

Abstract
If adenines and thymines in two mutually complementary mixed-base peptide nucleic acid (PNA) oligomers are substituted with diaminopurines and thiouracils, respectively, so-called pseudocomplementary PNAs (pcPNAs) are created. Pairs of pcPNAs have recently demonstrated an ability to highly selectively target essentially any designated site on double-stranded DNA (dsDNA) by forming very stable PNA–DNA strand-displacement complexes via double duplex invasion (helix invasion). These properties of pcPNAs make them unique and very promising ligands capable of denying the access of DNA-binding proteins to dsDNA. To elucidate the sequence-unrestricted mechanism of sequence-specific dsDNA recognition by pcPNAs, we have studied the kinetics of formation of corresponding PNA–DNA complexes at various temperatures by the gel-shift assay. In parallel, the conditions for possible self-hybridization of pcPNA oligomers have been assayed by mixing curve (Job plot) and thermal melting experiments. The data indicate that, at physiological temperatures (≈37°C), the equilibrium is shifted toward the pairing of corresponding pcPNAs with each other. This finding explains a linear concentration dependence, within the submicromolar range, of the pcPNA invasion rate into dsDNA at 37°C. At elevated temperatures (>50°C), the rather unstable pcPNA duplexes dissociate, yielding the expected quadratic dependence for the rate of pcPNA invasion on the PNA concentration. The polycationic character of pcPNA pairs, carrying the duplicated number of protonated terminal PNA residues commonly used to increase the PNA solubility and binding affinity, also explains the self-inhibition of pcPNA invasion observed at higher PNA concentrations. Melting of pcPNA duplexes occurs with the integral transition enthalpies ranged from −235 to −280 kJ⋅mol−1, contributing to an anomalously high activation energy of ≈150 kJ⋅mol−1 found for the helix invasion of pcPNAs carrying four different nucleobases. A simplified kinetic model for pcPNAs helix invasion is proposed that interprets all unusual features of pcPNAs binding to dsDNA. Our findings have important implications for rational use of pcPNAs.
Keywords: double-stranded DNA|pseudocomplementary PNA|sequence-selective recognition
Peptide nucleic acids (PNAs) and their derivatives are of significant biomedical and biotechnological interest as prospective biomolecular tools for highly selective manipulation of nucleic acids (1–8). Recently, a new modification of PNAs has been introduced for sequence-unrestricted targeting of double-stranded DNA (dsDNA). Along with ordinary guanines and cytosines, these PNAs, dubbed pseudocomplementary PNAs (pcPNAs; refs. 9 and 10), carry 2,6-diaminopurines (D) and 2-thiouracils (sU) instead of adenines and thymines, respectively.
Model building revealed a steric clash between the bulky thio group of sU (or similarly modified thymine) and one of the two amino groups of D within the sU-D pair (9, 11). This clashing effect must severely destabilize the pcPNA–pcPNA duplexes whereas binding of each pcPNA oligomer to its DNA complement should not be affected or may even become stronger, as compared with unmodified PNAs (ref. 9; Fig. 1a). As a result, under typical experimental conditions with temperatures near 37°C, two pcPNA oligomers invade the complementary dsDNA site with mixed sequence of purines and pyrimidines by forming very stable double-duplex invasion complexes (ref. 9; Fig. 1b).
Figure 1.

Schematics of dsDNA recognition by pcPNAs, which carry modified nucleobases, D instead of A and sU instead of T, along with the ordinary G and C. (a) Base pairing schemes showing that a steric clash departs D and sU from each other, hence significantly obstructing the complementary interactions between thus modified PNA nucleobases. Nonetheless, they can form stable pairs with normal DNA counterparts. (b) Outline of the double-duplex invasion process, which is based on the data presented in the this paper. A pair of pcPNAs can invade the dsDNA target site only in a free form after dissociation of pcPNA duplexes. As a result, the double-duplex invasion complex forms inside the DNA duplex via the strand displacement.
This unusual mode of dsDNA recognition, i.e., the double duplex (or helix) invasion, is very promising for advancement of the antigene strategy by selectively blocking binding sites for DNA-processing proteins (9, 10, 12). It may also have various biotechnological applications, including the PNA-assisted DNA rare cleavage (10, 13–17), duplex DNA capture (8, 18, 19), topological DNA labeling (8, 20–23), direct DNA sequencing (8, 17, 24), and hybridization of molecular beacons to dsDNA (8, 25, 26). Although the previous studies have provided convincing evidence for the double-duplex invasion mode of pcPNA binding to dsDNA (9, 10), the mechanism of this process is still not well characterized, and key factors controlling the formation of pcPNA–dsDNA complexes have yet to be revealed.
Understanding the mechanism of dsDNA helix invasion by pcPNAs is critically important for future design of DNA-targeting drugs capable of effective interference with proteins working on dsDNA, and for dsDNA manipulation. To reveal the helix invasion mechanism underlying the process of the pcPNA-dsDNA complex formation, we have performed a kinetic study of mixed-base pcPNA binding to dsDNA targets complemented by the mixing curves (Job plot) and thermal melting analyses of the stability of pcPNA homoduplexes. On the basis of our experimental findings, we propose a phenomenological model for the process of pcPNA invasion into the dsDNA target. Our model explains the unusual features of pcPNA binding to dsDNA observed in our experiments and yields substantial conclusions for prospective applications of this new generation of PNAs.
Materials and Methods
PNA Oligomers.
In the present study, we used the following pcPNA oligomers: PNA I, HLys2-sUsUGDsUCDD-LysNH2; PNA II, HLys2-sUCDDDCDsUGC-LysNH2; and PNA III, HLys2-GCDsUGsUsUsUGD-LysNH2. The PNAs were synthesized as described in ref. 9. As usual, the normally neutral PNA oligomers were appended with the cationic Lys residues to increase the PNA solubility and, in particular, binding efficacy (7, 10). Consequently, all PNAs carried +4 charges: protonated ɛ-amino groups of three conjugated Lys residues plus one extra amino group of N-terminal Lys (7). Molar PNA concentrations were determined spectrophotometrically as described before (10).
DNA Targets.
In the present study, we used plasmid DNAs carrying the PNA-binding sites that follow: pSD1, matched binding site for PNA I (TTGATCAA); pSD1/m2, mismatched binding site for PNA I (TAGATCAA; the mismatched nucleobase is underlined); and pSD2, matched binding site for PNAs II and III (GCATGTTTGA). All plasmids are pUC19 derivatives with the corresponding insert being at the polylinker cloning site.
Gel-Shift Assay.
Binding of pcPNAs to the PvuII-digested plasmids (PNA-binding sites are located within the ≈350-bp-long DNA fragment) was monitored by gel electrophoresis in 0.5 × TBE (45 mM Tris/45 mM boric acid/0.5 mM EDTA, pH 8.0)-containing, nondenaturing 7.5% polyacrylamide gels. To this end, the helix-invasion reactions were carried out at different temperatures and different PNA concentrations (PNA amount was in a severalfold excess over DNA). Aliquots were withdrawn at specified times, and reactions were stopped by immediately increasing the concentration of salt to 100 mM NaCl, followed by cooling to 0°C, or freezing. After electrophoresis, DNA was visualized by ethidium bromide staining and detected with a charge-coupled device (CCD) camera connected to the IS-1000 digital imaging system/software (Alpha Innotech, San Leandro, CA). Quantitative analysis of pcPNA–dsDNA complex formation was done by measuring the normalized intensities of faster and slower migrating bands (see Fig. 2), which correspond to free DNA fragment and to its complexes with PNA, respectively (10).
Figure 2.

Polyacrylamide gel mobility shift assay of pcPNA–dsDNA complex formation. (a) Binding of PNA I to matched (pSD1; lanes 1–3) vs. mismatched (pSD1/m2; lanes 4–6) dsDNA targets (see Materials and Methods for description of target plasmids). Conditions: 20 mM sodium phosphate buffer (pH 7.0), 37°C, 10 h. PNA concentration varies from 0.15 μM (lanes 1 and 4) through 0.63 μM (lanes 2 and 5) to 2.5 μM (lanes 3 and 6). Sometimes, in experiments with this PNA, we observed the appearance of minor upper bands (like in lanes 2 and 3) corresponding to the secondary pcPNA–dsDNA complexes. Their formation could be due to the palindromic character of this particular recognition sequence allowing the formation of alternative strand-displacement structures. We did not observe such additional bands in case of the pcPNAs targeting to a nonpalindromic dsDNA site (10). (b) Kinetics of PNA I binding to pSD1 target as monitored by gel-shift assay. Conditions: 20 mM sodium phosphate buffer (pH 7.0), 40°C, 2 μM PNA.
Study of pcPNA–pcPNA Association.
PNA mixing curves (Job plots) and thermal melting curves were obtained on a Cary-Varian (Palo Alto, CA) 4G UV-visible spectrophotometer equipped with a water-circulated temperature-controlled cell holder. The quasi-equilibrium absorbance vs. temperature profiles for pcPNA–pcPNA complexes were recorded at 275 and 272 nm for PNA I, and PNAs II and III, respectively, with the total PNA strand concentration about 10 μM, using a slow heating rate of ≈0.65°C per min. The normalized UV melting curves were calculated as the ratio of the difference between the absorbance at each temperature and lower baseline to the difference between the linearly sloping lower and upper baselines, as described in ref. 27.
Results and Discussion
The pcPNA–dsDNA Binding Kinetics.
We used a gel-shift assay to quantitatively study the formation of PNA–DNA complexes. Fig. 2a shows typical patterns demonstrating that the binding of the pseudo self-complementary 8-mer PNA I to the correct dsDNA target fragment results in its significant retardation (see lanes 1–3). Fig. 2a also shows that the pcPNA–dsDNA complex formation proceeds with a high sequence specificity: pcPNA I forms stable complexes with the complementary dsDNA target but not if there is a single mismatch inside the PNA-binding site (lanes 4–6). Similar data have previously been obtained by us for binding of pcPNAs I/II (both 10-mers) to the correct vs. mismatched dsDNA target: pcPNA–dsDNA complexes highly selectively formed with the complementary DNA target and only if both pcPNAs were present (9, 10).
Fig. 2b demonstrates that the gel-shift assay can be readily used to follow the time-course of the pcPNA–dsDNA complex formation. We then quantitatively studied the pcPNA–dsDNA binding kinetics under a variety of conditions. Fig. 3a shows that the binding of pcPNAs to correct dsDNA sites obeys pseudofirst-order kinetics:
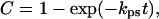 |
1 |
where C is the fraction of PNA–DNA complexes formed by the time t and kps is the pseudofirst-order rate constant, which can be determined from the slope of the kinetic curves linearized in a semilogarithmic plot. As it was expected for the double-duplex mode of pcPNA invasion, kps is not affected by the change of pH in the range from 6.4 to 7.6 (data not shown). This is in a sharp contrast to the case of pyrimidine (T,C-containing) PNAs, which bind to dsDNA via triplex invasion (28, 29).
Figure 3.

Kinetic analysis of pcPNAs binding to the complementary dsDNA targets (C is the fraction of PNA–DNA complexes formed at a certain time). (a) Semilogarithmic plot of the kinetic data on binding of PNA I (curve 1; open circles) and PNAs II/III (curve 2; filled circles) to pSD1 and pSD2 targets, respectively. Conditions: 3.5 μM pcPNA I, 10 mM sodium phosphate buffer (pH 7.0), 37°C; 1.8 μM pcPNAs II/III, 20 mM sodium phosphate buffer (pH 7.0), 45°C. Note that the apparent upward curvature observed for two kinetic curves presented here should be ascribed to the experimental uncertainties in the measurements at each point: other similar curves (not shown) exhibited the random distribution of points near a straight line or, occasionally, somewhat downward curvature. (b) Double-logarithmic plot of the fraction of PNA–DNA complexes formed by pcPNAs II/III with pSD2 targets for 20 h at 37°C (curve 1; filled circles) or for 1 h at 50°C (curve 2; open circles) at different PNA concentrations. Conditions: 20 mM sodium phosphate buffer (pH 7.0), PNA concentration varies from 0.06 μM to 0.8 μM. The slope of linear approximations is equal to 1.1 (correlation factor: 0.98) and 1.7 (correlation factor: 0.99) at 37°C or 50°C, respectively. (Inset) Percentage of PNA–DNA complexes formed by pcPNAs II/III at 50°C in a wider range of PNA concentrations.
To better understand the mechanism of the dsDNA recognition by pcPNAs, we have studied the concentration and temperature dependencies of the pcPNA invasion kinetics. The pseudofirst-order character of the pcPNA–dsDNA binding kinetics allows us to quantitatively analyze the effect of PNA concentration on the rate of PNA-to-DNA invasion by using a double-logarithmic plot. Varying the PNA concentration at a fixed incubation time and reasonably assuming that
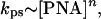 |
2 |
a straight line is expected when ln(−ln(1 − C)) is plotted vs. ln[PNA]. Indeed, by substituting Eq. 2 to Eq. 1, we obtain:
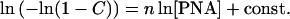 |
3 |
Eq. 3 shows that exponent n is equal to the slope of these linear plots.
We naturally expected the rate of the double-duplex invasion process performed by a pair of pcPNA oligomers to grow quadratically with the increasing total PNA concentration (when both pcPNAs are present at equal amounts). Surprisingly, our data showed that at 37°C the kps values for pcPNAs II/III demonstrated linear, rather than quadratic, dependence on the PNA concentration (line 1 in Fig. 3b) whereas the expected, nearly quadratic dependence of kps on PNA concentration was observed only at elevated temperature (line 2 in Fig. 3b). Also surprisingly, at high concentrations of pcPNAs II/III, a further increase of PNA concentration did not result in acceleration of PNA binding but, on the contrary, inhibited their binding to dsDNA (see inset in Fig. 3b). Note that a similar inhibitory effect had been previously observed for PNA I (10), as well as for polycationic pyrimidine bis-PNAs (P.E.N. and H. J. Larsen, unpublished data).
Our studies on temperature dependence of pcPNA invasion into dsDNA have also yielded unexpected results. Indeed, the value of the apparent activation energy E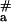 = 150 ± 22 kJ⋅mol−1 for binding of pcPNAs II/III (Fig. 4, line 2) is much higher than the value of E
= 150 ± 22 kJ⋅mol−1 for binding of pcPNAs II/III (Fig. 4, line 2) is much higher than the value of E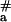 = 60–90 kJ⋅mol−1, which had been obtained for the dsDNA triplex invasion with pyrimidine PNAs (30, 31). It is also much higher than the value of E
= 60–90 kJ⋅mol−1, which had been obtained for the dsDNA triplex invasion with pyrimidine PNAs (30, 31). It is also much higher than the value of E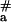 = 70 kJ⋅mol−1 reported recently for the binding of pcPNA consisting of the alternating D and sU nucleobases to [poly(dA-dT)]2 (32). Fig. 4 demonstrates that binding of pcPNA I to dsDNA at lower temperatures also occurred with very high energy of activation (E
= 70 kJ⋅mol−1 reported recently for the binding of pcPNA consisting of the alternating D and sU nucleobases to [poly(dA-dT)]2 (32). Fig. 4 demonstrates that binding of pcPNA I to dsDNA at lower temperatures also occurred with very high energy of activation (E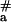 = 156 ± 16 kJ⋅mol−1; five points from the right part of curve 1 were used for this estimation), whereas a significantly lower value (E
= 156 ± 16 kJ⋅mol−1; five points from the right part of curve 1 were used for this estimation), whereas a significantly lower value (E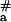 = 92 ± 29 kJ⋅mol−1; four points from the left part of the same curve) was observed at higher temperatures.
= 92 ± 29 kJ⋅mol−1; four points from the left part of the same curve) was observed at higher temperatures.
Figure 4.

The Arrhenius plots of kinetic data for binding of 0.8 μM pcPNA I (filled circles) and 0.6 μM pcPNAs II/III (open circles) to the complementary dsDNA targets in 20 mM sodium phosphate buffer (pH 7.0). To draw these graphs, the pseudofirst-order rate constants, kps, were determined at different temperatures, T. The slopes of thus obtained Arrhenius plots yield the apparent activation energies of 150 kJ⋅mol−1 for pcPNAs II/III(curve 2), and 156 or 92 kJ⋅mol−1 for pcPNA I (curve 1) at lower and higher temperatures, respectively.
To explain the surprising effects of PNA concentration and temperature on the helix invasion kinetics of mixed-base pcPNAs, we have hypothesized that, despite a steric clash between D and sU nucleobases, a pair of pcPNA oligomers can actually form a weak PNA–PNA duplex, similar to the case for pseudocomplementary mixed-base oligonucleotides at lower temperatures (11). As it is shown below, such an assumption explains the unexpected features of the pcPNAs binding to dsDNA. In the next section, we present our data that directly demonstrate the duplex formation between pcPNA oligomers.
Formation and Melting of pcPNA Duplexes.
Fig. 5a shows the mixing curves obtained for pcPNAs II/III at two different temperatures. It is clearly seen that at 24°C this pair of PNAs forms 1:1 complexes, whereas no complex formation is detected at 48°C. The hypochromicity observed for equimolar mixing of PNAs II and III lies in the range of 10–16%. We also noticed a similar although less pronounced minimum (≈7% hypochromicity) on the room temperature-mixing curve for pcPNAs 1495/1496 (data not shown), which were studied earlier (9). A weak hypochromic effect exhibited by pcPNA duplexes, which is smaller than usually observed with common PNA duplexes (33), is one of the reasons why this effect was previously overlooked (9).
Figure 5.

Formation and melting of pcPNA duplexes in 10 mM Tris⋅HCl buffer (pH 7.6) containing 10 mM NaCl. (a) Mixing curves for pcPNAs II/III obtained by variation of their molar ratios at 24°C (filled circles) and 48°C (open circles). (b) Temperature dependencies of the fraction of melted pcPNA duplexes obtained by the normalization of the absorbance vs. temperature profiles as described in Materials and Methods: PNA I, filled circles; equimolar ratio of PNAs II and III, open circles. (Inset) van't Hoff's plots obtained from the melting curves presented in the main figure (PNA I, right line; PNAs II/III, left line).
Fig. 5b shows the melting curves for PNAs I and PNAs II/III. These data additionally prove that pcPNA duplexes are rather unstable and cannot be observed at temperatures higher than 50°C. The temperature dependencies of the fraction of melted pcPNA duplexes, f, were analyzed to estimate the integral enthalpy (ΔH) of pcPNA duplex melting/formation. Considering the melting of these duplexes as the all-or-non transition, the values of the equilibrium constant of duplex formation, Kd, were determined at each temperature by using the equation (27, 33):
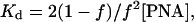 |
4 |
with [PNA] ≈ 10 μM (total strand concentration) in our experiments. Then, assuming that ΔH is a temperature-independent value, its meaning could be obtained from the slope of the van't Hoff's plots presented in the Inset of Fig. 5b. Consequently, it was found that ΔH ≈ −235 or −280 kJ⋅mol−1 for the 8-mer pcPNA I and for the 10-mer pcPNAs II/III, respectively.
Mechanism of the Double-Duplex Invasion.
On the basis of the obtained kinetic data along with the earlier evidence for the invasion of a pair of pcPNAs into the DNA duplex (9, 10), we assume that the rate-limiting step in this process consists in the formation of a transient tertiary “open” complex involving dsDNA and two pseudocomplementary PNA oligomers. We believe that such a complex is formed, as in the case of pyrimidine PNA triplex invasion (7, 28, 30, 31), because of a small-scale opening of dsDNA (DNA “breathing”; see the theoretical analysis below) and it may be schematically presented as in Fig. 1b.
Once the transient (pcPNA)2–dsDNA complex forms, the final structure in Fig. 1b, i.e., the complete double-duplex invasion complex, is formed very rapidly (within microsecond range) via synchronous zippering of two PNA–DNA duplexes. This rapid process goes downhill in the free energy landscape because at each step one DNA–DNA base pair is replaced by two PNA–DNA base pairs, each of which is at least as stable as the DNA–DNA base pair. Note that, although the latter argument is valid for both modified and normal base pairs, no temperature range exists in case of normal nucleobases, where the double-duplex invasion complexes (both transient and complete ones) are energetically favorable, as compared with two regular DNA–DNA and PNA–PNA duplexes.
We also assume that the double-duplex invasion complex should be kinetically stable even under conditions where its dissociation is thermodynamically favorable (say, after removal of pcPNAs from solution). In other words, we assume, in agreement with experiments (9, 10), the lifetime of this complex to be very large under conditions in which PNA–DNA complexes are normally studied. Therefore, we can consider the pcPNAs binding to dsDNA as virtually irreversible, which makes it possible to theoretically treat the course of pcPNA invasion into dsDNA by using a simplified scheme (see the next section).
This situation is analogous to the cases of the dissociation of linear DNA duplexes (34) or PNA–DNA triplex invasion complexes (35, 36). Indeed, the energetics of the elementary steps of these processes look very similar—dissociation of two PNA–DNA base pairs of the double-duplex invasion complex results in the restoration of one DNA base pair. Nevertheless, there are important differences between these cases. (i) PNA–DNA heteroduplex is normally more stable than DNA–DNA homoduplex (37, 38); (ii) PNA–PNA homoduplex can also form under the same conditions in case of pcPNAs.
Kinetic Consideration of the Double-Duplex Invasion Reaction.
Our phenomenological model of the pcPNA binding to dsDNA is shown in Fig. 1b. According to this model, the pcPNA–dsDNA complex formation can be described by a set of the two chemical equations (we neglect the short-living transient complexes):
 |
5 |
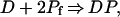 |
6 |
where Pf, Pd, D and DP represent free pcPNA oligomers, their “complementary” duplex, DNA target and the final helix invasion complex, respectively. In Eq. 6, we ignore the reverse reaction because, under our experimental conditions, this process is negligible, as compared with the forward reaction, due to the very high stability of pcPNA–dsDNA complexes (refs. 9, 10 and the previous section).
We also assume that only “free,” single-stranded pcPNAs (their equilibrium concentration being [PNA]f) can invade the DNA duplex, whereas association of pcPNAs into PNA duplexes prevents their binding to the dsDNA target. Typically, PNA is taken in excess over DNA, and, therefore, the PNA concentration can be considered as constant. Under these assumptions, Eqs. 5 and 6 yield the pseudofirst-order rate constant of the pcPNA binding to dsDNA, kps, as follows:
 |
7 |
where ko characterizes the energetics of helix invasion reaction determined predominantly by dsDNA opening (30, 31). We assume the temperature dependence of ko as exp(E′a/RT), where R is the universal gas constant and E′a is the activation energy of fluctuational openings of the DNA duplex.
Because the PNA-to-DNA helix invasion process described by Eq. 6 proceeds rather slowly, at least by one order of magnitude slower than the formation of PNA–PNA duplexes (39), we may treat the equilibrium process described by Eq. 5 as an uninterrupted one. Hence, at equilibrium we obtain:
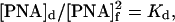 |
8 |
and
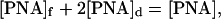 |
9 |
where Kd = Ko exp(ΔH/RT) is the pcPNA duplex stability constant (Ko is the temperature-independent coefficient), [PNA]d is the concentration of pcPNA duplexes, and [PNA] is the total pcPNA concentration. Eqs. 8 and 9 yield:
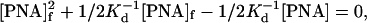 |
10 |
which can be solved with respect to [PNA]f as:
 |
11 |
Let us consider two different situations studied by us: the reaction temperature is (i) rather low and close to Tm (so that [PNA]Kd ≥ 1) or (ii) higher than Tm (so that [PNA]Kd ≪ 1). Then, for the “low-temperature” limit, Eq. 11 can be reduced to:
 |
12 |
Note that, because of a large numerical coefficient at the [PNA]Kd term in the nominator of the right part of Eq. 11, Eq. 12 is approximately valid even at T = Tm ([PNA]Kd = 1). At the “high-temperature” limit (when [PNA]Kd ≪ 1) we obtain from Eq. 11:
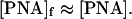 |
13 |
Substituting Eqs. 12 and 13 to Eq. 7, we see that a linear dependence of the invasion rate on the total PNA concentration is expected at the low-temperature limit:
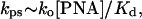 |
14 |
whereas a quadratic dependence of the invasion rate on the total PNA concentration is expected for the high-temperature limit:
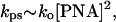 |
15 |
in full agreement with our experimental data presented in Fig. 3b.
Moreover, at the low-temperature limit, when Eq. 14 is valid, the apparent activation energy is:
 |
16 |
where ΔH is the melting enthalpy of pcPNA duplexes and E′a accounts mostly for the energetics of duplex DNA opening. Therefore, again in agreement with our experimental data presented in Fig. 4, the apparent activation energy for the process of pcPNA helix invasion at normal temperatures (T ≈ Tm = 30–40°C) should be much higher than that of pyrimidine PNAs strand-displacement reactions. The fact that, although anomalously high, E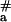 < −ΔH = 235–280 kJ⋅mol−1 in this temperature range (as estimated above) indicates that either partial melting of pcPNA duplexes is enough to form the transient PNA-DNA complex shown schematically in Fig. 1b or there may be other temperature-dependent processes that somewhat mask (or compensate) the entire effect of pcPNA duplex melting.
< −ΔH = 235–280 kJ⋅mol−1 in this temperature range (as estimated above) indicates that either partial melting of pcPNA duplexes is enough to form the transient PNA-DNA complex shown schematically in Fig. 1b or there may be other temperature-dependent processes that somewhat mask (or compensate) the entire effect of pcPNA duplex melting.
According to Eq. 15, the E value for the pcPNA invasion must be lower at higher temperatures approaching the activation energy, E′a, for small-scale openings of dsDNA. Because at normal conditions the equilibrium is strongly shifted toward the formation of the DNA duplex, only single base pairs are fluctuationally open (40–42). Therefore, the E′a value can be estimated on the basis of the DNA melting parameters as E′a ≥ |ΔHm| ≅ 30–40 kJ⋅mol−1, where ΔHm is the melting enthalpy per DNA base pair (43). The inequality takes into account the additional enthalpy (still unknown) necessary for the formation of an open region inside of a duplex as well as the fact that the activation enthalpy is normally larger than the enthalpy changes. The E′a value was also estimated from the NMR studies of dsDNA opening dynamics as E′a ≅ 40–100 kJ⋅mol−1 (44–46). Thus, it is reasonable to expect for the high-temperature pcPNA invasion the E
value for the pcPNA invasion must be lower at higher temperatures approaching the activation energy, E′a, for small-scale openings of dsDNA. Because at normal conditions the equilibrium is strongly shifted toward the formation of the DNA duplex, only single base pairs are fluctuationally open (40–42). Therefore, the E′a value can be estimated on the basis of the DNA melting parameters as E′a ≥ |ΔHm| ≅ 30–40 kJ⋅mol−1, where ΔHm is the melting enthalpy per DNA base pair (43). The inequality takes into account the additional enthalpy (still unknown) necessary for the formation of an open region inside of a duplex as well as the fact that the activation enthalpy is normally larger than the enthalpy changes. The E′a value was also estimated from the NMR studies of dsDNA opening dynamics as E′a ≅ 40–100 kJ⋅mol−1 (44–46). Thus, it is reasonable to expect for the high-temperature pcPNA invasion the E value to be around or lower than 100 kJ⋅mol−1, which is indeed the case for PNA I (see Fig. 4).
value to be around or lower than 100 kJ⋅mol−1, which is indeed the case for PNA I (see Fig. 4).
We did not observe a similar trend in case of PNAs II/III probably because the high-temperature limit for this pcPNA pair corresponds to significantly higher temperatures (≈55–60°C), which are close to the dsDNA melting transition. Note that Tuite et al. (32) studied pcPNA consisting only of D and sU. Hence, the corresponding PNA duplexes are expected to be less stable than those formed by GC-containing pcPNAs we study here. In case the pcPNA duplex is formed solely of D-sU base pairs, it must be completely melted under normal temperatures and no effect on binding kinetics (and the activation energy) is expected.
In addition to explaining the anomalous concentration and temperature dependencies of the rate of pcPNA invasion at normal temperatures, the process of pcPNA duplex formation explains also the effect of pcPNA invasion inhibition at high PNA concentrations. Indeed, pcPNA duplexes carry eight positive charges (each pcPNA oligomer contributes four positive charges; see Materials and Methods) and hence they should exhibit, like any polycation, a strong stabilizing effect on dsDNA. Therefore, the presence of heavily charged pcPNA duplexes, whose fraction increases with the growth of PNA concentration, causes self-inhibition of pcPNA invasion at some point (despite the overall increase in the concentration of “free” PNA; see Eq. 12).
Significance of the pcPNA Duplex Formation.
The formation of pcPNA duplexes that we demonstrate here entails several significant consequences for the rational use and further development of pcPNAs. First, this process should result in enhanced sequence specificity of pcPNA-dsDNA complex formation via the “stringency clamping” effect of structurally constrained probes, as was the case for site-specific DNA recognition by different DNA clamps (47, 48). Second, the pcPNA duplex formation causes the inhibition of the invasion process at elevated PNA concentration. Hence, optimization of pcPNA charges could be required in some cases to avoid these problems. Third, such a process may complicate the design of bis-pcPNAs, which is a next logical step in the pcPNA development to reduce the molecularity of strand-displacement reaction, as it was done in the case of pyrimidine bis-PNAs (28, 29, 49–51).
Evidently, the weak tendency of pcPNA oligomers toward association with each other is expected to be much more significant if they are linked into bis-pcPNA. As a result, the pcPNA oligomers may effectively clamp each other within the bis-pcPNA construct, thus hampering, instead of facilitating, their invasion. Yet, several possible solutions are envisioned to circumvent this potential problem. Synthesis of entirely modified pcPNAs carrying, besides D and sU, the pseudocomplementary analogs of G and C nucleobases (52), which should considerably decrease the associative tendency of pcPNA oligomers, is one of them.
Acknowledgments
We thank Dr. T. Yoshida for providing us the UV spectrometer, and Drs. H. Kuhn and N. E. Broude for critically reading this manuscript. This work was supported by a Human Frontier Science Program (HFSP) fellowship (E.P.), by the Lundbeck Foundation (P.E.N.) and by the National Institutes of Health.
Abbreviations
- dsDNA
double-stranded DNA
- PNA
peptide nucleic acid
- pcPNA
pseudocomplementary PNA
- D
2,6-diaminopurine
- sU
2-thiouracil
References
- 1.Nielsen P E, Egholm M, Berg R H, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 2.Uhlmann E, Peyman A, Breipohl G, Will D W. Angew Chem Int Ed Engl. 1998;37:2796–2823. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2796::AID-ANIE2796>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 3.Falkiewicz B. Acta Biochim Polon. 1999;46:509–529. [PubMed] [Google Scholar]
- 4.Winters T A. Curr Opin Mol Ther. 2000;2:670–681. [PubMed] [Google Scholar]
- 5.Ganesh K N, Nielsen P E. Curr Org Chem. 2000;4:916–928. [Google Scholar]
- 6.Ray A, Nordén B. FASEB J. 2000;14:1041–1060. doi: 10.1096/fasebj.14.9.1041. [DOI] [PubMed] [Google Scholar]
- 7.Demidov V V, Frank-Kamenetskii M D. Methods. 2001;23:108–122. doi: 10.1006/meth.2000.1112. [DOI] [PubMed] [Google Scholar]
- 8.Demidov V V. Expert Rev Mol Diagn. 2001;1:343–351. doi: 10.1586/14737159.1.3.343. [DOI] [PubMed] [Google Scholar]
- 9.Lohse J, Dahl O, Nielsen P E. Proc Natl Acad Sci USA. 1999;96:11804–11808. doi: 10.1073/pnas.96.21.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izvolsky K I, Demidov V V, Nielsen P E, Frank-Kamenetskii M D. Biochemistry. 2000;39:10908–10913. doi: 10.1021/bi000675e. [DOI] [PubMed] [Google Scholar]
- 11.Kutyavin I V, Rhinehart R L, Lukhtanov E A, Gorn V V, Meyer R B, Jr, Gamper H B., Jr Biochemistry. 1996;35:11170–11176. doi: 10.1021/bi960626v. [DOI] [PubMed] [Google Scholar]
- 12.Senior K. Drug Discov Today. 2000;5:538–540. doi: 10.1016/s1359-6446(00)01586-5. [DOI] [PubMed] [Google Scholar]
- 13.Veselkov A G, Demidov V V, Frank-Kamenetskii M D, Nielsen P E. Nature (London) 1996;379:214. doi: 10.1038/379214a0. [DOI] [PubMed] [Google Scholar]
- 14.Veselkov A G, Demidov V V, Nielsen P E, Frank-Kamenetskii M D. Nucleic Acids Res. 1996;24:2483–2488. doi: 10.1093/nar/24.13.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izvolsky K I, Demidov V V, Bukanov N O, Frank-Kamenetskii M D. Nucleic Acids Res. 1998;26:5011–5012. doi: 10.1093/nar/26.21.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demidov V V, Frank-Kamenetskii M D. In: Peptide Nucleic Acids: Protocols & Applications. Nielsen P E, Egholm M, editors. Wymondham, U.K.: Horizon Scientific Press; 1999. pp. 175–186. [Google Scholar]
- 17.Demidov V V, Frank-Kamenetskii M D. In: PNA: Methods and Protocols. Nielsen P E, editor. Totowa, NJ: Humana; 2002. , in press. [Google Scholar]
- 18.Bukanov N O, Demidov V V, Nielsen P E, Frank-Kamenetskii M D. Proc Natl Acad Sci USA. 1998;95:5516–5520. doi: 10.1073/pnas.95.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demidov V V, Bukanov N O, Frank-Kamenetskii M D. Curr Issues Mol Biol. 2000;2:31–35. [PubMed] [Google Scholar]
- 20.Kuhn H, Demidov V V, Frank-Kamenetskii M D. Angew Chem Int Ed Engl. 1999;38:1446–1449. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1446::AID-ANIE1446>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn H, Demidov V V, Frank-Kamenetskii M D. Proceedings of the 11th Conversation on Biomolecular Structure & Dynamics. 2000;2:221–225. doi: 10.1080/07391102.2000.10506625. [DOI] [PubMed] [Google Scholar]
- 22.Demidov V V, Kuhn H, Lavrentyeva-Smolina I V, Frank-Kamenetskii M D. Methods. 2001;23:123–131. doi: 10.1006/meth.2000.1113. [DOI] [PubMed] [Google Scholar]
- 23.Kuhn H, Demidov V V, Frank-Kamenetskii M D. Nucleic Acids Res. 2002;30:574–580. doi: 10.1093/nar/30.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demidov V V, Broude N E, Lavrentyeva-Smolina I V, Kuhn H, Frank-Kamenetskii M D. Chembiochem. 2001;2:133–139. doi: 10.1002/1439-7633(20010202)2:2<133::AID-CBIC133>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 25.Kuhn H, Demidov V V, Gildea B D, Fiandaca M J, Coull J M, Frank-Kamenetskii M D. Antisense Nucleic Acid Drug Dev. 2001;11:265–270. doi: 10.1089/108729001317022269. [DOI] [PubMed] [Google Scholar]
- 26.Kuhn H, Demidov V V, Coull J M, Gildea B D, Fiandaca M J, Frank-Kamenetskii M D. J Am Chem Soc. 2002;124:1097–1103. doi: 10.1021/ja0041324. [DOI] [PubMed] [Google Scholar]
- 27.Marky L A, Breslauer K J. Biopolymers. 1987;26:1601–1620. doi: 10.1002/bip.360260911. [DOI] [PubMed] [Google Scholar]
- 28.Demidov V V, Yavnilovich M V, Belotserkovskii B P, Frank-Kamenetskii M D, Nielsen P E. Proc Natl Acad Sci USA. 1995;92:2637–2641. doi: 10.1073/pnas.92.7.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn H, Demidov V V, Nielsen P E, Frank-Kamenetskii M D. J Mol Biol. 1999;286:1337–1345. doi: 10.1006/jmbi.1998.2578. [DOI] [PubMed] [Google Scholar]
- 30.Bentin T, Nielsen P E. Biochemistry. 1996;35:8863–8869. doi: 10.1021/bi960436k. [DOI] [PubMed] [Google Scholar]
- 31.Wittung P, Nielsen P, Nordén B. J Am Chem Soc. 1996;118:7049–7054. [Google Scholar]
- 32.Tuite E, Nielsen P, Norden B. J Biomol Struct Dyn. 1999;16:1361. [Google Scholar]
- 33.Tomac S, Sarkar M, Ratilainen T, Wittung P, Nielsen P E, Nordén B, Graslund A. J Am Chem Soc. 1996;118:5544–5552. [Google Scholar]
- 34.Anshelevich V V, Vologodskii A V, Lukashin A V, Frank-Kamenetskii M D. Biopolymers. 1984;23:39–58. doi: 10.1002/bip.360230105. [DOI] [PubMed] [Google Scholar]
- 35.Cherny D Y, Belotserkovskii B P, Frank-Kamenetskii M D, Egholm M, Buchardt O, Berg R H, Nielsen P E. Proc Natl Acad Sci USA. 1993;90:1667–1670. doi: 10.1073/pnas.90.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lomakin A, Frank-Kamenetskii M D. J Mol Biol. 1998;276:57–70. doi: 10.1006/jmbi.1997.1497. [DOI] [PubMed] [Google Scholar]
- 37.Egholm M, Buchardt O, Christensen L, Behrens C, Freier S M, Driver D A, Berg R H, Kim S K, Norden B, Nielsen P E. Nature (London) 1993;365:566–568. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- 38.Haaima G, Hansen H F, Christensen L, Dahl O, Nielsen P E. Nucleic Acids Res. 1997;25:4639–4643. doi: 10.1093/nar/25.22.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wittung P, Nielsen P E, Buchardt O, Egholm M, Nordén B. Nature (London) 1994;368:561–563. doi: 10.1038/368561a0. [DOI] [PubMed] [Google Scholar]
- 40.Frank-Kamenetskii M D. In: Structure & Motion: Membranes, Nucleic Acids & Proteins. Clementi E, Corongiu G, Sarma M H, Sarma R H, editors. Guilderland, NY: Adenine; 1985. pp. 417–432. [Google Scholar]
- 41.Guéron M, Kochoyan M, Leroy J L. Nature (London) 1987;328:89–92. doi: 10.1038/328089a0. [DOI] [PubMed] [Google Scholar]
- 42.Frank-Kamenetskii M D. Nature (London) 1987;328:17–18. doi: 10.1038/328017a0. [DOI] [PubMed] [Google Scholar]
- 43.Frank-Kamenetskii M D, Lazurkin Y S. Annu Rev Biophys Bioeng. 1974;3:127–150. doi: 10.1146/annurev.bb.03.060174.001015. [DOI] [PubMed] [Google Scholar]
- 44.Braunlin W H, Bloomfield V A. Biochemistry. 1988;27:1184–1191. doi: 10.1021/bi00404a018. [DOI] [PubMed] [Google Scholar]
- 45.Leroy J L, Kochoyan M, Huynh-Dinh T, Guéron M. J Mol Biol. 1988;200:223–238. doi: 10.1016/0022-2836(88)90236-7. [DOI] [PubMed] [Google Scholar]
- 46.Kochoyan M, Leroy J L, Guéron M. Biochemistry. 1990;29:4799–4805. doi: 10.1021/bi00472a008. [DOI] [PubMed] [Google Scholar]
- 47.Roberts R W, Crothers D M. Proc Natl Acad Sci USA. 1991;88:9397–9401. doi: 10.1073/pnas.88.21.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonnet G, Tyagi S, Libchaber A, Kramer F R. Proc Natl Acad Sci USA. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egholm M, Christensen L, Dueholm K L, Buchardt O, Coull J, Nielsen P E. Nucleic Acids Res. 1995;23:217–222. doi: 10.1093/nar/23.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griffith M C, Risen L M, Greig M J, Lesnik E A, Sprankle K G, Griffey R H, Kiely J S, Freier S M. J Am Chem Soc. 1995;117:831–832. [Google Scholar]
- 51.Kuhn H, Demidov V V, Frank-Kamenetskii M D, Nielsen P E. Nucleic Acids Res. 1998;26:582–587. doi: 10.1093/nar/26.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woo J, Meyer R B, Jr, Gamper H B. Nucleic Acids Res. 1996;24:2470–2475. doi: 10.1093/nar/24.13.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]