

Abstract
An ab initio protein structure prediction procedure, TOUCHSTONE, was applied to all 85 small proteins of the Mycoplasma genitalium genome. TOUCHSTONE is based on a Monte Carlo refinement of a lattice model of proteins, which uses threading-based tertiary restraints. Such restraints are derived by extracting consensus contacts and local secondary structure from at least weakly scoring structures that, in some cases, can lack any global similarity to the sequence of interest. Selection of the native fold was done by using the convergence of the simulation from two different conformational search schemes and the lowest energy structure by a knowledge-based atomic-detailed potential. Among the 85 proteins, for 34 proteins with significant threading hits, the template structures were reasonably well reproduced. Of the remaining 51 proteins, 29 proteins converged to five or fewer clusters. In the test set, 84.8% of the proteins that converged to five or fewer clusters had a correct fold among the clusters. If this statistic is simply applied, 24 proteins (84.8% of the 29 proteins) may have correct folds. Thus, the topology of a total of 58 proteins probably has been correctly predicted. Based on these results, ab initio protein structure prediction is becoming a practical approach.
The critical step in the utilization of the genome sequencing information is to assign the functions of the gene products of each genome; in this regard, protein structure can play an important role (1). With more than 60 genomes completely sequenced, several studies have predicted protein tertiary structures on a genome scale with the goal of annotating the function of the ORFs (2, 3) and investigating the distribution of protein folds among organisms (4, 5). These studies used sequence comparison methods (2, 4, 5), threading (3, 6), or homology modeling (7–9). Although powerful, they will inevitably fail if the structure of the sequence of interest has not been seen before. In contrast, an ab initio method does not explicitly use a previously determined structure, so that it in principle could predict novel protein structures. This category of protein structure prediction has become more feasible in recent years especially for small proteins, which are up to 150 residues. One of the main reasons is the development of sophisticated ways of using known protein structures. For example, the ROSETTA method proposed by Baker and coworkers (10) explicitly uses structural fragments extracted from the structural database, whereas TOUCHSTONE uses tertiary restraints derived from threading results (11). Both methods have been tested with large test sets and also in blind prediction contests (12, 13). In a similar spirit, we apply our ab initio structure prediction procedure, TOUCHSTONE, to predict the tertiary structure of all the small proteins in the Mycoplasma genitalium genome.
Methods
The TOUCHSTONE Procedure.
TOUCHSTONE uses the SICHO (side chain only) model, where the protein is represented as a lattice chain connecting the side-chain centers of mass (14). TOUCHSTONE uses two kinds of restraints derived from threading to restrict the conformational search space. The first and most important are the side-chain contact predictions obtained from a threading algorithm, PROSPECTOR (15), where consensus contacts are extracted from multiple templates even if the associated score significance is weak. As shown in CASP4 (13), the methodology can assemble novel folds from contacts extracted from sequences that do not have the same global fold as its native state. The second class of restraints are the local distance restraints derived from multiple sequence alignments and the threading of short sequence fragments that play the role of a kind of geometric type of secondary structure prediction scheme. To search conformational space, the replica exchange (RE) Monte Carlo method (16) is used. Fifty to 70 independent simulations were run for each protein, and the trajectories were clustered to establish the degree of convergence (17).
We also use a Monte Carlo sampling scheme, parallel hyperbolic sampling (PHS), to obtain another independent set of trajectories (18); subsequently, consensus cluster centroids obtained by RE and PHS are reported, thereby increasing the ability to identify the correct tertiary fold. In the PHS Monte Carlo sampling, the energy of a conformation of the protein is transformed in the following way during the simulation to smooth the energy landscape:
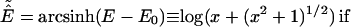 |
1 |
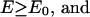 |
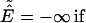 |
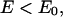 |
where E0 and E are the original protein energy of the current and next structure, respectively.
Forty replicas are run at a distinct fixed temperature, and two replicas are swapped at the acceptance probability:
 |
2 |
Finally, a heavy-atom knowledge-based potential (19) is used to rank-order the structures generated in the simulation. The structures generated from the simulations are rebuilt at atomic detail. Then the lowest energy structure in each cluster and nonclustered structures are subject to rank by the atomic potential.
If ab initio protein structure prediction is to be useful, it is critical to establish a confidence level for the prediction quality. Previously, using a representative 65-test protein set, we have shown that the number of residue contact restraints derived from threading and the number of obtained clusters are reliable indicators of the likelihood of a successful prediction (11). In the test cases, 84.8% of the cases (28 of 33 proteins) when the number of obtained clusters is five or fewer, there is a correct fold [rms deviation (rmsd) 6.5 Å or less to the native structure] obtained as one of the centroids of the clusters. Moreover, in 26 of the 33 cases when the number of predicted contacts is 100% or more relative to the length of the protein, 23 cases (88.5%) had correct folds.
Selection of the best structure (the one closest to the native fold) out of all of the generated clusters was done by using the convergence of the simulation of PHS and RE and the atomic-detailed potential. In our previous study designed to benchmark the method (18), choosing the closest cluster generated from RE and PHS sampling was an effective way to pick up the best fold from a pool of cluster centroid. For the 65 benchmark proteins, in the 43 cases (66.2%), the closest consensus cluster between PHS and RE corresponded to the best cluster (in terms of rmsd to the native structure). This pair of clusters has the minimum relative rmsd (mrrmsd) between them. As for the atomic-detailed potential, in the benchmark set, when the near-native structures do form a cluster, in 47 proteins, this procedure also picked those good candidates in the top five; furthermore it identified three near-native structures that don't belong to any cluster for three other proteins. We emphasize that the approach is fully automated and does not require any manual intervention for structure generation or selection.
Structural Comparison to Representative Proteins.
For all of the obtained clusters, structurally similar fragments in representative proteins in the Protein Data Bank (PDB) (20) were searched. The representative protein set is selected by the sequence identity criteria (35%), including 2,927 entries. The dynamic programming algorithm was used in iterative way to superimpose two protein structures to extract structurally similar fragments. Therefore the selected fragments by the algorithm can have gaps. The Z score of the rrmsd, which is less sensitive to protein size, was used to measure statistical significance of the structural similarity (21).
Results and Discussion
M. genitalium is predicted to have 484 proteins and has the smallest known genome of any free-living organism (22). Here, we have examined the 85 small proteins less than 150 amino residues in length because our ab initio prediction method performs best when applied to such small proteins. Of the 85 small proteins, 51 have annotated functions in the KEGG (Kyoto Encyclopedia of Genes and Genomes) (23) and GTOP (Genomes TO Protein structures and functions) (24) databases, whereas 50 are annotated in COG (Clusters of Orthologous Groups of proteins) (25) (KEGG and GTOP annotates M211.1 as “holosynthase” but COG doesn't give any annotation). These databases include 37 ribosomal proteins and two subunits of ATP synthase. These subunit proteins were also folded by the same procedure, but problems might be expected for those proteins whose structure in the isolated monomer differs from that in the complex.
Fold Assignments by Sequence Comparison and Threading.
Fig. 1 shows the number of obtained clusters for each of the 85 proteins, and Fig. 2 shows the number of predicted contact restraints and the number of clusters obtained in the simulation. Among the 85 proteins, the tertiary structure of 34 can be also assigned by sequence comparison or by threading with rather high confidence. These 34 are listed separately in Table 1 and Figs. 1 and 2. psi-blast (26) and fasta (27) were used as the sequence comparison methods to search the PDB with an E value of 0.01 as the threshold. For threading, PROSPECTOR (15) was used with a Z-score threshold of 10.0, with the more positive Z score being the more significant. Using these criteria, fasta and psi-blast combined identified homologous PDB structures for 27 proteins, and PROSPECTOR obtained folds for 34 proteins of which the fasta- or psi-blast-identified proteins are a subset.
Figure 1.

The number of clusters obtained for the 85 proteins. The subset of proteins whose fold can be also assigned by sequence comparison (fasta and psi-blast, using an E value 0.01) or a threading method (PROSPECTOR, Z score >10) are shown separately: cross hatched, by threading; black bar, by fasta or psi-blast. All of the latter cases are included in the former.
Figure 2.

The number of obtained clusters by RE with respect to the number of the predicted contacts from threading results. Nc, the number of contacts; L, the length of the chain. ●, proteins whose fold can be assigned by fasta or psi-blast; ▵, those whose fold can be assigned by threading but not by fasta nor psi-blast; □, the rest of the proteins.
Table 1.
Summary of the proteins that have a significant threading template hit
| ID | Template | Restraints, % | Clusters | mrrmsd, Å | Coverage, % |
|---|---|---|---|---|---|
| MG041 | 1hdn | 194.3 | 4 | 0.56 | 100.0 |
| MG052 | 1af2A | 200.0 | 5 | 0.84 | 91.5 |
| MG081 | 1eg0K | 48.9 | 6 | 2.0 | 68.4 |
| MG087 | 1fjfL | 84.9 | 3 | 3.0 | 59.7 |
| MG092 | 1fjfR | 43.8 | 4 | 1.2 | 79.5 |
| MG093 | 1div | 140.7 | 5 | 3.7 | 63.1 |
| MG124 | 1dbyA | 232.4 | 4 | 0.39 | 100.0 |
| MG129 | 1iba | 82.1 | 8 | 1.8 | 93.6 |
| MG132 | 3rhn | 237.6 | 3 | 1.5 | 96.5 |
| MG150 | 1fjfJ | 95.3 | 6 | 1.5 | 72.4 |
| MG155 | 1fjfS | 133.3 | 6 | 0.49 | 86.3 |
| MG156 | 1bxeA | 108.3 | 4 | 2.2 | 93.5 |
| MG160 | 1rip | 62.4 | 8 | 1.0 | 81.5 |
| MG161 | 487dM | 186.9 | 3 | 1.0 | 100.0 |
| MG164 | 1fjfN | 63.9 | 9 | 1.1 | 71.7 |
| MG165 | 1seiA | 155.3 | 9 | 1.6 | 100.0 |
| MG173 | 1ah9 | 192.9 | 2 | 0.45 | 100.0 |
| MG174 | 1dfeA | 97.3 | 4 | 0.24 | 100.0 |
| MG175 | 1fjfM | 39.5 | 11 | 2.6 | 54.8 |
| MG176 | 1fjfK | 160.3 | 4 | 1.4 | 71.4 |
| MG219 | 1ghc* | 33.8 | 3 | 7.3 | 92.0 |
| MG287 | 1acp* | 182.1 | 8 | 0.61 | 100.0 |
| MG325 | 1ef4A* | 98.1 | 4 | 0.86 | 88.7 |
| MG353 | 1hueA* | 84.4 | 4 | 1.6 | 71.1 |
| MG362 | 1ctf | 100.0 | 7 | 1.4 | 100.0 |
| MG363.1 | 1fjfT* | 84.1 | 6 | 0.34 | 79.5 |
| MG393 | 1aonO | 130.0 | 4 | 1.5 | 83.5 |
| MG398 | 1aqt* | 162.4 | 6 | 0.64 | 100.0 |
| MG404 | 1a91* | 88.2 | 4 | 0.69 | 63.3 |
| MG417 | 1fjfI | 70.5 | 5 | 4.3 | 70.9 |
| MG424 | 1a32 | 89.8 | 4 | 0.39 | 75.3 |
| MG446 | 1fjfP | 169.7 | 8 | 1.2 | 73.5 |
| MG449 | 1b70B | 140.3 | 9 | 2.7 | 100.0 |
| MG465 | 1a6f | 160.2 | 3 | 0.87 | 100.0 |
Template: PDB code of the protein hit by PROSPECTOR. Those with asterisks are not detected by fasta and psi-blast with the threshold (E value 0.01) used in the search. Restraints: percentage of the number of the contact restraints relative to the length of the protein. Cluster: the number of obtained clusters from RE. mrrmsd: the smallest rmsd between the clusters obtained by RE and PHS.
Although none of the native structures of the 85 proteins have been experimentally determined, it is possible to examine the performance of the prediction procedure for the 34 cases identified from threading. Among them, 20 proteins converged to five or fewer clusters. As shown in Table 1, in all but two cases (MG087 and MG175), when a protein has a threading template hit, at least one of the cluster centroids obtained from the simulations has the same fold as the predicted threaded structure with at least 60% of the structures aligned, or 60% coverage (the ratio of the maximum length of the fragment whose rmsd to the threading template structure is not larger than 6.5 Å, relative to the entire length of the protein).
One of the two cases, the C-terminal 60 residues of 1fjfM, which is the predicted template structure of MG175, does not have a stable structure. The best rmsd of the N-terminal domain (66 residues) is 6.1 Å. For another, 1fjfL, the template hit of MG087, has a 26-residue-long dangling N-terminal tail and C-terminal six-residue-long tail that is similarly impossible to reproduce. Excluding both tails, the best coverage is 78.3%. Note that 1fjfM and 1fjfL, together with several other proteins, are part of the 30S ribosomal subunit. Besides these three proteins, the 1div (MG093), 1fjf I (MG417), J (MG150), K (MG176), N (MG164), S (MG155), 1a91 (MG404), 1eg0K (081), 1hueA (MG353), 1aonO (MG393), 1a32 (MG424), and 1bxeA (MG156) templates also have dangling structures that were not reproduced well (the corresponding genes are shown in parentheses). Furthermore, 1div, the template hit of MG093, is a two-domain structure. Because our ab initio potential contains a compactness term that forces the protein to adopt a single domain structure, it is impossible to predict a two-domain structure using the current methodology. But the structures of both of the two domains are independently reproduced quite well (rmsd of the N-terminal domain of 50 residues is 4.0 Å, and rmsd of C-terminal domain 85 residues is 4.5 Å).
In many cases, the obtained cluster centroids include pairs of topological mirror-image structures, where the chirarity of turns is reversed, but helices, if present, are right-handed. The radius of gyration of the cluster centroids does not differ by much, most of the cases differ by ±1%. Usually, these cluster centroids share the same local substructures, but their global assembly is different.
We also examined the relationship between the mrrmsd from PHS and RE sampling and the coverage of these 34 proteins. The average coverage of 23 proteins whose mrrmsd is not more than 1.5 Å is 87.6%. The average coverage increases to 91.1% when 1.0 Å was taken as threshold of mrrmsd (15 proteins). If the number of clusters is combined, namely, the average coverage of 14 proteins having no more than five obtained clusters, and whose mrrmsd is not more 1.5 Å, improves to 89.2%. The average percentage of the number of the contact restraints relative to the length of the proteins in these three cases is 137.6%, 144.3%, and 150.8%, respectively, indicating that the more contact restraints are the better the simulations converge, and in many cases, they converge to the correct fold. The correlation coefficient of the percentage of restraints and the coverage is 0.56. There are four proteins with an unknown function included in these 34 proteins, namely MG129, MG219, MG353, and MG449. In Fig. 3, the predicted structures of these four proteins are shown.
Figure 3.

The predicted structures of MG129 (A), MG132 (B), MG353 (C), and MG449 (D) where the cluster centroid that has the largest overlap to the threading template is shown. The structures shown for MG132 and MG353 have the lowest energy in the entire simulation by the knowledge-based atomic-detailed potential, and that of MG449 has the second lowest energy by the potential. N terminus of the protein is colored blue, and the C terminus is red.
Proteins Without Threading Templates.
Also shown in Figs. 1 and 2 are 51 proteins for which our threading method doesn't find a structural match. Among them, 29 proteins have five or fewer clusters obtained from RE sampling. Predicted structures of four representative proteins (MG059, MG158, MG232, MG335.1) are shown in Fig. 4 together with the list of structurally similar fragments of real proteins in Table 2. The atomic-detailed potential was also used to select a probable correct fold among the obtained clusters. The number of predicted contact restraints of MG158 and MG232 are more than 100% relative to their sequence length, which may indicate that one of the obtained clusters is correct. The simulation of MG158 by RE converged to three clusters, and the structure shown in Fig. 4 has the lowest energy by the atomic-detailed potential among the cluster centroids. MG232 converged to four clusters, and the structure shown also has the lowest energy by the atomic-detailed potential and the smallest mrrmsd (2.1 Å). Both MG059 and MG198 converged to three clusters and their structures shown have the lowest energy by the atomic-detailed potential and the smallest mrrmsd (2.2 Å and 1.5 Å, respectively). MG335.1 converged to four clusters, and the structure shown has the second lowest energy by the atomic-detailed potential and the smallest mrrmsd (0.6 Å). The N-terminal 28 residues (the first helix and its N-terminus franking region) of MG335.1 are predicted to be transmembrane region by tseg (28), so that this region may have a stretched rather than the bent form in the picture. The predicted structures for all 85 proteins can be found at http://bioinformatics.danforthcenter.org/services/mgabinitio.html.
Figure 4.

Predicted structures of MG059 (small protein B homolog) (A), MG158 (50S ribosomal protein L16) (B), MG198 (50S ribosomal protein L20) (C), MG232 (50S ribosomal protein L21) (D), and MG335.1 (function unknown) (E). The functional annotations in parentheses are according to KEGG database. N terminus of the protein is colored blue, and the C terminus is red.
Table 2.
Functions of top five structurally similar fragments in PDB
| ID | Length | Structurally matched protein | Function of the protein | CATH code | Length of the fragment | rmsd (Å) of the fragment |
|---|---|---|---|---|---|---|
| MG059 | 106 | 1c9uA (444) | Soluble quinoprotein glucose dehydrogenase | 2.120.10.30 | 99 | 4.9 |
| 1iov (306) | d-Ala ligase | 2.30.35.30 | 100 | 4.9 | ||
| 1fiqC (745) | Xanthine oxidase | 8.1.51.1 | 104 | 5.6 | ||
| 1fsz (334) | Cell division protein ftsz | 3.40.50.1440 | 99 | 4.9 | ||
| 1a8l (226) | Protein disulfide oxidoreductase | 3.40.30.10 | 100 | 4.8 | ||
| MG158 | 138 | 1qfmA (705) | Prolyl oligopeptidase | 3.40.50.950 | 105 | 3.5 |
| 1qmuA (380) | Carboxypeptidase D domain II | 8.1.24.1 | 109 | 4.1 | ||
| 1obr (323) | Carboxypeptidase t. chain | 3.40.630.10 | 114 | 4.7 | ||
| 1a4sA (503) | Betaine aldehyde dehydrogenase | 3.40.605.10 | 103 | 4.2 | ||
| 1ai2 (414) | Isocitrate dehydrogenase | 3.40.718.10 | 111 | 4.7 | ||
| MG198 | 124 | 1axb (263) | Tem-1 β-lactamase | 3.40.710.10 | 89 | 3.9 |
| 1ecrA (305) | Replication terminator protein | 3.50.14.10 | 78 | 4.1 | ||
| 2gsaA (427) | Glutamate-1-semialdehyde aminomutase | 3.40.640.10 | 105 | 6.2 | ||
| 1fwyA (326) | N-acetylglucosamine 1-phosphate uridyltransferase | 8.1.72.1 | 89 | 5.0 | ||
| 2aacA (163) | Gene regulatory protein arac | 2.60.120.280 | 76 | 3.7 | ||
| MG232 | 100 | 1c8zA (265) | Tubby protein | 3.20.90.10 | 81 | 4.2 |
| 1apa (261) | Pokeweed antiviral protein | 3.40.420.10 | 89 | 4.8 | ||
| 1epaA (160) | Epididymal retinoic acid binding protein | 2.40.128.20 | 74 | 4.1 | ||
| 1thtA (294) | Myristoyl-ACP-specific thioesterase | 3.40.50.950 | 83 | 4.8 | ||
| 1smlA (266) | Metallo β lactamase | 3.60.15.10 | 86 | 5.0 | ||
| MG335.1 | 73 | 1g99A (398) | Acetate kinase | — | 59 | 2.7 |
| 1csmA (256) | Chorismate mutase | 1.10.590.10 | 66 | 3.3 | ||
| 1pbwA (184) | Phosphatidylinositol 3-kinase | 1.10.555.10 | 66 | 3.4 | ||
| 1bluA (80) | 2[4Fe-4S] ferredoxin | 3.30.70.20 | 64 | 3.1 | ||
| 1f7cA (182) | Rho GTPase regulator | — | 73 | 3.9 |
Similar structures to the consensus cluster centroid between RE and PHS are searched in PDB. Detected fragments were sorted by their relative rmsd Z scores. A representative set of proteins that do not have more than 35% sequence similarity between each other was used. The numbers in parentheses next to the PDB code are the length of the chain. CATH code: protein fold classification code in CATH database (12). — denotes that the protein has not been included in CATH yet. Proteins that have the same first three numbers in CATH code were eliminated in this table.
Membrane Proteins.
There are 15 membrane proteins [MG055 (1), MG055.2 (3), MG074 (1), MG076 (2), MG129 (1), MG131 (2), MG149.1 (2), MG233 (2), MG267 (2), MG335.1 (1), MG384.1 (3), MG389 (1), MG404 (2), MG406 (3), MG441 (1)] (the number of predicted transmembrane regions is shown in parentheses) predicted by tseg (28). In principle, the current method cannot be applied to transmembrane proteins because all of the potentials are extracted from water-soluble, globular proteins. But all but two membrane proteins (MG233 and MG129) have distinct long (presumably transmembrane) helices that consistently occur in all of the cluster centroids. Two proteins have significant threading hits: MG129 has 1iba as its threading hit for its nontransmembrane domain, and MG404 has 1a91, which is a transmembrane subunit C of F1F0 ATPase. Note that some of the transmembrane segments are too long so that they are bent in our predicted structure because of the compactness term in the potential.
Structural Match in the PDB.
Fig. 5 shows the distribution of the structurally similar fragments found in the PDB for each of the cluster centroids (both from RE and PHS). Strikingly, most of the cluster centroids have similar fragments in the PDB of significant length (on average 84.7 residues), regardless of how accurate (closest to native) the predicted structure is. In most cases, a given cluster centroid has structurally similar fragments that cover more than 60% of the given (and not necessarily correct, namely native) structure. This observation has two important consequences: First, our methodology produces protein-like structures even if the global topology is incorrect (which implies that the generic potentials in our force field generate protein-like environments). Moreover, even if the predicted structure has very similar fragments (60–120 residues in length) in the PDB, this fact does not allow us to conclude that the predicted structure is correct or that the protein with that predicted structure has a functional similarity with the proteins found in the PDB. We also have analyzed the structural similarity between real protein structures in the PDB and obtained a similar plot, as in Fig. 5. Indeed, one could see from Table 2 that functions of structurally similar proteins are diverse. By the same reason, we could not assign biological function of 34 proteins with no annotation with confidence. To infer function from the structural similarity of a part of its global structure, one has to identify the functionally important residues (1, 29).
Figure 5.

Similar fragments found in PDB for all of the clusters of 85 proteins. Top five fragments are selected for each cluster centroid (both from RE and PHS) according to the Z score of rrmsd (19). The rmsd of the fragments with respect of the fraction of their length to that of the proteins is shown.
Conclusion
We have applied our ab initio protein structure prediction procedure, TOUCHSTONE, to all of the small proteins in the M. genitalium genome. For the 85 proteins, 34 have obvious structure templates found by the threading method. For the remaining 51 proteins, RE simulation trajectories for 29 proteins converged to five or fewer clusters. If we naively apply the statistics of the test proteins (11), 84.8% of them, namely 24 proteins, may have correct folds. Thus, the topology of a total of 58 proteins (24 plus 34 proteins with threading templates) probably have been correctly predicted as one of the cluster centroids.
As the international structural genomics projects (30) progress, eventually almost all protein folds will be solved experimentally. At face value, there is an argument that threading methods may become predominant under such circumstances. Nevertheless, as this process occurs, ab initio folding will also benefit from the expansion of the structural database by improvement in the potentials and the contact prediction. In addition, in the interim it can be used to identify proteins having novel folds. As shown in CASP3 (12) and more recently in CASP4 (13) for the difficult targets, ab initio folding methods produce better models than threading method. Along this line, sparse NMR-derived restraints crosslinks (e.g., disulfide bonds) or other rapidly obtained experimental information can be effectively used to increase the yield and quality of native-like structures (14).
Based on the studies here, as well as previous benchmarking (8, 14), it is safe to conclude that our ab initio folding method is now of practical use. Although the method may not always be successful, it does yield native clusters a significant fraction of the time. However, because it does a good job of generating protein-like environments, just because a structurally related fragment is found in the structural database does not imply that it can be used for fold identification, much less for function assignment. Thus, selection of the structurally similar fragment cannot be solely used for the fold selection. Therefore, we have used a knowledge-based atomic-detailed potential and also checked the convergence of the two different series (RE and PHS) of simulations.
Acknowledgments
We thank Ms. Julie Heger for invaluable assistance in the preparation of this paper. This research was supported in part by National Institutes of Health (Division of General Medical Sciences) Grants GM-37408 and GM-48834.
Abbreviations
- RE
replica exchange
- PHS
parallel hyperbolic sampling
- rmsd
rms deviation
- mrrmsd
minimum relative rmsd
- PDB
Protein Data Bank
References
- 1.Fetrow J S, Godzik A, Skolnick J. J Mol Biol. 1998;282:703–711. doi: 10.1006/jmbi.1998.2061. [DOI] [PubMed] [Google Scholar]
- 2.Rychlewski L, Zhang B, Godzik A. Protein Sci. 1999;8:614–624. doi: 10.1110/ps.8.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones D T. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 4.Wolf Y I, Brenner S E, Bash P A, Koonin E V. Genome Res. 1999;9:17–26. [PubMed] [Google Scholar]
- 5.Gerstein M. Proteins. 1998;33:518–534. doi: 10.1002/(sici)1097-0134(19981201)33:4<518::aid-prot5>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 6.Fischer D, Eisenberg D. Proc Natl Acad Sci USA. 1997;94:11929–11931. doi: 10.1073/pnas.94.22.11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez R, Sali A. Proc Natl Acad Sci USA. 1998;95:13597–13602. doi: 10.1073/pnas.95.23.13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bates P A, Kelley L A, MacCallum R M, Sternberg M J. Proteins. 2001;45, Suppl. 5:39–46. doi: 10.1002/prot.1168. [DOI] [PubMed] [Google Scholar]
- 9.Ogata K, Umeyama H. J Mol Graph Model. 2000;18:258–272. doi: 10.1016/s1093-3263(00)00037-1. , 305–306. [DOI] [PubMed] [Google Scholar]
- 10.Simons K T, Strauss C, Baker D. J Mol Biol. 2001;306:1191–1199. doi: 10.1006/jmbi.2000.4459. [DOI] [PubMed] [Google Scholar]
- 11.Kihara D, Lu H, Kolinski A, Skolnick J. Proc Natl Acad Sci USA. 2001;98:10125–10130. doi: 10.1073/pnas.181328398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orengo C A, Bray J E, Hubbard T, LoConte L, Sillitoe I I. Proteins. 1999;37:149–170. doi: 10.1002/(sici)1097-0134(1999)37:3+<149::aid-prot20>3.3.co;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Skolnick J, Kolinski A, Kihara D, Betancourt M, Rotkiewicz P, Boniecki M. Proteins. 2001;45, Suppl. 5:149–156. doi: 10.1002/prot.1172. [DOI] [PubMed] [Google Scholar]
- 14.Kolinski A, Skolnick J. Proteins. 1998;32:475–494. [PubMed] [Google Scholar]
- 15.Skolnick J, Kihara D. Proteins. 2001;42:319–331. [PubMed] [Google Scholar]
- 16.Swendsen R H, Wang J S. Phys Rev Lett. 1986;57:2607–2609. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 17.Betancourt M R, Skolnick J. J Comp Chem. 2001;22:339–353. [Google Scholar]
- 18. Zhang, Y., Kihara, D. & Skolnick, J. (2002) Proteins, in press.
- 19.Lu H, Skolnick J. Proteins. 2001;44:223–232. doi: 10.1002/prot.1087. [DOI] [PubMed] [Google Scholar]
- 20.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betancourt M R, Skolnick J. Biopolymers. 2001;59:305–309. doi: 10.1002/1097-0282(20011015)59:5<305::AID-BIP1027>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Fraser C M, Gocayne J D, White O, Adams M D, Clayton R A, Fleischmann R D, Bult C J, Kerlavage A R, Sutton G, Kelley J M, et al. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawabata T, Fukuchi S, Homma K, Ota M, Araki J, Ito T, Ichiyoshi N, Nishikawa K. Nucleic Acids Res. 2002;30:294–298. doi: 10.1093/nar/30.1.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatusov R L, Natale D A, Garkavtsev I V, Tatusova T A, Shankavaram U T, Rao B S, Kiryutin B, Galperin M Y, Fedorova N D, Koonin E V. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson W R, Lipman D J. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kihara D, Shimizu T, Kanehisa M. Protein Eng. 1998;11:961–970. doi: 10.1093/protein/11.11.961. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann K, Bucher P, Falquet L, Bairoch A. Nucleic Acids Res. 1999;27:215–219. doi: 10.1093/nar/27.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burley S K. Nat Struct Biol. 2000;7,Suppl.:932–934. doi: 10.1038/80697. [DOI] [PubMed] [Google Scholar]