented a single hybridizing fragment 1.2 kb longer than in the wild-type (WT) strain (Fig. 1B). This 1.2-kb increase corresponded to the size of the aph gene that was inserted into the mutated allele. The selected glbN mutant thereafter was designated ΔHbN. Western blot analysis confirmed that ΔHbN cells no longer express trHbN (Fig. 1C).
trHbN Is Not Essential for Cell Growth.
Growth characteristics of ΔHbN cells were compared with those of its parental strain. No difference in colony morphology was visible when the strains were plated on solid medium. When grown in liquid medium with shaking, no difference was observed in the growth curves of the mutant versus the WT strains at 30 or 37°C. No significant difference in survival was noted between the two strains throughout the growth phase. These results suggest that trHbN is not essential at least under laboratory growth conditions.
trHbN Is Critical for NO Metabolism in Vivo.
To determine whether trHbN is necessary for NO metabolism, we examined NO consumption by stationary phase cells of WT and ΔHbN strains. As shown in Fig. 2A, WT cells readily consumed NO in comparison to ΔHbN cells, the latter displaying only activity that was quite similar to buffer alone. Although these results strongly suggested that trHbN was responsible for the NO-metabolizing activity, it still remained possible that lprI, not glbN, was metabolizing NO. Indeed, insertion of the kan gene in glbN may have interfered with the transcriptional or translational machinery and abolished synthesis of the LprI protein. To verify this possibility, we transformed ΔHbN cells with the replicative plasmid pMG-12 bearing glbN and inactivated lprI. As shown in Fig. 2A, NO-consuming activity was restored fully in complemented ΔHbN cells (ΔHbN:HbN). In fact, the activity was even greater than in WT cells (Fig. 2A) because of elevated levels of trHbN (Fig. 2C). Identical results were obtained when the ΔHbN strain was transformed with plasmid pMG-11 bearing WT glbN and lprI (data not shown). These results thus demonstrate that trHbN and not LprI metabolizes NO. Interestingly, as shown in Fig. 2B, when NO concentration was lowered below 1 μM, an additional NO-consuming activity was detected reproducibly in ΔHbN cells. NO concentration used in Fig. 2A (1 μM) exceeded that of trHbN in the reaction chamber (20–40 nM), indicating that NO scavenging by trHbN is not restricted to a single catalytic cycle.
Figure 2.

trHbN is required for aerobic metabolism of NO in M. bovis BCG (ATCC 35734). NO (A, 1.0 μM; B, 0.5 μM) was added to buffer or cell suspensions. NO was monitored with an NO electrode. (C) Aliquots of cell suspensions were taken for Western blot analysis of soluble proteins by using the polyclonal antibodies raised against trHbN.
TyrB10 Is Necessary for NO Detoxification by trHbN.
The role of the highly conserved B10Tyr residue in regulating the NO-consuming activity of trHbN was examined. trHbN has a unique distal heme pocket structure that allows strong stabilization of the bound oxygen molecule (koff = 0.2 s−1), largely by hydrogen bonding to the tyrosine residue at the B10 position (35, 36). Mutation of the B10Tyr residue to phenylalanine increased the oxygen dissociation rate 150-fold, indicating the importance of the tyrosine hydroxyl for tight oxygen binding (35). Fig. 2 A and B shows that when plasmid pMG-13 bearing glbN with the B10Tyr → Phe mutation was transformed into ΔHbN cells (ΔHbN:YB10F), the NO-consuming activity was not enhanced, which demonstrates that the B10Tyr hydroxyl is essential for the NO-detoxification by trHbN. Western blot analysis confirmed that failure to restore NO activity could not be attributed to a reduced expression or instability of the mutant protein in ΔHbN:YB10F cells (Fig. 2C).
trHbN Protects Aerobic Respiration in Stationary Phase Cells of M. bovis BCG.
To understand better the metabolic function of trHbN, we determined the sensitivity of WT and ΔHbN cell respiration to NO under various O2 and NO concentrations. In the absence of NO, the respiration rates of WT, ΔHbN, and complemented cells were not significantly different, indicating that trHbN does not contribute significantly to O2 uptake. NO is known to inhibit reversibly mitochondrial and bacterial respiration by competing with O2 for the active sites of the terminal oxidases. For these experiments, cell suspensions were allowed to consume O2 until the desired O2 concentration was reached. At that point, NO was injected and O2 consumption was measured. The addition of NO to a final concentration of 1 μM in a respiring WT cell suspension in which the O2 tension was ≈15 μM caused immediate inhibition of respiration (Fig. 3A). After ≈2 min, respiration resumed at its initial rate and proceeded until O2 was depleted. In contrast, inhibition elicited by 1 μM NO was much greater in ΔHbN cells, and respiration never returned to the initial rate (Fig. 3A). When NO was added at progressively higher dissolved O2 tensions, the extent and duration of inhibition was decreased in both WT and ΔHbN cells (Fig. 3 B and C). On the other hand, raising the NO concentration (Fig. 3D) increased the duration of inhibition. The inhibitory effect of NO was reversed when ΔHbN cells were complemented with plasmid pMG-12 (ΔHbN:HbN), which restores expression of trHbN (Fig. 3 A–D). Taken together, these results demonstrate that NO reversibly inhibits aerobic respiration by quiescent BCG cells, and trHbN protects respiration from NO inhibition under biologically relevant O2 concentrations.
Figure 3.

trHbN affords protection for M. bovis BCG (ATCC 35734) respiration from NO. Respiration of washed cell suspensions was measured with an oxygen electrode apparatus. A solution of NO was added at the times indicated by the arrows to give a final concentration of 1 (A–C) and 2 (D) μM NO. The O2 concentration at the times of injection is shown alongside each addition. The experiments were repeated at least three times with similar results. The duration of inhibition was determined as the period between the addition of NO and the point obtained by extrapolation of the residual respiration rate (after inhibition was relieved) and the inhibited rate.
trHbN Oxidizes NO to Nitrate.
Several Hbs have been shown to protect cells against nitrosative stress by converting NO into nitrate in the presence of oxygen. As shown in Fig. 4, titration of trHbN⋅O2 with NO in vitro resulted in immediate stoichiometric oxidation of the protein. Nitrate, the principal product of the reaction, was formed in exact molar equivalence with the trHbN⋅O2 consumed (Table 1).
Figure 4.

Titration of NO against trHbN⋅O2. Repeated additions of a saturated aqueous NO solution were made to trHbN⋅O2 (15 μM heme content). (A) Spectra were recorded after equilibrium was reached. The addition of NO led to the rapid appearance of a partial oxidized spectrum. A spectrum of the fully oxidized proteins was obtained after 14 additions of NO (14 μM NO). Subsequent additions of NO resulted in the formation of the ferric–NO complex of the proteins (data not shown). 1, trHbN⋅O2; 2, 3, and 4, 4, 8, and 12 μM NO, respectively. (B) Fraction (%) of oxidized protein was plotted against NO concentration.
Table 1.
Stoichiometric formation of nitrate in the reaction of NO with M. bovis BCG trHbN⋅O2
| Protein | NO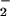 , μM , μM |
NO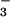 , μM , μM |
|---|---|---|
| Mb⋅O2 (25 μM) | 0.2 ± 0.1 | 25.8 ± 0.32 |
| TrHbN⋅O2 (25 μM) | 0.2 ± 0.2 | 26.0 ± 0.25 |
Horse heart Mb⋅O2 serves as a control. Aqueous NO (to a final concentration of 25 μM) was added to a solution of either trHbN⋅O2 or Mb⋅O2 (25 μM heme content). Spectra were recorded after equilibrium was reached to ensure that the reaction was completed. Final concentrations of nitrite and nitrate were determined as described in Material and Methods.
Time Courses and Rate Constant for the Oxidation of trHbN⋅O2.
The NO-induced reactions of trHbN⋅O2 and horse heart Mb⋅O2 were studied by stopped-flow spectrophotometry. Because accurate determination of NO concentration is difficult to achieve, the proteins were present in a 10-fold excess over NO to achieve reaction kinetics that were pseudo first-order with respect to trHbN⋅O2 or Mb⋅O2. Fig. 5A shows the time courses of the reactions of trHbN⋅O2 at pH 7.5 and 23°C (average of six traces). Kinetic traces measured at 409 and 404 nm resulted in the rapid appearance of aquomet form of the Mb and trHbN, respectively, without any evidence for intermediate(s). trHbN⋅O2 and Mb⋅O2 showed time courses that can be fitted to a single exponential expression. Plots of the pseudo first-order rate constants, kobs, versus [trHbN⋅O2] or [Mb⋅O2] were linear with a y intercept ≈ 0 (Fig. 5B). The second-order rate constants, obtained from the linear plots, were 46.1 and 745 μM−1⋅s−1 for the NO-mediated oxidation of Mb⋅O2 and trHbN⋅O2, respectively, showing that in vitro trHbN⋅O2 detoxifies NO ≈20-fold more rapidly than Mb⋅O2. The rate constant measured for Mb is in good agreement with those reported previously (40, 41).
Figure 5.

Time course and rate constant for the oxidation of trHbN⋅O2. (A) Time course (average of six traces) measured at 404 nm for the reaction of 0.8 μM trHbN⋅O2 with ≈0.08 μM NO at pH 7.5 and 23°C. The solid line corresponds to the best fit to the data with one exponential. (B) Plots of the rates (s−1) versus trHbN⋅O2 and horse heart Mb⋅O2 concentrations at pH 7.5 and 23°C.
Discussion
In this study we demonstrate that M. bovis trHbN, in situ in living cells, rapidly removes NO and protects aerobic respiration from NO inhibition under biologically relevant O2 tensions. By metabolizing NO to the innocuous product nitrate, trHbN also may prevent NO-mediated modification of thiol and metal centers of other critical enzymes and regulatory proteins (3, 42). In addition, it may reduce the formation of the highly reactive peroxinitrite, thereby preventing the irreversible oxidation and nitration of proteins. trHbN thus might be important to the bacilli under conditions where O2 supply is limited and NO concentration is elevated such as is anticipated in the caseous granuloma or during the reactivation of the bacilli.
Significant NO-metabolizing activity was detected in resting M. bovis BCG cells not synthesizing trHbN (Fig. 2B). A likely candidate for this additional activity is trHbO, the other trHb in M. bovis and M. tuberculosis. trHbO is expressed throughout the growth period and also readily converts NO into nitrate (unpublished results).
We observed no increase in trHbN protein levels with NO-donor treatments [nitrite, 2,2′-(hydroxynitrosohydrazono) bis-ethanimine and S-nitroso-N-acetylpenicillamine] in logarithmic or stationary phase under aerobic conditions (unpublished results). Chemicals that impose an oxidative stress such as peroxide and menadione were also without effect on trHbN expression. These results may indicate the ubiquitous presence of NO in the natural environment of M. bovis and/or that trHbN function goes beyond NO detoxification as exemplified by Mb, which both scavenges NO and transports oxygen. trHbN may constitute a first line of defense against NO, allowing cells to adjust adequately to a nitrosative stress.
The second-order rate constant (K = 745 μM−1⋅s−1) measured for the NO-mediated oxidation of trHbN⋅O2 at 23°C and pH 7.5 is close to that of E. coli FHb-O2 (850 μM−1⋅s−1; ref. 23) and is 20-fold greater than that of reaction of NO with Mb⋅O2 (35–45 μM−1⋅s−1; refs. 40 and 41). Based on their resonance Raman studies, Yeh et al. (36, 43) have postulated that the active site structure of trHbN and E. coli FHb is tailored to promote O2/NO chemistry instead of O2 delivery, which could explain their high activity with respect to Mb. In both of these bacterial Hbs, B10Tyr strongly interacts with the bound O2 and may provide a strong electronic pull for the O—O bond activation, just as in peroxidases. In line with these observations, we found that the B10Tyr is necessary for the reaction of trHbN⋅O2 with NO. Similarly, Gardner et al. (23) demonstrated that changing the B10Tyr in E. coli FHb for a Phe decreases the reaction rate by a factor of ≈30-fold while increasing the O2 dissociation rate constant 80-fold.
Dioxygenation of NO by Mb⋅O2 does not require hydrogen bonding between E7His and the bound O2 (41). In fact, substitution of the E7His by amino acids, which do not form hydrogen bonds to the bound O2, accelerated NO-mediated oxidation of Mb (41). It has been proposed that this hydrogen bond must be disrupted to allow for the entry of NO in the distal heme pocket, the process that is considered to be the rate-determining step for this reaction. In trHbN, as in other trHbs studied, x-ray (44, 45) and resonance Raman spectroscopy (31, 35, 36) indicated a very crowded pocket in which solvent access through the E7 gate is impaired totally. However, a route for ligand diffusion to and from the heme is present as a hydrophobic tunnel or cavity network through the protein matrix (31, 44, 45). Such a guiding tunnel may allow efficient diffusion of ligands to the active site. It also may accelerate the NO detoxification reaction by transiently storing several reactant NO molecules. The presence of a large and unobstructed distal heme pocket in the crystal structures of Alcaligenes FHb (46) and Vitreoscilla Hb (47) suggests that diffusion of ligand may also not be a rate-determining step in FHbs.
The conservation of the B10Tyr in trHbs, and possibly the tunnel, points to a similar function for other trHbs. It will be important to determine whether all trHbs show significant NO-scavenging activity and whether some catalyze novel reactions with specialized functions. It is noteworthy that the B10Tyr is also highly conserved in FHbs (48). Such conservation indicates a very ancient origin for the B10Tyr as well as a conserved function. Apart from a few exceptions, B10Tyr is absent in almost all modern animal and plant Hbs. The loss of the B10Tyr and therefore of a less reactive heme-bound O2 may have been mandatory to evolve a protein capable of transport and diffusion of O2.
Acknowledgments
This work was supported by Natural Sciences and Engineering Research Council Grant 46306-01 (to M.G.).
Abbreviations
- FHb
flavohemoglobin
- Mb
myoglobin
- trHbs
truncated Hbs
- BCG
bacillus Calmette-Guérin
- WT
wild type
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Styblo K. Adv Tuberc Res. 1980;20:1–63. [PubMed] [Google Scholar]
- 2.Flynn J L, Chan J. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 3.Stamler J S, Lamas S, Fang F C. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 4.Chan E D, Morris K R, Belisle J T, Hill P, Remigio L K, Brennan P J, Riches D W. Infect Immun. 2001;69:2001–2010. doi: 10.1128/IAI.69.4.2001-2010.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan E D, Chan J, Schluger N W. Am J Respir Cell Mol Biol. 2001;25:606–612. doi: 10.1165/ajrcmb.25.5.4487. [DOI] [PubMed] [Google Scholar]
- 6.MacMicking J D, North R J, LaCourse R, Mudgett J S, Shah S K, Nathan C F. Proc Natl Acad Sci USA. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan J, Tanaka K, Carroll D, Flynn J, Bloom B R. Infect Immun. 1995;63:736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan J, Xing Y, Magliozzo R S, Bloom B R. J Exp Med. 1992;175:1111–1122. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nathan C, Shiloh M U. Proc Natl Acad Sci USA. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiloh M U, Nathan C F. Curr Opin Microbiol. 2000;3:35–42. doi: 10.1016/s1369-5274(99)00048-x. [DOI] [PubMed] [Google Scholar]
- 11.Garcia I, Guler R, Vesin D, Olleros M L, Vassalli P, Chvatchko Y, Jacobs M, Ryffel B. Lab Invest. 2000;80:1385–1397. doi: 10.1038/labinvest.3780146. [DOI] [PubMed] [Google Scholar]
- 12.Brunori M. Trends Biochem Sci. 2001;26:21–23. doi: 10.1016/s0968-0004(00)01698-4. [DOI] [PubMed] [Google Scholar]
- 13.Brunori M, Giuffre A, Sarti P, Stubauer G, Wilson M T. Cell Mol Life Sci. 1999;56:549–557. doi: 10.1007/s000180050452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevanin T M, Ioannidis N, Mills C E, Kim S O, Hughes M N, Poole R K. J Biol Chem. 2000;275:35868–35875. doi: 10.1074/jbc.M002471200. [DOI] [PubMed] [Google Scholar]
- 15.Cleeter M W, Cooper J M, Darley-Usmar V M, Moncada S, Schapira A H. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 16.Brown G C, Cooper C E. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 17.Gardner P R, Costantino G, Salzman A L. J Biol Chem. 1998;273:26528–26533. doi: 10.1074/jbc.273.41.26528. [DOI] [PubMed] [Google Scholar]
- 18.Gardner P R, Costantino G, Szabo C, Salzman A L. J Biol Chem. 1997;272:25071–25076. doi: 10.1074/jbc.272.40.25071. [DOI] [PubMed] [Google Scholar]
- 19.Kissner R, Nauser T, Bugnon P, Lye P G, Koppenol W H. Chem Res Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 20.Pfeiffer S, Gorren A C, Schmidt K, Werner E R, Hansert B, Bohle D S, Mayer B. J Biol Chem. 1997;272:3465–3470. doi: 10.1074/jbc.272.6.3465. [DOI] [PubMed] [Google Scholar]
- 21.Gardner P R, Gardner A M, Martin L A, Salzman A L. Proc Natl Acad Sci USA. 1998;95:10378–10383. doi: 10.1073/pnas.95.18.10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hausladen A, Gow A J, Stamler J S. Proc Natl Acad Sci USA. 1998;95:14100–14105. doi: 10.1073/pnas.95.24.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner A M, Martin L A, Gardner P R, Dou Y, Olson J S. J Biol Chem. 2000;275:12581–12589. doi: 10.1074/jbc.275.17.12581. [DOI] [PubMed] [Google Scholar]
- 24.Gardner P R, Gardner A M, Martin L A, Dou Y, Li T, Olson J S, Zhu H, Riggs A F. J Biol Chem. 2000;275:31581–31587. doi: 10.1074/jbc.M004141200. [DOI] [PubMed] [Google Scholar]
- 25.Hausladen A, Gow A, Stamler J S. Proc Natl Acad Sci USA. 2001;98:10108–10112. doi: 10.1073/pnas.181199698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Zeng M, Hausladen A, Heitman J, Stamler J S. Proc Natl Acad Sci USA. 2000;97:4672–4676. doi: 10.1073/pnas.090083597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flogel U, Merx M W, Godecke A, Decking U K, Schrader J. Proc Natl Acad Sci USA. 2001;98:735–740. doi: 10.1073/pnas.011460298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wennmalm A, Benthin G, Petersson A S. Br J Pharmacol. 1992;106:507–508. doi: 10.1111/j.1476-5381.1992.tb14365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minning D M, Gow A J, Bonaventura J, Braun R, Dewhirst M, Goldberg D E, Stamler J S. Nature (London) 1999;401:497–502. doi: 10.1038/46822. [DOI] [PubMed] [Google Scholar]
- 30.Poole R K, Hughes M N. Mol Microbiol. 2000;36:775–783. doi: 10.1046/j.1365-2958.2000.01889.x. [DOI] [PubMed] [Google Scholar]
- 31.Wittenberg J B, Bolognesi M, Wittenberg B A, Guertin M. J Biol Chem. 2002;277:871–874. doi: 10.1074/jbc.R100058200. [DOI] [PubMed] [Google Scholar]
- 32.Moens L, Vanfleteren J, Van de Peer Y, Peeters K, Kapp O, Czeluzniak J, Goodman M, Blaxter M, Vinogradov S. Mol Biol Evol. 1996;13:324–333. doi: 10.1093/oxfordjournals.molbev.a025592. [DOI] [PubMed] [Google Scholar]
- 33. Pathania, R., Navani, N. K., Rajamohan, G. & Dikshit, K. L. (Jan. 16, 2002) J. Biol. Chem., 10.1074/jbc.M111478200. [DOI] [PubMed]
- 34.Cole S T, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon S V, Eiglmeier K, Gas S, Barry C E, III, et al. Nature (London) 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 35.Couture M, Yeh S R, Wittenberg B A, Wittenberg J B, Ouellet Y, Rousseau D L, Guertin M. Proc Natl Acad Sci USA. 1999;96:11223–11228. doi: 10.1073/pnas.96.20.11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh S R, Couture M, Ouellet Y, Guertin M, Rousseau D L. J Biol Chem. 2000;275:1679–1684. doi: 10.1074/jbc.275.3.1679. [DOI] [PubMed] [Google Scholar]
- 37.Pelicic V, Jackson M, Reyrat J M, Jacobs W R, Jr, Gicquel B, Guilhot C. Proc Natl Acad Sci USA. 1997;94:10955–10960. doi: 10.1073/pnas.94.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stover C K, de la Cruz V F, Fuerst T R, Burlein J E, Benson L A, Bennett L T, Bansal G P, Young J F, Lee M H, Hatfull G F, et al. Nature (London) 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 39.Miranda K M, Espey M G, Wink D A. Nitric Oxide. 2001;5:62–71. doi: 10.1006/niox.2000.0319. [DOI] [PubMed] [Google Scholar]
- 40.Herold S, Exner M, Nauser T. Biochemistry. 2001;40:3385–3395. doi: 10.1021/bi002407m. [DOI] [PubMed] [Google Scholar]
- 41.Eich R F, Li T, Lemon D D, Doherty D H, Curry S R, Aitken J F, Mathews A J, Johnson K A, Smith R D, Phillips G N, Jr, Olson J S. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 42.Gardner P R. Biosci Rep. 1997;17:33–42. doi: 10.1023/a:1027383100936. [DOI] [PubMed] [Google Scholar]
- 43.Mukai M, Mills C E, Poole R K, Yeh S R. J Biol Chem. 2001;276:7272–7277. doi: 10.1074/jbc.M009280200. [DOI] [PubMed] [Google Scholar]
- 44.Pesce A, Couture M, Dewilde S, Guertin M, Yamauchi K, Ascenzi P, Moens L, Bolognesi M. EMBO J. 2000;19:2424–2434. doi: 10.1093/emboj/19.11.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milani M, Pesce A, Ouellet Y, Ascenzi P, Guertin M, Bolognesi M. EMBO J. 2001;20:3902–3909. doi: 10.1093/emboj/20.15.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ermler U, Siddiqui R A, Cramm R, Friedrich B. EMBO J. 1995;14:6067–6077. doi: 10.1002/j.1460-2075.1995.tb00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarricone C, Galizzi A, Coda A, Ascenzi P, Bolognesi M. Structure (London) 1997;5:497–507. doi: 10.1016/s0969-2126(97)00206-2. [DOI] [PubMed] [Google Scholar]
- 48.Bolognesi M, Bordo D, Rizzi M, Tarricone C, Ascenzi P. Prog Biophys Mol Biol. 1997;68:29–68. doi: 10.1016/s0079-6107(97)00017-5. [DOI] [PubMed] [Google Scholar]