

Abstract
Titanium binding peptides are useful tools for material functionalization in both biomedical and nanotechnology applications because of their ability to attach selectively to titanium surfaces. In this work, we investigate the adsorption behavior of a series of 360 six amino acids long peptides obtained by permutations of titanium binding peptide residues, RKLPDA, on hydroxylated anatase 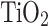 (101) surfaces using extensive atomistic molecular dynamics (MD) simulations, with the purpose identifying sequences with stronger adsorption affinity to titanium. Our results show that small changes in amino acid order can significantly affect both binding strength and structural conformations. Peptides with arginine at the N-terminus and lysine or aspartic acid near the C-terminus tended to exhibit more stable adsorption. The clustering and radial distribution function (RDF) analyzes revealed different binding modes and key atomic interactions, with nitrogen-containing groups and, in some cases,
(101) surfaces using extensive atomistic molecular dynamics (MD) simulations, with the purpose identifying sequences with stronger adsorption affinity to titanium. Our results show that small changes in amino acid order can significantly affect both binding strength and structural conformations. Peptides with arginine at the N-terminus and lysine or aspartic acid near the C-terminus tended to exhibit more stable adsorption. The clustering and radial distribution function (RDF) analyzes revealed different binding modes and key atomic interactions, with nitrogen-containing groups and, in some cases,  ions playing a significant role in the anchoring of peptides to the surface. These findings suggest a detailed sequence-level understanding of peptide-
ions playing a significant role in the anchoring of peptides to the surface. These findings suggest a detailed sequence-level understanding of peptide-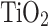 interactions and can guide the design of improved peptides for titanium functionalization.
interactions and can guide the design of improved peptides for titanium functionalization.
Keywords: Titanium dioxide, Peptides, Adsorption, Molecular dynamics
Subject terms: Computational chemistry, Molecular dynamics, Atomistic models, Organic-inorganic nanostructures
Introduction
Solid binding peptides (SBPs) are short amino acid sequences that can specifically recognize and bind to a wide range of solid surfaces. In general, SBPs bind to solid surfaces via multiple non-covalent interactions. In physiological environments, SBPs can enhance the biocompatibility of hybrid materials and be efficiently engineered to select and recognize specific interfaces. SBPs can be used to initiate and control the synthesis of composite materials or as molecular linkers to guide the simple and directed immobilization of biomolecules on solid surfaces1–3. These features make SBPs attractive for manufacturing bioinspired materials in diagnostic and therapeutic applications. SBPs have been extensively developed for a diverse range of applications in the areas of biotechnology, bio-sensing, biocatalysis, drug delivery, vaccine development, biomedical implants, bioimaging, and cell capture4–6.
Standard inert materials used in the production of orthopedic and dental implants include titanium (Ti) and titanium alloys. Due to titanium’s its excellent mechanical qualities, biocompatibility, and corrosion resistance, it is the perfect material for implantable devices. However, the absence of recognizable moieties on the implant prevents cell activities from being regulated and may induce a foreign body reaction to the implant, causing infections due to biofilm formation and bacterial adhesion. Achieving adhesion and selectivity at the interface of biomacromolecules and inorganic materials is vital to the construction of well-organized hybrid materials. The natural adhesion of peptides and proteins is the result of the evolutionary process. As such, these solid-binding peptides may not possess the required properties for biomedical applications.
Titanium-binding peptides have gained significant interest because of their potential applications in various fields, particularly biomedicine and material science. No specific binders have evolved for titanium in nature. Therefore, it is important to design novel peptides that specifically bind to titanium. There are several techniques to identify and tailor SBPs, such as isolating the functional domains of naturally occurring proteins, creating customized peptides with amino acid sequences that are not present in nature and have desired binding properties to the solid using biophysical and biological principles and computational methods (de novo design), and combinatorial phage display method, where a diverse set of random amino acid sequences are tested and a small number of candidates selected based on how well they bind to the target material. Other designed peptides are inspired by biomineralization, where the interaction of precursor ions interacts with biomacromolecules, which results in the growth of some inorganic layers. Moreover, throughput computational screening tools like Rosetta, alphafold, etc., are increasingly used to guide the design of peptides and proteins.
Many studies have shown the adsorption of different peptides on titanium7. An established titanium-binding peptide (TBP), RKLPDA (min TBP-1), was introduced by Sano and Shiba8. First, they discovered a dodecamer sequence, RKLPDAPGMHTW, using the phage display method. They realized that the first six residues (RKLPDA), or the so-called min TBP-1, play a central role in the binding of dodecamer to Ti, and, by isolating the hexamer RKLPDA, they found that it binds as strongly as dodecamer. Min TBP-1 is known for its strong affinity for titanium surfaces, which makes it useful to improve the properties and biocompatibility of titanium-based materials. One end of the TBP specifically recognizes the surface of titanium, while the other end can be used to link with various bioactive molecules. Watanabe et al., by inserting min TBP-1 into the silk fibroin (SF) protein, prepared a titanium functionalizing material (TiBP-SF), which improved bone formation activity in osteoblasts-like cells (bone synthesizing) when coated on the surface of Ti plates9. In another study, Yoshinari et al. evaluated the binding behavior of conjugated molecules consisting of min TBP-1 and antimicrobial peptides on titanium surfaces in order to investigate the bioactivity of Porphyromonas gingivalis bacteria involved in inflammatory gum and peri-implant diseases. Their results indicated that modifying the titanium surface improved the antimicrobial activity of the surface against P. gingivalis and is a promising method to reduce biofilm formation on Ti surfaces10. Over the past few decades, several peptide tags and chemical modifications have been created torotein immobilize protein molecules. They have been used to purify proteins and monitor enzymatic reactions on lab-on-a-chip devices11–13. The orientation of the immobilized polymerase enzyme on a solid-state device has a significant impact on its enzymatic activity14. By coupling min TBP-1 with DNA polymerase, Hirokazu et al. designed a min TBP-1-tagged DNA polymerase that could not only stably immobilize DNA polymerase on the 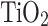 surface but also maintain its enzymatic activity15.
surface but also maintain its enzymatic activity15.
In general, SBPs bind to solid surfaces via multiple non-covalent interactions. The success of finding the most suitable peptide for the given task depends on understanding the interactions between amino acids and the target material, and the great challenge with effectively utilizing SBPs is in controlling the relationship between peptide sequences and their functionality. Various factors, including the surface topography of the solid and surrounding solution conditions, influence the binding strength of solid binding peptides. Sampath et al. highlighted the importance of the surface chemistry of  on the binding strength of peptides and, in particular, the distribution of hydroxyl groups due to the dissociation of water molecules at the interface16. They calculated the binding free energies of the 6mer, min TBP-1 (RKLPDA), and 12mer (RKLPDAPGMHTW) on neutral hydroxylated (NH) (
on the binding strength of peptides and, in particular, the distribution of hydroxyl groups due to the dissociation of water molecules at the interface16. They calculated the binding free energies of the 6mer, min TBP-1 (RKLPDA), and 12mer (RKLPDAPGMHTW) on neutral hydroxylated (NH) (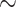 −10 and
−10 and  −12 kJ/mol) and negative non-hydroxylated (NeNH) (−23 and 21 kJ/mol) of rutile surfaces using molecular dynamics (MD) simulations. In agreement with prior experiments and simulations, they found that these two peptides bind with almost the same strength to the same surface. However, their binding free energies are significantly different when it comes to different surfaces of rutile with different charges. The peptides adsorbed weaker to the roxylatedsurface (NeNH) when they bound primarily through the arginine (R) and lysine (K) residues, whereas on the surface with higher affinity of peptides (NH), they bound primarily through the arginine and aspartate (D) amino acids. This aligns with previous experimental studies showing that the binding free energy is lower when the residues involved are arginine and lysine compared to binding with arginine and aspartate, which results in higher binding strengths. This study denoted the importance of having detailed information about the surface (such as charge, coverage with hydroxyl, etc.) of reactive materials such as titania. However, a pivotal determinant in the binding process is the composition of these peptides, encompassing the sequence of amino acids, structural conformation, and charge. Hayashi et al. elucidated the underlying mechanism of binding of ferritin molecules whose N-terminal domain is endowed with min TBP-1 and titanium surface as substrate. They showed that min TBP-1 maintains its specificity towards the titanium surface even after being introduced into the N-terminal domain of ferritin. Also, by conducting adhesion force analysis using atomic force microscopy (AFM), they showed that electrostatic interactions between R and D groups of min TBP-1 and the titanium surface, and they strongly bound to charges originating from the protonation and deprotonation of the surface groups of a Ti substrate, in agreement with the mechanism proposed by Sano and Shiba17. Mirau et al., using Nuclear Overhauser Effects (NOEs) NMR spectroscopy, determined the three-dimensional structure of the bound peptide at the surface of titania , which adopts a C-shaped conformation (in agreement with Sano and Shiba), realizing that lysine has little or no effect on this bound state8,18. By performing Saturation Transfer Difference (STD) NMR studies, Suzuki et al. characterized the interactions of a min TBP-1 with
−12 kJ/mol) and negative non-hydroxylated (NeNH) (−23 and 21 kJ/mol) of rutile surfaces using molecular dynamics (MD) simulations. In agreement with prior experiments and simulations, they found that these two peptides bind with almost the same strength to the same surface. However, their binding free energies are significantly different when it comes to different surfaces of rutile with different charges. The peptides adsorbed weaker to the roxylatedsurface (NeNH) when they bound primarily through the arginine (R) and lysine (K) residues, whereas on the surface with higher affinity of peptides (NH), they bound primarily through the arginine and aspartate (D) amino acids. This aligns with previous experimental studies showing that the binding free energy is lower when the residues involved are arginine and lysine compared to binding with arginine and aspartate, which results in higher binding strengths. This study denoted the importance of having detailed information about the surface (such as charge, coverage with hydroxyl, etc.) of reactive materials such as titania. However, a pivotal determinant in the binding process is the composition of these peptides, encompassing the sequence of amino acids, structural conformation, and charge. Hayashi et al. elucidated the underlying mechanism of binding of ferritin molecules whose N-terminal domain is endowed with min TBP-1 and titanium surface as substrate. They showed that min TBP-1 maintains its specificity towards the titanium surface even after being introduced into the N-terminal domain of ferritin. Also, by conducting adhesion force analysis using atomic force microscopy (AFM), they showed that electrostatic interactions between R and D groups of min TBP-1 and the titanium surface, and they strongly bound to charges originating from the protonation and deprotonation of the surface groups of a Ti substrate, in agreement with the mechanism proposed by Sano and Shiba17. Mirau et al., using Nuclear Overhauser Effects (NOEs) NMR spectroscopy, determined the three-dimensional structure of the bound peptide at the surface of titania , which adopts a C-shaped conformation (in agreement with Sano and Shiba), realizing that lysine has little or no effect on this bound state8,18. By performing Saturation Transfer Difference (STD) NMR studies, Suzuki et al. characterized the interactions of a min TBP-1 with  nanoparticles, which have a negative zeta potential on the surface. They found that the positively charged guanidyl group of arginine (R) and the amino group of lysine (K) play key roles in interaction with the
nanoparticles, which have a negative zeta potential on the surface. They found that the positively charged guanidyl group of arginine (R) and the amino group of lysine (K) play key roles in interaction with the  surface due to electrostatic interactions between the negative surface and positive sidechains. However, they did not find the aspartic acid (D) residue binding to the surface significantly different from the binding mechanism that Sano and Shiba initially proposed19. Brandt et al. performed unbiased MD and Metadynamics simulations of amino acids’ side chains and min TBP-1 on the rutile (100) surface to investigate the collective effects of amino acids on peptide adsorption. They found out that the total adsorption free energy of the peptide is −11.3 kJ/mol, which is stronger than the individual contributions of side chain amino acids20. They have also mentioned that the compact binding mode is stabilized on the surface due to the salt bridge formation between ARG and ASP amino acids. Schneider et al. manifested that the reason behind the selectivity and binding preference of min TBP-1 in Ti over Si is primarily influenced by solvent-mediated interactions at the interfaces. Using molecular dynamics simulations, they revealed that the min TBP-1 peptide binds selectively to titanium over silicon due to variations in local solvent structure. Adsorption free energies and adhesion forces on rutile and silicon surfaces were quantified using Metadynamics and steered MD simulations, aligning well with experimental results. Unlike previous assumptions focusing on electrostatics, the study emphasized the peptide’s ability to sense the interfacial solvent structure at an atomic scale21. Polimeni et al. investigated the dynamics of the AMRKLPDAPGMHC peptide sequence at the anatase (
surface due to electrostatic interactions between the negative surface and positive sidechains. However, they did not find the aspartic acid (D) residue binding to the surface significantly different from the binding mechanism that Sano and Shiba initially proposed19. Brandt et al. performed unbiased MD and Metadynamics simulations of amino acids’ side chains and min TBP-1 on the rutile (100) surface to investigate the collective effects of amino acids on peptide adsorption. They found out that the total adsorption free energy of the peptide is −11.3 kJ/mol, which is stronger than the individual contributions of side chain amino acids20. They have also mentioned that the compact binding mode is stabilized on the surface due to the salt bridge formation between ARG and ASP amino acids. Schneider et al. manifested that the reason behind the selectivity and binding preference of min TBP-1 in Ti over Si is primarily influenced by solvent-mediated interactions at the interfaces. Using molecular dynamics simulations, they revealed that the min TBP-1 peptide binds selectively to titanium over silicon due to variations in local solvent structure. Adsorption free energies and adhesion forces on rutile and silicon surfaces were quantified using Metadynamics and steered MD simulations, aligning well with experimental results. Unlike previous assumptions focusing on electrostatics, the study emphasized the peptide’s ability to sense the interfacial solvent structure at an atomic scale21. Polimeni et al. investigated the dynamics of the AMRKLPDAPGMHC peptide sequence at the anatase ( ) 100 surface using unbiased MD simulations. The adsorption process begins with the peptide diffusing from the bulk water phase toward the
) 100 surface using unbiased MD simulations. The adsorption process begins with the peptide diffusing from the bulk water phase toward the  surface, followed by its attachment to the surface . This attachment is facilitated by interfacial water layers, which interact with the charged side chain groups of the peptide22.
surface, followed by its attachment to the surface . This attachment is facilitated by interfacial water layers, which interact with the charged side chain groups of the peptide22.
In this study, we address two major gaps in studies of the adsorption of titanium binding peptides on titanium surfaces. Although previous investigations have focused extensively on the adsorption behavior of the RKLPDA (minTBP-1) peptide on rutile surfaces, adsorption on anatase has been much less studied. This is significant given the structural and surface chemistry differences between rutile and anatase, as well as the fact that the anatase form is prevalent in small nano-sized  nanoparticles. Furthermore, while the role of individual amino acids in the RKLPDA sequence has been explored using different experimental approaches and alanine substitution, the effect of sequence order on adsorption strength has not been systematically studied. Here, we explore 360 different permutations of the six amino acids in RKLPDA to identify those with stronger adsorption than the original sequence, RKLPDA. We first generated all 720 possible permutations of the six amino acids in RKLPDA (R, K, L, P, D, A). To avoid redundancy, we treated reverse sequences as equivalent in terms of composition. For example, RKLPDA and ADPLKR contain the same residues in opposite order. However, because the peptide backbone is not symmetric upon reversion and because the peptides in our simulations are modeled in zwitterionic form with charged N- and C-termini, reversed sequences are not structurally identical. Despite this distinction, we chose to include only one sequence from each reverse pair for computational feasibility. The selection was made arbitrarily during sequence building. This resulted in a final dataset of 360 unique, and directionally distinct sequences, with about uniform distribution of amino acids over the six positions in the peptide. Although many studies of adsorption involved biased simulations such as umbrella sampling or metadynamics to compute the potential of mean force and adsorption free energies, in this work we utilized unbiased atomistic MD. Unbiased simulations have the advantages of more straightforward evaluation of canonical averages such as radial distribution functions, statistics of different events, kinetic properties, and their comparison across different systems. Through this approach, we aim to provide a comprehensive understanding of sequence-dependent adsorption behavior and to identify peptides with enhanced affinity for
nanoparticles. Furthermore, while the role of individual amino acids in the RKLPDA sequence has been explored using different experimental approaches and alanine substitution, the effect of sequence order on adsorption strength has not been systematically studied. Here, we explore 360 different permutations of the six amino acids in RKLPDA to identify those with stronger adsorption than the original sequence, RKLPDA. We first generated all 720 possible permutations of the six amino acids in RKLPDA (R, K, L, P, D, A). To avoid redundancy, we treated reverse sequences as equivalent in terms of composition. For example, RKLPDA and ADPLKR contain the same residues in opposite order. However, because the peptide backbone is not symmetric upon reversion and because the peptides in our simulations are modeled in zwitterionic form with charged N- and C-termini, reversed sequences are not structurally identical. Despite this distinction, we chose to include only one sequence from each reverse pair for computational feasibility. The selection was made arbitrarily during sequence building. This resulted in a final dataset of 360 unique, and directionally distinct sequences, with about uniform distribution of amino acids over the six positions in the peptide. Although many studies of adsorption involved biased simulations such as umbrella sampling or metadynamics to compute the potential of mean force and adsorption free energies, in this work we utilized unbiased atomistic MD. Unbiased simulations have the advantages of more straightforward evaluation of canonical averages such as radial distribution functions, statistics of different events, kinetic properties, and their comparison across different systems. Through this approach, we aim to provide a comprehensive understanding of sequence-dependent adsorption behavior and to identify peptides with enhanced affinity for  anatase surfaces.
anatase surfaces.
Results and discussions
Surface separation distance and end-to-end distance analysis
We first analyze the behavior of the peptides near the 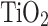 surface in terms of the distance between the center of mass of the peptides and the
surface in terms of the distance between the center of mass of the peptides and the 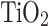 surface (Surface Separation Distance, SSD), and End-to-End Distances (EED) of the peptides. The
surface (Surface Separation Distance, SSD), and End-to-End Distances (EED) of the peptides. The  surface was determined as the layer of the outmost Ti atoms of the slab. EED was calculated as the distance the between
surface was determined as the layer of the outmost Ti atoms of the slab. EED was calculated as the distance the between  atoms of the first and last amino acids in the sequence, as shown in Fig. 1.
atoms of the first and last amino acids in the sequence, as shown in Fig. 1.
Fig. 1.

Components of the simulated systems for RKLPDA (min TBP-1) peptide. (a) simulation box; slab of anatase 101 hydroxylated surface, water shown as ice blue area), RKLPDA peptide, 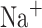 and
and  ions represented as green and blue spheres. (b) sketch of the RKLPDA peptide (c) respresentation of composing amino acids of RKLPDA peptide.
ions represented as green and blue spheres. (b) sketch of the RKLPDA peptide (c) respresentation of composing amino acids of RKLPDA peptide.
An example of the time evolution of SSD and EED for some selected peptides is shown in Fig. 2.
Fig. 2.

Examples of SSD and EED as function of the simulation time for permanently bound (PLARKD and AKPLDR), repeatedly binding and unbinding (KPALDR and LRPAKD), and weakly binding (DPAKRL and RDLKAP) peptides.
We conventionally determined the adsorbed (bound) state of the peptide if SSD is less than 1 nm. When the peptide is not bound to the surface, it changes SSD between close (within 1 nm) and far (about 2.5 nm) states on a nanosecond time scale, repeatedly approaching the surface during 600 ns simulation. We observed that, depending on the sequence, some peptides already become bound to the surface after first contact, other peptides adsorb and desorb several times, and some peptides do not stay near the surface any longer. We have ranked the binding potency of the peptides by determining the percentage of time when they stay bound to the surface with SSD below 1 nm during the 600 ns trajectory. The complete ranking list of all 360 peptides according to this criterion is given in Table S1 of the Supporting Information. The maximum value of approximately ∼2.5 nm in the SSD plots corresponds to half the simulation box length in the z-direction (excluding the slab thickness). It is worth mentioning that even when peptides travel to the opposite side of the box, they effectively interact with the other top surface of the slab, which is identical.
Figure S1 of the Supporting Information represents the SSD and EED data for all 360 peptides, along with the percentage of simulation time during which the SSD remains below 1 nm during the 600 ns trajectory. The analysis reveals that 138 out of 360 sequences spent more than 50% of the simulation time close to  .
.
To gain insight into how adsorption affects the conformation of peptides, we analyzed their end-to-end distance separately for bound and unbound states. Table 1 presents the average EED values of peptides adsorbed to the surface (SSD < 1 nm, denoted 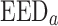 ) and desorbed (SSD
) and desorbed (SSD 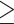 1 nm,
1 nm,  ), as well as standard deviations of EEDs for 50 top-ranking peptides according to their adsorption potency. The comparison of
), as well as standard deviations of EEDs for 50 top-ranking peptides according to their adsorption potency. The comparison of 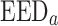 and
and  reveals several interesting trends. In many sequences,
reveals several interesting trends. In many sequences, 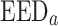 is noticeably lower than
is noticeably lower than  , indicating that peptides tend to adopt more compact conformations when adsorbed. This compaction likely reflects structural rearrangement upon surface binding, where residues come closer together to maximize interaction with the surface, either through side-chain contacts or terminal interactions. For example, PAKRLD and RPLAKD both show low EED values in the bound state consistent with the compact structures observed in cluster analysis (
, indicating that peptides tend to adopt more compact conformations when adsorbed. This compaction likely reflects structural rearrangement upon surface binding, where residues come closer together to maximize interaction with the surface, either through side-chain contacts or terminal interactions. For example, PAKRLD and RPLAKD both show low EED values in the bound state consistent with the compact structures observed in cluster analysis ( 0.5–0.65 nm, see below). In contrast, peptides such as ADKPLR and KALPDR maintain higher EED values during adsorption (
0.5–0.65 nm, see below). In contrast, peptides such as ADKPLR and KALPDR maintain higher EED values during adsorption ( 1.4 nm), reflecting more extended or linear conformations even when in close proximity to the surface. These differences likely stem from sequence-specific properties: for instance, proline is known to introduce rigidity in the peptide backbone because of its cyclic structure, which restricts rotation and can prevent compact folding. Likewise, the location of charged residues (such as ARG, LYS, or ASP) can influence electrostatic interactions and stabilize certain conformations over others. Interestingly, a few sequences show little change in EED between bound and unbound states (e.g., RPKADL or KPLDAR), suggesting that some peptides retain similar structural characteristics regardless of their proximity to the surface. This suggests that some peptides may have intrinsically rigid structures that naturally favor surface binding without requiring major conformational changes. Overall, our results highlight that adsorption-induced structural changes are strongly influenced by the peptide sequence. Measurement of end-to-end distance provides a straightforward yet informative way to capture these conformational differences. Peptides that adopt more compact structures upon adsorption may interact more effectively with the surface by allowing multiple residues or termini to make contact simultaneously with the suitable fragment of the surface structure. These insights can support the rational design of peptides with enhanced surface-binding properties for material functionalization and related applications.
1.4 nm), reflecting more extended or linear conformations even when in close proximity to the surface. These differences likely stem from sequence-specific properties: for instance, proline is known to introduce rigidity in the peptide backbone because of its cyclic structure, which restricts rotation and can prevent compact folding. Likewise, the location of charged residues (such as ARG, LYS, or ASP) can influence electrostatic interactions and stabilize certain conformations over others. Interestingly, a few sequences show little change in EED between bound and unbound states (e.g., RPKADL or KPLDAR), suggesting that some peptides retain similar structural characteristics regardless of their proximity to the surface. This suggests that some peptides may have intrinsically rigid structures that naturally favor surface binding without requiring major conformational changes. Overall, our results highlight that adsorption-induced structural changes are strongly influenced by the peptide sequence. Measurement of end-to-end distance provides a straightforward yet informative way to capture these conformational differences. Peptides that adopt more compact structures upon adsorption may interact more effectively with the surface by allowing multiple residues or termini to make contact simultaneously with the suitable fragment of the surface structure. These insights can support the rational design of peptides with enhanced surface-binding properties for material functionalization and related applications.
Table 1.
Peptides binding ranking according to the percentage of time to have SSD value less than 1 nm over production part of the simulation (% bound) together with average EED in adsorption ( 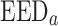 ) and desorption (
) and desorption (  ) region. The standard deviation of EED values in adsorption
) region. The standard deviation of EED values in adsorption  and desorption
and desorption  region are shown as well.
region are shown as well.
| Sequence | % bound |  |
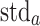 |
 |
 |
Sequence | % bound |  |
 |
 |
 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PLARKD | 100.00 | 1.16 | 0.22 | 1.15 | - | RPKLDA | 87.53 | 0.74 | 0.16 | 0.74 | 0.20 |
| RLDKAP | 99.56 | 0.98 | 0.21 | 1.30 | 0.25 | PDKRLA | 87.53 | 1.21 | 0.25 | 1.39 | 0.15 |
| DPKLRA | 99.14 | 0.94 | 0.17 | 1.49 | 0.15 | KRALDP | 87.47 | 1.37 | 0.10 | 1.09 | 0.22 |
| PKRALD | 98.36 | 1.15 | 0.11 | 1.32 | 0.18 | KLRPDA | 87.30 | 1.15 | 0.19 | 1.26 | 0.19 |
| RPLDKA | 98.09 | 0.79 | 0.20 | 1.20 | 0.21 | APDKLR | 87.26 | 0.87 | 0.21 | 1.07 | 0.23 |
| PRLKAD | 97.90 | 1.31 | 0.26 | 1.46 | 0.13 | DPALKR | 87.04 | 0.92 | 0.18 | 1.21 | 0.23 |
| KRPLAD | 97.76 | 1.05 | 0.07 | 1.23 | 0.17 | RKALDP | 86.32 | 0.96 | 0.36 | 1.14 | 0.32 |
| AKPLDR | 97.52 | 0.68 | 0.15 | 1.46 | 0.13 | RKAPLD | 86.03 | 1.00 | 0.16 | 1.34 | 0.21 |
| RLAPKD | 96.76 | 1.17 | 0.21 | 1.46 | 0.10 | KPDLAR | 85.32 | 0.69 | 0.11 | 0.73 | 0.15 |
| ADKPLR | 96.70 | 1.39 | 0.17 | 1.52 | 0.15 | RPALDK | 83.82 | 0.78 | 0.11 | 0.76 | 0.22 |
| RKLPDA | 96.51 | 1.17 | 0.16 | 1.48 | 0.14 | LPAKRD | 83.22 | 1.21 | 0.19 | 1.24 | 0.24 |
| LPDARK | 96.41 | 0.84 | 0.23 | 1.34 | 0.16 | AKLPRD | 83.08 | 1.19 | 0.19 | 1.43 | 0.19 |
| RADPKL | 96.10 | 1.26 | 0.22 | 1.52 | 0.10 | LKRDAP | 83.05 | 1.56 | 0.10 | 1.32 | 0.17 |
| KPRLDA | 95.93 | 1.04 | 0.26 | 1.33 | 0.15 | PKLDRA | 81.84 | 0.80 | 0.32 | 1.24 | 0.32 |
| PAKRLD | 95.63 | 0.84 | 0.39 | 1.35 | 0.20 | KLPADR | 81.09 | 1.12 | 0.22 | 1.31 | 0.21 |
| RPLAKD | 95.55 | 0.66 | 0.10 | 0.92 | 0.27 | LDRPKA | 80.73 | 1.30 | 0.21 | 1.46 | 0.13 |
| RADPLK | 94.34 | 1.30 | 0.22 | 1.43 | 0.14 | RPLKDA | 80.72 | 0.75 | 0.21 | 0.75 | 0.19 |
| RPDLAK | 93.99 | 0.76 | 0.13 | 0.82 | 0.15 | KDRAPL | 80.71 | 1.21 | 0.25 | 1.49 | 0.13 |
| RLAKPD | 93.02 | 0.96 | 0.17 | 1.16 | 0.30 | LAKRDP | 80.69 | 0.94 | 0.19 | 1.37 | 0.26 |
| DAKRPL | 92.74 | 1.15 | 0.17 | 1.40 | 0.17 | KARPDL | 80.49 | 1.31 | 0.19 | 1.38 | 0.15 |
| RAKPLD | 92.64 | 1.22 | 0.19 | 1.46 | 0.10 | KLDRPA | 80.47 | 1.39 | 0.20 | 1.20 | 0.22 |
| RKAPDL | 91.96 | 1.31 | 0.27 | 1.46 | 0.14 | APRDKL | 79.95 | 0.79 | 0.23 | 1.09 | 0.32 |
| KALPDR | 91.73 | 1.33 | 0.19 | 1.44 | 0.13 | DRKALP | 79.90 | 1.26 | 0.23 | 1.30 | 0.29 |
| PDKRAL | 91.40 | 1.24 | 0.27 | 1.31 | 0.22 | ADRLKP | 79.79 | 1.01 | 0.24 | 1.19 | 0.23 |
| RADKPL | 88.43 | 1.11 | 0.26 | 1.36 | 0.19 | AKDPLR | 78.40 | 1.11 | 0.30 | 1.24 | 0.31 |
Effect of aminoacids sequence on the binding potency
We first analyze the effect of the positioning of each amino acid in the peptide sequence. From the data in Table 1 one can observe that of the 25 peptides showing the highest binding ratio, 13 peptides have arginine (R) as the first amino acid, which in most cases is followed by alanine (A) or proline (P). The last peptide in the sequence for the strongest binding peptides is most often aspartic acid (D). However, among the 25 least binding peptides, there are only two peptides with arginine in the first position and 2 with aspartic acid in the last position. One can suggest that arginine in the first position and the N-terminal group, create the best conditions for binding to the 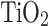 surface.
surface.
We have looked further at the occurrence of different subsequences of peptides that show stronger binding to 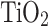 . Here, to obtain better statistics, we analyzed peptides that have SSD within 1 nm more than 50% of the observed time. Fig. 3 illustrates the frequency of pairs and triplets of amino acids in the sequences of these peptides. Fig. 3a presents the most common pairs of amino acids, considering both their sequential order (left) and total occurrence (right) but ignoring their position within the sequence. The pairs identified the most frequently are LP, KP, DL, PR, and DR. In contrast, Fig. 3b and Fig. 3c differentiate between sequence regions. Fig. 3b focuses on the middle section of sequences between positions 2 and 5, while Fig. 3c highlights the most frequent amino acids at the N-terminal (positions 1 and 2) and C-terminal (positions 5 and 6). The analysis reveals that sequences commonly start with
. Here, to obtain better statistics, we analyzed peptides that have SSD within 1 nm more than 50% of the observed time. Fig. 3 illustrates the frequency of pairs and triplets of amino acids in the sequences of these peptides. Fig. 3a presents the most common pairs of amino acids, considering both their sequential order (left) and total occurrence (right) but ignoring their position within the sequence. The pairs identified the most frequently are LP, KP, DL, PR, and DR. In contrast, Fig. 3b and Fig. 3c differentiate between sequence regions. Fig. 3b focuses on the middle section of sequences between positions 2 and 5, while Fig. 3c highlights the most frequent amino acids at the N-terminal (positions 1 and 2) and C-terminal (positions 5 and 6). The analysis reveals that sequences commonly start with 
 and end with either
and end with either 
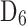 or
or 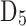
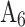 . Additionally, Fig. 4 displays the most frequent positional pairs and triplets within the strongly adsorbed sequences.
. Additionally, Fig. 4 displays the most frequent positional pairs and triplets within the strongly adsorbed sequences.
Fig. 3.

Pair occurrence counts for amino acids in peptides bound to the surface more than 50% of time: (a) across all positions (b) excluding terminal residues (c) at positions 1 and 2 (left) and 5 and 6 (right), (d) top 50 most frequent triplets.
Fig. 4.

The 50 most common amino acid pairs (top) and triplets (bottom) are labeled by their positions within the sequence, for peptides bound to the surface more than 50% of time.
Clustering analysis
Cluster analysis has been performed in order to investigate whether peptides adopt a single dominant binding conformation or multiple binding modes. Fig. 5 and Fig. S2 of the Supporting Information show the representative bound-state conformations from different clusters for sequences that remained adsorbed (SSD < 1 nm) for over 90% of the simulation time. The corresponding SSD and EED values for each cluster are summarized in Table 2. Some sequences, such as PLARKD, DPKLRA, RLAPKD, ADKPLR, LPDARK, RADPKL, KPRLDA, PAKRLD, RPLAKD, RADPLK, RLAKPD and RAKPLD, were found to bind in a single conformation. Others, such as RLDKAP, PKRLAD, RPLDKA, KRPLAD, AKPLDR, RKLPDA, RPDLAK, DAKRPL, and KALPDR, exhibited two or even three distinct binding conformations, each with populations above 10%. These peptides displayed a range of structural preferences, from compact conformations such as PAKRLD to more extended ones like KALPDR. The structures in Fig. 5 reveal how diverse peptide-surface interactions can be. Some peptides adopt tightly packed, looped conformations that likely maximize favorable contacts with the surface, while others bind in more stretched-out forms. This suggests that there is no single preferred binding motif; instead, different sequences have their own best way of interacting with the surface, depending on their flexibility and residue composition. In several cases, such as RKLPDA, RLDKAP, and KRPLAD, we observed multiple distinct binding conformations, each representing a stable adsorption mode. This indicates that some sequences are conformationally adaptable, possibly shifting their orientation or anchoring residues in response to local electrostatics or hydration effects at the surface. Sodium ions ( ) also play a noticeable role in these interactions. In many structures,
) also play a noticeable role in these interactions. In many structures,  acts as a bridge between negatively charged groups, like the carboxylate side chain of ASP or the C-terminus and the surface for some of peptides. For example, AKPLDR adsorbs via ARG, with coordination by
acts as a bridge between negatively charged groups, like the carboxylate side chain of ASP or the C-terminus and the surface for some of peptides. For example, AKPLDR adsorbs via ARG, with coordination by  , while in RKLPDA (min TBP-1), one cluster shows adsorption through ARG and LYS, with ALA involvement mediated by
, while in RKLPDA (min TBP-1), one cluster shows adsorption through ARG and LYS, with ALA involvement mediated by 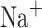 . These observations align well with previous studies by Shiba et al., who emphasized electrostatic interactions between ARG and hydroxyl groups, as well as ASP anchoring via its carboxylate19. Our RKLPDA conformations also closely resemble those reported by Sampath et al. on hydroxylated
. These observations align well with previous studies by Shiba et al., who emphasized electrostatic interactions between ARG and hydroxyl groups, as well as ASP anchoring via its carboxylate19. Our RKLPDA conformations also closely resemble those reported by Sampath et al. on hydroxylated  surfaces16, though ALA was not involved in their work due to terminal capping. Another factor influencing binding is the zwitterionic form of the peptides used in our simulations. Because the terminals are not capped, both N- and C-terminal groups are available for interaction. For instance, in RPDLAK and DAKRPL, LYS at the C-terminus anchors the peptide to the surface through interactions with negatively charged surface oxygens or hydroxyls. Side chains with oxygen atoms, such as ASP, can directly coordinate with titanium, while basic residues like ARG may bind through their nitrogen atoms.
surfaces16, though ALA was not involved in their work due to terminal capping. Another factor influencing binding is the zwitterionic form of the peptides used in our simulations. Because the terminals are not capped, both N- and C-terminal groups are available for interaction. For instance, in RPDLAK and DAKRPL, LYS at the C-terminus anchors the peptide to the surface through interactions with negatively charged surface oxygens or hydroxyls. Side chains with oxygen atoms, such as ASP, can directly coordinate with titanium, while basic residues like ARG may bind through their nitrogen atoms. 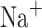 ions further facilitate these interactions by bridging between the peptide and the surface. Moreover, due to the zwitterionic nature of the peptides, surface binding can occur regardless of the intrinsic polarity of individual side chains. For instance, although leucine (LEU) is non-polar and hydrophobic, when located at the terminal position–as in RADPKL–it can still interact electrostatically with the surface via its carboxyl group and a coordinating
ions further facilitate these interactions by bridging between the peptide and the surface. Moreover, due to the zwitterionic nature of the peptides, surface binding can occur regardless of the intrinsic polarity of individual side chains. For instance, although leucine (LEU) is non-polar and hydrophobic, when located at the terminal position–as in RADPKL–it can still interact electrostatically with the surface via its carboxyl group and a coordinating 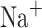 ion. Similarly, in DAKRPL, although ASP typically binds through its side-chain carboxylate, when present at the N-terminus, it adsorbs via its amino group instead. Finally, there seems to be a connection between structural compactness and stronger or more stable binding. Peptides with tighter, more folded conformations often correspond to higher-population clusters, indicating that these compact structures might allow for enhanced cooperative interaction with the surface that through several binding groups that match the structure of the surface, which in turn stabilize the compact peptide structure.
ion. Similarly, in DAKRPL, although ASP typically binds through its side-chain carboxylate, when present at the N-terminus, it adsorbs via its amino group instead. Finally, there seems to be a connection between structural compactness and stronger or more stable binding. Peptides with tighter, more folded conformations often correspond to higher-population clusters, indicating that these compact structures might allow for enhanced cooperative interaction with the surface that through several binding groups that match the structure of the surface, which in turn stabilize the compact peptide structure.
Fig. 5.

Bound-state conformations for the sequences with SSD < 1 nm for more than 90% of the simulation time (first 24 sequences in Table 2) obtained from clustering analysis. Only the most populated clusters (more than 10%) for each sequence is shown together with their populations. The 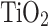 surface atoms–including titanium, oxygen, and the oxygen and hydrogen atoms of surface hydroxyl groups–as well as sodium ions (
surface atoms–including titanium, oxygen, and the oxygen and hydrogen atoms of surface hydroxyl groups–as well as sodium ions (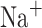 ), are depicted in ball representation and marked with circle shapes, following a consistent color scheme. The peptide backbones are shown in yellow, while only the amino acid residues in direct contact with the surface are displayed in cylinder representation, color-coded and indicated with square shapes. Dashed lines denote possible interactions, such as hydrogen bonding or coordination, between peptide atoms and surface atoms.
), are depicted in ball representation and marked with circle shapes, following a consistent color scheme. The peptide backbones are shown in yellow, while only the amino acid residues in direct contact with the surface are displayed in cylinder representation, color-coded and indicated with square shapes. Dashed lines denote possible interactions, such as hydrogen bonding or coordination, between peptide atoms and surface atoms.
Table 2.
Clusters populations together with their corresponding SSD and EED for the sequences with SSD < 1 nm for more than 90% of the simulation time (first 24 sequences in Table 1) obtained from clustering. Only the most populated clusters (more than 10%) for each sequence are shown.
| Sequences | Cluster | Cluster Population(%) | SSD (nm) | EED (nm) | time (ns) |
|---|---|---|---|---|---|
| PLARKD | Cluster 1 | 44.75 | 0.54 | 1.06 | 584505 |
| Cluster 1 | 26.28 | 0.80 | 1.01 | 29375 | |
| RLDKAP | Cluster 2 | 15.56 | 0.70 | 0.73 | 586165 |
| Cluster 3 | 12.30 | 0.69 | 1.25 | 392490 | |
| DPKLRA | Cluster 1 | 25.12 | 0.80 | 0.90 | 320860 |
| PKRALD | Cluster 1 | 39.79 | 0.69 | 1.17 | 371580 |
| Cluster 2 | 12.70 | 0.68 | 1.14 | 437050 | |
| RPLDKA | Cluster 1 | 51.95 | 0.84 | 0.68 | 466035 |
| Cluster 2 | 12.22 | 0.89 | 0.96 | 486065 | |
| PRLKAD | - | - | - | - | - |
| KRPLAD | Cluster 1 | 63.37 | 0.77 | 1.05 | 181940 |
| Cluster 2 | 12.60 | 0.84 | 1.07 | 265030 | |
| AKPLDR | Cluster 1 | 75.23 | 0.69 | 0.65 | 463870 |
| Cluster 2 | 11.78 | 0.73 | 0.59 | 121370 | |
| RLAPKD | Cluster 1 | 36.49 | 0.65 | 1.02 | 369450 |
| ADKPLR | Cluster 1 | 12.66 | 0.55 | 1.43 | 39610 |
| RKLPDA | Cluster 1 | 32.61 | 0.65 | 1.05 | 100460 |
| Cluster 2 | 14.92 | 0.61 | 1.33 | 475740 | |
| LPDARK | Cluster 1 | 45.20 | 0.79 | 0.73 | 348135 |
| RADPKL | Cluster 1 | 15.51 | 0.71 | 1.24 | 399515 |
| KPRLDA | Cluster 1 | 10.76 | 0.72 | 0.93 | 358450 |
| PAKRLD | Cluster 1 | 53.91 | 0.84 | 0.50 | 368775 |
| RPLAKD | Cluster 1 | 89.10 | 0.77 | 0.65 | 433175 |
| RADPLK | Cluster 1 | 13.21 | 0.73 | 0.69 | 533940 |
| RPDLAK | Cluster 1 | 61.87 | 0.73 | 0.74 | 392785 |
| Cluster 2 | 11.25 | 0.70 | 0.74 | 98135 | |
| RLAKPD | Cluster 1 | 44.31 | 0.74 | 0.83 | 284395 |
| DAKRPL | Cluster 1 | 19.85 | 0.79 | 1.09 | 459725 |
| Cluster 2 | 10.06 | 0.63 | 1.17 | 141345 | |
| RAKPLD | Cluster 1 | 17.89 | 0.65 | 1.38 | 367355 |
| RKAPDL | - | - | - | - | - |
| KALPDR | Cluster 1 | 14.35 | 0.85 | 1.51 | 563730 |
| Cluster 2 | 11.08 | 0.73 | 1.23 | 377925 | |
| PDKRAL | - | - | - | - | - |
Radial distribution functions
To get further insight into the atomistic details of the peptide binding, we calculated radial distribution functions (RDF) between selected atoms of the peptides: N atoms of basic amino acids (ARG and LYS) and amino acids at the N-terminus, oxygen atoms of the acidic amino acid (ASP) and amino acids at C-terminus, 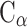 for non-charged amino acids (ALA, LEU and PRO), and oxygen atoms of
for non-charged amino acids (ALA, LEU and PRO), and oxygen atoms of 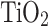 surface: bridge oxygens (OB) and oxygens of the hydroxyl groups (OF). On the anatase-101 surface, oxygen atoms are more exposed to the environment than Ti atoms and provide favorable possibilities for binding of polar peptide atoms through hydrogen bonds. Fig. 6 explains the notations used for RDFs between different atoms in the peptide sequences and
surface: bridge oxygens (OB) and oxygens of the hydroxyl groups (OF). On the anatase-101 surface, oxygen atoms are more exposed to the environment than Ti atoms and provide favorable possibilities for binding of polar peptide atoms through hydrogen bonds. Fig. 6 explains the notations used for RDFs between different atoms in the peptide sequences and 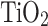 surface oxygens. The list of RDFs with the highest RDF maximum for each peptide is provided in Tables S1 and S2 of the Supporting Information, and RDFs for 24 highest ranking binding peptides are shown in Fig. 7. In this figure, only RDFs with the maxima occurring at distances less than 4 Å are shown. The complete RDF dataset is available as stated in the Data Availability section.
surface oxygens. The list of RDFs with the highest RDF maximum for each peptide is provided in Tables S1 and S2 of the Supporting Information, and RDFs for 24 highest ranking binding peptides are shown in Fig. 7. In this figure, only RDFs with the maxima occurring at distances less than 4 Å are shown. The complete RDF dataset is available as stated in the Data Availability section.
Fig. 6.

Notation guide for RDF of different atoms in amino acids. Notations for atoms of amino acids at different positions including N-terminal, middle (SCA), and C-terminal are shown.
Fig. 7.


RDFs of the top 24 sequences in the Table 1.
Our analysis shows that the highest RDF peaks are observed for atoms binding to the oxygen of hydroxyl groups on the surface (atom type OF), while lower (but still high) maximum RDF values are often observed for bridging oxygens (atom type OB). This indicates that adsorption is driven mostly by the hydroxyl groups at the surface, and thus is expected to be dependent on pH. This conclusion is in agreement with the experimental findings of pH-dependent amino acids adsorption on the 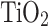 surface23, and in particular with the fact that Lysine adsorption increases with pH when the amount of OH groups is expected to increase.
surface23, and in particular with the fact that Lysine adsorption increases with pH when the amount of OH groups is expected to increase.
Among the peptides atoms that show the highest RDF peaks, more than 85% are nitrogen atoms, either from the N-terminus or the side chains of amino acids. This can also be seen in Fig. 5, where in most sequences the basic N-containing amino acids (ARG and LYS) are anchored or close to the surface, together with the amino acids at the N-terminus (ALA in ADKPLR, PRO in PKRALD and PLARKD, LEU in LPDARK, etc.). N-terminus atoms show systematically high RDF peaks even for less strongly bound peptides. Furthermore, examining other high peaks in each sequence reveals that other atoms in amino acids also exhibit first RDF maxima close to the surface. As shown in Fig. 7, each of the strongly bound peptides has multiple amino acids that contribute to surface adsorption. This observation is consistent with the bound-state conformations presented in Fig. 5. For example, PLARKD displays high RDF peaks within 0.4 nm for four amino acids, including K, P, D, and R, corresponding to their proximity to the surface in the bound state (see Fig. 5). Similarly, in RKLPDA, two amino acids, R and D, have peaks within 0.4 nm, which is consistent with the peptide conformation in the bound state, Fig. 5. One can note that, although the A residue also regularly appears close to the surface, it is anchored to the surface via  resulting in a slightly larger distance from the surface oxygen atoms OB and OF. This is further analyzed in the next Section.
resulting in a slightly larger distance from the surface oxygen atoms OB and OF. This is further analyzed in the next Section.
Effect of  on adsorption
on adsorption
To investigate the role of 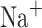 ions in the adsorption of peptides on the
ions in the adsorption of peptides on the  surface, we analyzed conditional radial distribution functions between sodium ions on the surface (
surface, we analyzed conditional radial distribution functions between sodium ions on the surface ( ) and oxygen atoms of peptides and
) and oxygen atoms of peptides and  surface. For these calculations,
surface. For these calculations,  ions were filtered based on a dual distance criterion: only ions located within 3 Å of either of the surface oxygen atoms (OB and OF), and simultaneously within 3 Å of any peptide oxygen (e.g., backbone or sidechain oxygens), were selected for the analysis. Fig. 8 shows these RDFs calculated for 24 peptides that remained adsorbed to the surface (SSD < 1 nm) for more than 90% of the simulation time. Compared with the RDFs in Fig. 8 with the RDFs in Fig. 7 and the bound-state conformations shown in Fig. 5, we observe that in some sequences,
ions were filtered based on a dual distance criterion: only ions located within 3 Å of either of the surface oxygen atoms (OB and OF), and simultaneously within 3 Å of any peptide oxygen (e.g., backbone or sidechain oxygens), were selected for the analysis. Fig. 8 shows these RDFs calculated for 24 peptides that remained adsorbed to the surface (SSD < 1 nm) for more than 90% of the simulation time. Compared with the RDFs in Fig. 8 with the RDFs in Fig. 7 and the bound-state conformations shown in Fig. 5, we observe that in some sequences, 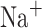 ions appear to play a significant role in mediating peptide adsorption. For example, the sequences DPKLRA, PKRALD, and PRLKAD show strong RDF peaks for peptide oxygen atoms (especially from ASP side chains or C-terminal groups) that overlap in position and intensity with those of
ions appear to play a significant role in mediating peptide adsorption. For example, the sequences DPKLRA, PKRALD, and PRLKAD show strong RDF peaks for peptide oxygen atoms (especially from ASP side chains or C-terminal groups) that overlap in position and intensity with those of 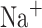 . This spatial overlap and comparable peak intensity suggest that these peptides are likely stabilized through
. This spatial overlap and comparable peak intensity suggest that these peptides are likely stabilized through 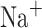 bridging interactions with the surface. In contrast, other peptides, such as RPLDKA, RLAPKD, ADKPLR, LPDARK, RPDLAK, and DAKRPL, show much weaker or negligible RDF peaks for peptide oxygen atoms near
bridging interactions with the surface. In contrast, other peptides, such as RPLDKA, RLAPKD, ADKPLR, LPDARK, RPDLAK, and DAKRPL, show much weaker or negligible RDF peaks for peptide oxygen atoms near  . This implies that
. This implies that 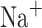 is not directly involved in mediating adsorption for the dominant bound-state conformation of these sequences. These observations are consistent with the conformations in Fig. 5, where sodium ions are absent or distant from the key adsorption sites. Furthermore, the specific types of oxygen atoms that contribute to the RDF peaks vary between sequences. In most cases, the high peak of
is not directly involved in mediating adsorption for the dominant bound-state conformation of these sequences. These observations are consistent with the conformations in Fig. 5, where sodium ions are absent or distant from the key adsorption sites. Furthermore, the specific types of oxygen atoms that contribute to the RDF peaks vary between sequences. In most cases, the high peak of 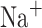 - peptide oxygen RDF is seen for ASP oxygen atoms, which are known to interact strongly with cations. However, there are notable exceptions. For example, in DPKLRA and KPRLDA, the dominant contribution comes from the C-terminal carboxyl oxygen in ALA, while in RADPKL this contribution comes from LEU. These results indicate that, although ASP is a primary anchor point, other residues, particularly at the terminal positions, can also participate in
- peptide oxygen RDF is seen for ASP oxygen atoms, which are known to interact strongly with cations. However, there are notable exceptions. For example, in DPKLRA and KPRLDA, the dominant contribution comes from the C-terminal carboxyl oxygen in ALA, while in RADPKL this contribution comes from LEU. These results indicate that, although ASP is a primary anchor point, other residues, particularly at the terminal positions, can also participate in 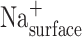 -mediated adsorption, depending on their spatial proximity and accessibility. In general, the comparison between Fig. 8 and Fig. 5 highlights the sequence-specific nature of
-mediated adsorption, depending on their spatial proximity and accessibility. In general, the comparison between Fig. 8 and Fig. 5 highlights the sequence-specific nature of 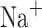 -facilitated adsorption. Some peptides rely on sodium ions to stabilize their interaction with the surface, while others bind directly via side chains or terminal groups without significant ionic mediation. This distinction is reflected not only in the RDF profiles but also in the clustering and structural conformations observed in the bound states. Together, these findings highlight the importance of considering both direct and ion-mediated contacts when characterizing peptide-surface interactions at the atomic level.
-facilitated adsorption. Some peptides rely on sodium ions to stabilize their interaction with the surface, while others bind directly via side chains or terminal groups without significant ionic mediation. This distinction is reflected not only in the RDF profiles but also in the clustering and structural conformations observed in the bound states. Together, these findings highlight the importance of considering both direct and ion-mediated contacts when characterizing peptide-surface interactions at the atomic level.
Fig. 8.


RDFs of oxygen atoms in 24 top-ranking sequences and surface  .
.
The observed sequence-dependent binding behavior can be understood from the chemical interactions between the peptides and the hydroxylated  surface. Positively charged residues such as arginine (R) exhibits strong surface affinity, particularly when located at the N-terminus, due to the ability to form electrostatic interactions and multiple hydrogen bonds with the surface hydroxyl groups through the side-chain NH groups as it can be seen in the representative snapshots in Figure 5. As noted in the RDF plots (Figures 7), the oxygen atoms of surface hydroxyl groups (OF) play a significant role in facilitating these interactions. Arginine’s guanidinium group, in particular, contributes to stable binding by anchoring the peptide, as seen in sequences such as RLDKAP, PAKRLD, and RPLAKD. Lysine, with its positively charged side chain, can also form hydrogen bonds with surface OH groups, as observed in KPRLDA, RKLPDA, and related sequences. Another important factor is the presence of the free amino group at the N-terminus, which is positively charged at physiological pH and can directly interact with the surface so as the carboxyl groups at C-terminus which bind through sodium ions. This is evident in sequences such as DAKRPL, PKRLAD, and PLARKD, which, despite not starting with R or K side chains, still high in the ranking due to contributions from the terminal amine group. The negatively charged residue aspartic acid (D) also participates in surface binding, typically through sodium ion (
surface. Positively charged residues such as arginine (R) exhibits strong surface affinity, particularly when located at the N-terminus, due to the ability to form electrostatic interactions and multiple hydrogen bonds with the surface hydroxyl groups through the side-chain NH groups as it can be seen in the representative snapshots in Figure 5. As noted in the RDF plots (Figures 7), the oxygen atoms of surface hydroxyl groups (OF) play a significant role in facilitating these interactions. Arginine’s guanidinium group, in particular, contributes to stable binding by anchoring the peptide, as seen in sequences such as RLDKAP, PAKRLD, and RPLAKD. Lysine, with its positively charged side chain, can also form hydrogen bonds with surface OH groups, as observed in KPRLDA, RKLPDA, and related sequences. Another important factor is the presence of the free amino group at the N-terminus, which is positively charged at physiological pH and can directly interact with the surface so as the carboxyl groups at C-terminus which bind through sodium ions. This is evident in sequences such as DAKRPL, PKRLAD, and PLARKD, which, despite not starting with R or K side chains, still high in the ranking due to contributions from the terminal amine group. The negatively charged residue aspartic acid (D) also participates in surface binding, typically through sodium ion (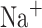 )-mediated interactions. This can be seen in sequences such as RAKPLD and PAKRLD, as well as in others like RKLPDA and AKPLDR, which do not possess D at C-terminus but still exhibit binding via their C-terminal carboxyl groups. Due to the zwitterionic nature of the peptides, the terminal carboxyl group can coordinate with
)-mediated interactions. This can be seen in sequences such as RAKPLD and PAKRLD, as well as in others like RKLPDA and AKPLDR, which do not possess D at C-terminus but still exhibit binding via their C-terminal carboxyl groups. Due to the zwitterionic nature of the peptides, the terminal carboxyl group can coordinate with  ions and contribute to surface adsorption. This bridging mechanism is evident in the RDF profiles (Fig. 8), where
ions and contribute to surface adsorption. This bridging mechanism is evident in the RDF profiles (Fig. 8), where  ions accumulate near carboxylate oxygen atoms, and is consistent with the binding conformations illustrated in Fig. 5. Overall, these findings highlight the key roles of surface chemistry, charge complementarity, and specific residue–side chain interactions in governing peptide adsorption.
ions accumulate near carboxylate oxygen atoms, and is consistent with the binding conformations illustrated in Fig. 5. Overall, these findings highlight the key roles of surface chemistry, charge complementarity, and specific residue–side chain interactions in governing peptide adsorption.
Conclusions
In this study, we systematically investigated the adsorption behavior of 360 possible permutations of the titanium-binding peptide RKLPDA on the surfaces of hydroxylated anatase 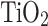 (101) using extensive atomistic molecular dynamics simulations. Our findings highlight the strong sequence dependence of peptide adsorption, with 138 peptides showing more than 50% binding over the simulation time and several sequences exhibiting enhanced binding compared to the original RKLPDA. We identified sequence motifs and residue positions, particularly arginine at the N-terminus and lysine or aspartic acid at the C-terminus, that contribute to stronger and more stable adsorption. Clustering analysis revealed that high-binding peptides often adopt compact conformations to provide favorable contact with the surface, while RDF analysis confirmed the importance of basic nitrogen-containing residues (ARG, LYS) and the N-terminus of peptides in binding to
(101) using extensive atomistic molecular dynamics simulations. Our findings highlight the strong sequence dependence of peptide adsorption, with 138 peptides showing more than 50% binding over the simulation time and several sequences exhibiting enhanced binding compared to the original RKLPDA. We identified sequence motifs and residue positions, particularly arginine at the N-terminus and lysine or aspartic acid at the C-terminus, that contribute to stronger and more stable adsorption. Clustering analysis revealed that high-binding peptides often adopt compact conformations to provide favorable contact with the surface, while RDF analysis confirmed the importance of basic nitrogen-containing residues (ARG, LYS) and the N-terminus of peptides in binding to 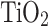 . We further found that hydroxyl groups on the surface facilitate binding of peptides by nitrogen-containing groups, while
. We further found that hydroxyl groups on the surface facilitate binding of peptides by nitrogen-containing groups, while 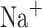 ions were found to facilitate adsorption in a sequence-specific manner, bridging between negatively charged side chains and the
ions were found to facilitate adsorption in a sequence-specific manner, bridging between negatively charged side chains and the  surface in several peptides. Additionally, the zwitterionic form of peptides resulted in the adsorbance through amino acids that are intrisically non-polar and more hydrophobic so that the presence of amino group at N-terminus and carboxyl groups at C-terminus improved the adsorption tendency to the surface.
surface in several peptides. Additionally, the zwitterionic form of peptides resulted in the adsorbance through amino acids that are intrisically non-polar and more hydrophobic so that the presence of amino group at N-terminus and carboxyl groups at C-terminus improved the adsorption tendency to the surface.
We should further note that the length of our simulations, 600 ns for each peptide, can provide reliable distinguishing between strongly and weakly binding peptides and a qualitative ranking of their binding potency; however, it is likely not enough to quantitatively characterize the strength of binding of strongly binding peptides in terms of binding free energies. Such computations need to be carried out by biased simulations such as Metadynamics, and we are planning to carry out such computations for strongly bound peptides identified in this study in the follow-up work.
Furthermore, the large amount of data obtained in this work, including non-biased molecular dynamics trajectories, instant interaction energies, RDFs, other structural and dynamical information obtained for each amino acid sequence, can contribute to subsequent data-driven analysis to develop machine learning models which would greatly facilitate investigation of various aspects of peptide-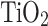 interactions.
interactions.
Together, our work provides detailed insights into the molecular mechanisms of peptide-surface interactions and offers a computational framework for optimizing solid-binding peptide sequences for surface functionalization applications. These insights can be extended to the design of next-generation biomaterials with improved specificity and binding affinity.
Materials and methods
Titania surface
In this work, we investigate the binding of peptides on a fully hydrated and partially hydroxylated 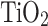 anatase (101) surface. The (101) anatase surface has the lowest surface energy and prevails in typical anatase nanoparticles24. Furthermore, Grote et al. reported that the density profile of water around the
anatase (101) surface. The (101) anatase surface has the lowest surface energy and prevails in typical anatase nanoparticles24. Furthermore, Grote et al. reported that the density profile of water around the  nanoparticle is the most similar to the previously obtained density profile for anatase (101) plane surface, both computed by ab initio molecular dynamics simulations25,26.
nanoparticle is the most similar to the previously obtained density profile for anatase (101) plane surface, both computed by ab initio molecular dynamics simulations25,26.
The 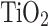 atoms were arranged in a two-dimensional periodic slab with (101) surface exposed to water. We set the fraction of hydroxyl groups on the surface of anatase (101) as 30% to match the experimentally observed surface charge of
atoms were arranged in a two-dimensional periodic slab with (101) surface exposed to water. We set the fraction of hydroxyl groups on the surface of anatase (101) as 30% to match the experimentally observed surface charge of 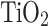 nanoparticle at neutral pH of −0.62 e/
nanoparticle at neutral pH of −0.62 e/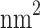 27. The hydroxyl groups were randomly attached to 5-coordinated Ti surface atoms. Each surface has dimensions of approximately 4.92
27. The hydroxyl groups were randomly attached to 5-coordinated Ti surface atoms. Each surface has dimensions of approximately 4.92  4.94
4.94  3.05
3.05  including 6 layers of Ti atoms. In this work, the force field parameterized by Rouse et al.28 based on previously performed ab initio molecular dynamics (DFT) simulations of several
including 6 layers of Ti atoms. In this work, the force field parameterized by Rouse et al.28 based on previously performed ab initio molecular dynamics (DFT) simulations of several 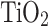 surfaces in water26 was used.
surfaces in water26 was used.
Peptides
We performed MD simulations of 360 six amino acids long peptides 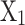
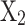
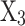

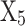
 , where
, where  ={R, K, L, P, D, A} and R is arginine (ARG), K lysine (LYS), L leucine (LEU), P proline (PRO), D aspartic acid (ASP) and A is alanine (ALA,) obtained by permutations of amino acids present in titanium binding peptide RKLPDA. The peptides were in zwitterionic form, so the two ends of the peptides known as the N-terminus and C-terminus are not capped and are charged to mimic physiological conditions at pH=7.5 and 37
={R, K, L, P, D, A} and R is arginine (ARG), K lysine (LYS), L leucine (LEU), P proline (PRO), D aspartic acid (ASP) and A is alanine (ALA,) obtained by permutations of amino acids present in titanium binding peptide RKLPDA. The peptides were in zwitterionic form, so the two ends of the peptides known as the N-terminus and C-terminus are not capped and are charged to mimic physiological conditions at pH=7.5 and 37  C. At neutral pH, ARG (pKa = 12.4) and LYS (pKa = 10.5) carry a positive charge, and ASP (pK=3.7) has a negative charge. The amino acids and peptides were built using Pymol29 and simulated using the Amber03 force field30. Lorentz-Berthelot combining rules were used for cross-interactions.
C. At neutral pH, ARG (pKa = 12.4) and LYS (pKa = 10.5) carry a positive charge, and ASP (pK=3.7) has a negative charge. The amino acids and peptides were built using Pymol29 and simulated using the Amber03 force field30. Lorentz-Berthelot combining rules were used for cross-interactions.
The system
Each simulated system consisted of anatase  slab, one of peptides and water, which was presented by the TIP3P model. Furthermore,
slab, one of peptides and water, which was presented by the TIP3P model. Furthermore, 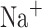 and
and  ions were added to neutralize the system and provide a physiologically relevant ion concentration of 0.15 M. The simulation systems composed in this way, as well as their components, are shown in Fig. 1. Each of the systems consisted of
ions were added to neutralize the system and provide a physiologically relevant ion concentration of 0.15 M. The simulation systems composed in this way, as well as their components, are shown in Fig. 1. Each of the systems consisted of  25000 atoms. An example of system compositions is provided in Table 3. For visualizing the systems, the Visual Molecular Dynamics (VMD) package has been used31.
25000 atoms. An example of system compositions is provided in Table 3. For visualizing the systems, the Visual Molecular Dynamics (VMD) package has been used31.
Table 3.
Size and composition of the simulated system (an example).
Box size (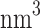 ) ) |
5.11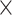 4.95 4.95 10.70 10.70 |
| Slab thickness (nm) | 3.05 |
 |
2340 Ti, 4680 O |
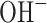 (bound to Ti-surface) (bound to Ti-surface) |
78 |
 O O |
6106 |
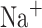 |
57 |
 |
26 |
| Peptide | 1 |
Simulations
All molecular dynamics simulations were performed using Gromacs simulation package, version 202232. The SSD and EED distances were extracted from the simulation trajectories using the PLUMED v.2.7 plugin33. The parameters of all MD simulations were the following. A time step was set to 2 fs. The LINCS algorithm was used to constrain the bonds to hydrogen atoms34. Electrostatic interactions were computed using the Particle Mesh Ewald (PME) algorithm with a real-space cutoff of 1.0 nm and a grid resolution of 0.12 nm. The same cutoff was used to calculate short-range Lennard-Jones interactions35. After the initial configuration of all components, the steepest descent algorithm was used for energy minimization to remove unfavorable contacts. This was followed by an equilibration run at 300 K and 1 bar using the Berendsen thermostat and barostat36 for 3 ns. The last frame of that simulation was used for the subsequent NVT with the Nosé–Hoover thermostat, which lasted 600 ns37,38.
Clustering algorithm
We performed clustering analysis to investigate the distinct conformations that contribute to the adsorbed state of the peptides. The clustering process was carried out as follows. Initially, frames corresponding to the adsorbed state, that is, when the SSD is less than 1 nm were extracted. Clustering was performed using the gmx cluster tool in GROMACS with an RMSD cutoff of 0.15 nm applied to the entire peptide structure. The Gromos clustering algorithm was employed, which iteratively identifies the clusters. The algorithm selects the first cluster as the structure with the largest number of neighbors within the cutoff distance. This structure and its neighbors are then excluded from subsequent iterations, and the process is repeated until all frames are assigned to certain clusters39. This analysis enabled us to quantify the structural diversity of the adsorbed conformations.
Supplementary Information
Acknowledgements
This work has been supported by the NanoSolveIt Horizon2020 project and by the Swedish Research Council (Vetenskapsrådet), grant no. 2021-04474. The computations were performed using resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at the Center for Parallel Computing (PDC, Stockholm) and the National Supercomputer Center (NSC, Linköping) partially funded by the Swedish Research Council through grant agreements no. 2022-06725.
Author contributions
R.R.: formulations the goals of the study; methodology; performing computations; data analysis; preparing figures; writing the text A.P.L.: formulations the study concept and goals; methodology, data analysis, writing the text All authors reviewed the manuscript.
Funding
Open access funding provided by Stockholm University.
Data availability
All relevant data, including files to start and run simulations, initial and final configurations, data files for SSD and EED as function of time, RDFs, Jupyther notebooks, are provided in an archive deposited at Zenodo repository, https://doi.org/10.5281/zenodo.15267234
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-10966-3.
References
- 1.Pushpavanam, K., Ma, J., Cai, Y., Naser, N. Y. & Baneyx, F. Solid-binding proteins: Bridging synthesis, assembly, and function in hybrid and hierarchical materials fabrication. Annu. Rev. Chem. Biomol. Eng.12(1), 333–357 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Yucesoy, D. T., Akkineni, S., Tamerler, C., Hinds, B. J. & Sarikaya, M. Solid-binding peptide-guided spatially directed immobilization of kinetically matched enzyme cascades in membrane nanoreactors. ACS omega6(41), 27129–27139 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho, C. H. et al. Re-engineering of peptides with high binding affinity to develop an advanced electrochemical sensor for colon cancer diagnosis. Anal. Chim. Acta1146, 131–139 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Care, A., Bergquist, P. L. & Sunna, A. Solid-binding peptides: Smart tools for nanobiotechnology. Trends Biotechnol.33(5), 259–268 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Coppage, R. et al. Exploiting localized surface binding effects to enhance the catalytic reactivity of peptide-capped nanoparticles. J. Am. Chem. Soc.135(30), 11048–11054 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Riool, M., Breij, A., Drijfhout, J. W., Nibbering, P. H. & Zaat, S. A. Antimicrobial peptides in biomedical device manufacturing. Front. Chem.5, 63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sultan, A. M. et al. Aqueous peptide-TiO2 interfaces: Isoenergetic binding via either entropically or enthalpically driven mechanisms. ACS Appl. Mater. Interfaces8(28), 18620–18630 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Sano, K.-I. & Shiba, K. A hexapeptide motif that electrostatically binds to the surface of titanium. J. Am. Chem. Soc.125(47), 14234–14235 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Watanabe, M. et al. Bio-functionalized titanium surfaces with modified silk fibroin carrying titanium binding motif to enhance the ossific differentiation of MC3T3-E1. Biotechnol. Bioeng.118(7), 2585–2596 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Yoshinari, M., Kato, T., Matsuzaka, K., Hayakawa, T. & Shiba, K. Prevention of biofilm formation on titanium surfaces modified with conjugated molecules comprised of antimicrobial and titanium-binding peptides. Biofouling26(1), 103–110 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Hodneland, C. D., Lee, Y.-S., Min, D.-H. & Mrksich, M. Selective immobilization of proteins to self-assembled monolayers presenting active site-directed capture ligands. Proc. atl. Acad. Sci.99(8), 5048–5052 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee, H., Rho, J. & Messersmith, P. B. Facile conjugation of biomolecules onto surfaces via mussel adhesive protein inspired coatings. Adv. Mater.21(4), 431 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong, P. & Grainger, D. W. Comparison of DNA immobilization efficiency on new and regenerated commercial amine-reactive polymer microarray surfaces. Surf. Sci.570(1–2), 67–77 (2004). [Google Scholar]
- 14.Guo, W., Lu, T., Gandhi, Z. & Chen, Z. Probing orientations and conformations of peptides and proteins at buried interfaces. J. Phys. Chem. Lett.12(41), 10144–10155 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Hirokazu, N. et al. Self-oriented immobilization of DNA polymerase tagged by titanium-binding peptide motif. Langmuir31, 732–740 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Sampath, J., Kullman, A., Gebhart, R., Drobny, G. & Pfaendtner, J. Molecular recognition and specificity of biomolecules to titanium dioxide from molecular dynamics simulations. NPJ Comput. Mater.6(1), 34 (2020). [Google Scholar]
- 17.Hayashi, T. et al. Mechanism underlying specificity of proteins targeting inorganic materials. Nano Lett.6(3), 515–519 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Mirau, P. A., Naik, R. R. & Gehring, P. Structure of peptides on metal oxide surfaces probed by nmr. J. Am. Chem. Soc.133(45), 18243–18248 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Suzuki, Y., Shindo, H. & Asakura, T. Structure and dynamic properties of a Ti-binding peptide bound to tio2 nanoparticles as accessed by 1H NMR spectroscopy. J. Phys. Chem. B120(20), 4600–4607 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Brandt, E. G. & Lyubartsev, A. P. Molecular dynamics simulations of adsorption of amino acid side chain analogues and a titanium binding peptide on the TiO2 (100) surface. J. Phys. Chem. C119(32), 18126–18139 (2015). [Google Scholar]
- 21.Schneider, J. & Colombi Ciacchi, L. Specific material recognition by small peptides mediated by the interfacial solvent structure. J. Am. Chem. Soc.134(4), 2407–2413 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Polimeni, M., Petridis, L., Smith, J. C. & Arcangeli, C. Dynamics at a peptide-TiO2 anatase (101) interface. J. Phys. Chem. B121(38), 8869–8877 (2017). [DOI] [PubMed] [Google Scholar]
-
23.Ustunol, I. B., Gonzalez-Pech, N. I. & Grassian, V. H. ph-dependent adsorption of
 -amino acids, lysine, glutamic acid, serine and glycine, on tio2 nanoparticle surfaces. J. Colloid Interface Sci.554, 362–375 (2019). [DOI] [PubMed]
-amino acids, lysine, glutamic acid, serine and glycine, on tio2 nanoparticle surfaces. J. Colloid Interface Sci.554, 362–375 (2019). [DOI] [PubMed] -
24.Lazzeri, M., Vittadini, A. & Selloni, A. Structure and energeticsof stoichiometric
 anatase surfaces. Phys. Rev. B63, 155409 (2001).
anatase surfaces. Phys. Rev. B63, 155409 (2001).
- 25.Grote, F. & Lyubartsev, A. P. Water structure, dynamics and reactivity on a TiO2-nanoparticle surface: New insights from ab initio molecular dynamics. Nanoscale14(44), 16536–16547 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Agosta, L., Brandt, E. G. & Lyubartsev, A. P. Diffusion and reaction pathways of water near fully hydrated TiO2 surfaces from ab initio molecular dynamics. J. Chem. Phys.10.1063/1.4991381 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Vakurov, A., Drummond-Brydson, R., Ugwumsinachi, O. & Nelson, A. Significance of particle size and charge capacity in TiO2 nanoparticle-lipid interactions. J. Colloid Interface Sci.473, 75–83 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Rouse, I. et al. First principles characterisation of bio-nano interface. Phys. Chem. Chem. Phys.23(24), 13473–13482 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Schrödinger, LLC: The PyMOL Molecular Graphics System, Version 2.3.0 (2015)
- 30.Duan, Y. et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem.24(16), 1999–2012 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Humphrey, W., Dalke, A. & Schulten, K. VMD - Visual Molecular Dynamics. J. Mol. Graph.14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Abraham, M. J. et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX1–2, 19–25. 10.1016/j.softx.2015.06.001 (2015). [Google Scholar]
- 33.The PLUMED Consortium. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods16(8), 670–673 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Hess, B., Bekker, H., Berendsen, H. J. & Fraaije, J. G. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem.18(12), 1463–1472 (1997). [Google Scholar]
-
35.Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An
 method for ewald sums in large systems. J. Chem. Phys.98(12), 10089–10092 (1993).
method for ewald sums in large systems. J. Chem. Phys.98(12), 10089–10092 (1993).
- 36.Berendsen, H.J., Postma, J.v., Van Gunsteren, W.F., DiNola, A., & Haak, J.R.Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81(8), 3684–3690 (1984)
- 37.Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys.81(1), 511–519 (1984). [Google Scholar]
- 38.Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A31(3), 1695 (1985). [DOI] [PubMed] [Google Scholar]
- 39.Daura, X. et al. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed.38(1–2), 236–240 (1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data, including files to start and run simulations, initial and final configurations, data files for SSD and EED as function of time, RDFs, Jupyther notebooks, are provided in an archive deposited at Zenodo repository, https://doi.org/10.5281/zenodo.15267234