

Abstract
Background
Lung adenocarcinoma (LUAD) exhibits significant cellular heterogeneity, yet the precise interactions between epithelial and stromal cells remain unclear. This study integrates single-cell and spatial transcriptomics to delineate tumor microenvironment dynamics, aiming to uncover key cellular subpopulations and their roles in LUAD progression.
Methods
We analyzed single-cell RNA sequencing (scRNA-seq) data from 21 LUAD patients and performed spatial transcriptomic deconvolution. Epithelial and fibroblast subpopulations were identified using Seurat and Harmony. Cell-cell communication was inferred via CellChat, while metabolic interactions were assessed using MEBOCOST. Copy number variation (CNV) analysis distinguished malignant cells, and trajectory inference mapped differentiation states. Spatial colocalization was examined via CellTrek. Prognostic signatures were derived from Cox regression, and a six-gene MCI score was validated using survival analysis.
Results
We identified eight epithelial (e.g., MUC21 + Epi, ASCL1 + Epi) and nine fibroblast subpopulations (e.g., Fb_IGFBP4, Fb_COL11A1), with tumor-enriched subsets showing elevated CNVs and metabolic crosstalk. Fb_IGFBP4 correlated with poor prognosis, while MUC21 + Epi exhibited amplified COL1A1/SDC4-mediated interactions with fibroblasts. Pathway analysis highlighted tumor-specific MK and collagen signaling between fibroblasts and epithelial cells, suggesting stromal-epithelial synergy drives progression. Spatial analysis revealed colocalization of epithelial and fibroblast subclusters in tumors, contrasting with normal tissue. The MCI score, derived from six genes (e.g., ADAM10, MARVELD1), independently predicted survival and stratified high-risk patients (AUC > 0.6).
Conclusion
This study identifies key stromal-epithelial subset interactions in LUAD, proposing prognostic biomarkers and therapeutic targets.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00432-025-06250-6.
Keywords: Lung adenocarcinoma, Single-cell transcriptomics, Tumor microenvironment, Epithelial cell, Fibroblast
Introduction
Lung cancer remains the leading cause of cancer-related mortality worldwide, accounting for approximately 1.8 million deaths annually [1]. Non-small cell lung cancer (NSCLC), which includes lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), constitutes the majority of cases. At present, LUAD has replaced LUSC as the main pathological subtype of NSCLC, causing serious medical burden. In recent years, the advent of single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of tumor heterogeneity by enabling transcriptomic profiling at single-cell resolution [2]. Several studies have employed scRNA-seq to explore the cellular composition of LUAD [2–4]. However, the comprehensive mapping of immune and stromal components, as well as the interactions between malignant and non-malignant cells, may still lack precision. Therefore, it is crucial to further delineate the intercellular communications within the tumor microenvironment to uncover potential mechanisms and therapeutic targets.
Previous single-cell studies have characterized various epithelial subpopulations in lung cancer, revealing distinct transcriptional programs associated with tumor initiation, progression, and metastasis [2, 5, 6]. Similarly, fibroblast heterogeneity has been increasingly recognized, with subsets such as inflammatory CAFs (iCAFs) and myofibroblastic CAFs (myCAFs) exhibiting diverse roles in modulating the tumor microenvironment [7–9]. Notably, specific epithelial and fibroblast subtypes have been linked to clinical outcomes and therapeutic resistance [10, 11]. Despite these advances, a systematic understanding of the functional diversity and spatial organization of epithelial and fibroblast subclusters in LUAD remains incomplete.
However, studies specifically focusing on the interactions between epithelial and fibroblast cells, especially within the spatial context of LUAD, are still limited. Moreover, the integration of spatial transcriptomics with single-cell data to decipher the intercellular communication landscape has been rarely explored. To address these gaps, we conducted an integrated single-cell and spatial transcriptomic analysis of LUAD, aiming to elucidate the cellular interactions within the tumor microenvironment and identify potential therapeutic targets.
Materials and methods
Data collection
The scRNA-seq data were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/, accession number GSE131907) [3] and the Code Ocean platform (https://codeocean.com/capsule/8321305/tree/v1). In total, samples from 21 patients were included in this study, comprising 18 LUAD tumor tissue samples and 18 normal tissue samples.
Single-cell data processing and analysis
Single-cell data preprocessing, dimensionality reduction, clustering, and visualization were performed using the Seurat package (v4.3.0.1) [12]. Batch effect correction across different patients and conditions was achieved using the Harmony package (v0.1.1) [13]. Cells were clustered based on principal component analysis (PCA) followed by uniform manifold approximation and projection (UMAP) for visualization [14].
Trajectory inference
Cellular trajectory analysis was conducted to explore differentiation dynamics. The CytoTRACE2 package (v1.0.0) [15] was used to estimate cellular differentiation potential, while lineage trajectories were inferred using the Slingshot package (v2.8.0) [16].
Copy number variation analysis
To infer large-scale chromosomal copy number variations (CNVs) from the single-cell transcriptomic data, we applied the inferCNV tool [17]. Malignant and non-malignant cells were distinguished based on inferred CNV profiles.
Functional enrichment analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the clusterProfiler package [18]. Marker genes identified from different cell clusters were subjected to enrichment analysis to reveal biological functions and signaling pathways.
Cell-cell communication analysis
Cell-cell communication networks were analyzed using the CellChat package (v2.1.2) [19]. Ligand-receptor interactions between different cell subtypes were systematically inferred to map intercellular communication patterns within the tumor microenvironment.
Metabolic flow analysis
Metabolic interaction analysis between fibroblast and epithelial subclusters was conducted using the MEBOCOST framework [20], which allows the prediction of metabolite-mediated cell-cell interactions based on single-cell transcriptomic profiles.
Spatial transcriptomics analysis
Spatial transcriptomic deconvolution and spatial localization analyses were performed using the CellTrek package (v0.0.94) [21]. Reference-based deconvolution integrated scRNA-seq profiles to spatial transcriptomics data to map the distribution and colocalization of cell types in both tumor and normal lung tissues.
MCI score and prognostic model construction
Univariate and multivariate Cox regression analyses were performed on the marker genes of three key cell subsets: MUC21 + Epi, Fb_COL11A1, and Fb_IGFBP4. Furthermore, six genes closely associated with patient survival were identified in the LUAD and LUSC datasets of the TCGA database, and MCI scores were built based on the corresponding indices of these genes:
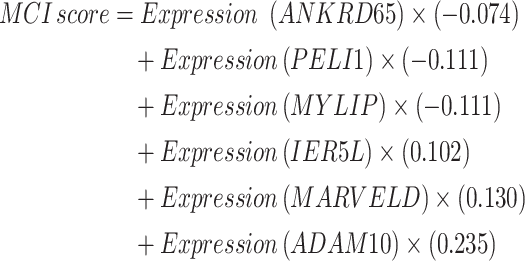 |
This score was used together with clinical information such as TNM stage, age, and gender to construct a nomogram and predict the 1-year, 3-year, and 5-year survival rates of lung cancer patients.
Statistical analysis
All statistical analyses were performed using R software (version 4.2.1). Group comparisons were conducted using the Wilcoxon rank-sum test. Kaplan–Meier survival analysis and Cox proportional hazards regression were applied to assess the prognostic significance, using the survival and survminer packages. Nomograms were constructed to predict overall survival based on Cox regression results, and their performance was evaluated using time-dependent receiver operating characteristic (ROC) curves generated by the timeROC package. A two-sided p value < 0.05 was considered statistically significant.
Results
Single-cell transcriptomic profiling reveals the cellular landscape of LUAD
To delineate the cellular heterogeneity of LUAD, we analyzed the scRNA-seq data from 21 patients, including 18 LUAD tumor tissues and 18 adjacent normal tissues. After quality control, dimensionality reduction, and batch effect correction, a total of 10 major cell populations were identified and visualized using UMAP (Fig. 1A). These populations were annotated as epithelial cells, fibroblasts, myeloid cells, T cells, B cells, NK cells, plasma cells, endothelial cells, mast cells, and neutrophils based on canonical marker gene expression.
Fig. 1.

Single-cell atlas of lung adenocarcinoma and adjacent normal tissues. A Uniform manifold approximation and projection (UMAP) visualization of 10 major cell populations in LUAD. B Stacked bar plot showing the distribution of 10 cell types across 21 patients. C Bar plots comparing cell type proportions between tumor (left) and normal tissues (right). D Bubble plot displaying marker genes for each cell type. E Feature plots illustrating spatial distribution of marker genes in UMAP space
The distribution of these 10 cell types across individual patients was consistent, although certain cell types displayed inter-patient variability (Fig. 1B). We next compared the cellular composition between tumor and normal tissues. As shown in Fig. 1C, tumor tissues exhibited a marked enrichment of epithelial cells, T cells and myeloid cells, whereas immune populations (such as T cell and B cells), and myeloid cells were relatively more abundant in normal tissues.
To further validate the identified cell types, we analyzed the expression patterns of known marker genes. A bubble plot summarized the top markers for each cell population, confirming their identity (Fig. 1D). Furthermore, feature plots illustrated the spatial distribution of key marker genes within the UMAP space, highlighting distinct transcriptional profiles across different cell types (Fig. 1E). These findings collectively provide a comprehensive single-cell atlas of LUAD tissues and their adjacent normal counterparts.
Heterogeneity and developmental potential of epithelial cell
We next performed the deep clustering of epithelial cells, revealing eight distinct subpopulations (MUC21 + Epi, SFTPC + Epi, MKI67 + Epi, C11orf97 + Epi, TMEM45A + Epi, RTKN2 + Epi, ASCL1 + Epi, and TFF2 + Epi) through UMAP visualization (Fig. 2A). Marker gene analysis confirmed distinct epithelial subpopulations: SFTPC+ (alveolar-like), MKI67+ (proliferative), ASCL1+ (neuroendocrine-like), and tumor-enriched TMEM45A+ (extracellular matrix remodeling) and RTKN2+ (cell adhesion), underscoring functional specialization within the tumor microenvironment (Fig. 2B).
Fig. 2.

Epithelial cell heterogeneity and differentiation dynamics. A UMAP plot of eight epithelial subclusters. B Bubble plot showing marker genes for epithelial subpopulations. C Bar plots comparing epithelial subset proportions (left) and cell counts (right) between tumor and normal tissues. D Violin plots of copy number variation (CNV) scores across epithelial subclusters. E Ro/e index heatmap depicting tissue-specific enrichment (red: tumor; blue: normal). F–G Potency score distribution and bar plot highlighting differentiation potential. H Trajectory analysis illustrating five evolutionary paths of epithelial subsets
Tumor tissues showed a pronounced expansion of MUC21 + Epi and MIK67 + Epi subpopulations, while SFTPC + Epi and C11orf97 + Epi subsets were relatively enriched in normal tissues (Fig. 2C). CNV scores further distinguished malignant subpopulations, with MUC21 + Epi and ASCL1 + Epi cells displaying elevated CNV levels, indicative of genomic instability (Fig. 2D). Ro/e index analysis highlighted the tumor-specific enrichment of TFF2 + Epi and MUC21 + Epi subpopulations, contrasting with the normal tissue dominance of RTKN2 + Epi and SFTPC + Epi subsets (Fig. 2E).
Developmental potential analysis revealed heterogeneous potency scores across subpopulations, with SFTPC + Epi exhibiting unipotent-like states, while C11orf97 + Epi subsets were predominantly differentiated (Fig. 2F–H). Trajectory inference delineated five evolutionary paths, linking SFTPC + Epi subpopulations to various differentiated states (RTKN2 + Epi, MKI67 + Epi and C11orf97 + Epi), suggesting dynamic plasticity during tumor progression (Fig. 2I). Collectively, these findings uncover the molecular and functional diversity of epithelial subpopulations, implicating specific subsets in LUAD pathogenesis.
Heterogeneity and prognostic significance of fibroblast subpopulations
Clustering of fibroblasts identified nine distinct subsets (Fb_MFAP5, Fb_GPC3, Fb_IGFBP4, Fb_CD52, Fb_RERGL, Fb_COL11A1, Fb_HIGD1B, Fb_WIF1, Fb_SERPINB2) with unique spatial distributions in UMAP (Fig. 3A). These subsets exhibited marker gene expression profiles (Fig. 3B), including MFAP5 (matrix organization), GPC3 (Wnt signaling), and IGFBP4 (metabolic regulation). Tumor tissues exhibited a significant increase in Fb_COL11A1 and Fb_IGFBP4 subpopulations, while Fb_MFAP5 and Fb_GPC3 were enriched in normal tissues (Fig. 3C).
Fig. 3.

Fibroblast subpopulations and prognostic significance. A UMAP plot of nine fibroblast subsets. B Bubble plot of fibroblast marker genes. C Bar plots comparing fibroblast subset proportions (left) and cell counts (right). D–F Enrichment analysis for Fb_MFAP5, Fb_GPC3, and Fb_IGFBP4. G Kaplan-Meier survival curves in lung adenocarcinoma patients with high and low Fb_IGFBP4 group
Functional enrichment analysis revealed subtype-specific roles: Fb_MFAP5 was linked to extracellular matrix organization and extracellular matrix organization (Fig. 3D), Fb_GPC3 to epithelial cell migration and cell − cell signaling by wnt (Fig. 3E), as well as Fb_IGFBP4 to extracellular structure organization and Golgi transport (Fig. 3F). Strikingly, LUAD patients with high Fb_IGFBP4 group showed markedly worse survival compared to the low group in LUAD (p = 0.0024), implicating this subset in tumor progression (Fig. 3G). These findings delineate fibroblast heterogeneity and highlight prognostically relevant subpopulations in LUAD.
Tumor-specific MUC21 + Epi-fibroblast crosstalk
Intercellular communication analysis revealed significant differences in interaction numbers between tumor and normal tissues, with tumor samples showing heightened CD99, FN1, COLLGENA and so on (Fig. 4A and S1A). Ligand-receptor pair analysis identified tumor-specific enrichment of fibroblast-epithelial crosstalk, notably between Fb_COL11A1/Fb_IGFBP4 and MUC21 + Epi subset, driven by different interactions, such as COL1A1/SDC4 and COL1A2/SDC4 (Figs. 4B and S1B).
Fig. 4.

Tumor-specific epithelial-fibroblast crosstalk. A Bar plots comparing interaction numbers between tumor (top) and normal tissues (bottom). B Bubble plots showing differential ligand-receptor interactions between fibroblast subsets and MUC21 + Epi. C Circos plots depicting MK pathway interactions from fibroblast subsets (senders) to MUC21 + Epi (receivers) in tumor (top) versus normal tissues (bottom). Line thickness reflects interaction strength. D Circos plots illustrating COLLAGEN pathway interactions between epithelial subclusters and fibroblast subsets in tumor (top) versus normal tissues (bottom)
Pathway-centric mapping highlighted tumor-dominant MK signaling from fibroblasts (Fb_COL11A1, Fb_IGFBP4, Fb_SERPINB2) to MUC21 + Epi subset, with thickened interaction lines indicating enhanced pro-tumorigenic signaling (Fig. 4C). In addition, COLLAGEN pathway interactions between epithelial cells and fibroblasts (Fb_COL11A1, Fb_IGFBP4) were amplified in tumors, implicating matrix remodeling in invasion (Fig. 4D). Reverse MK signaling from MUC21 + Epi to fibroblasts further underscored the dynamic reciprocity of tumor-stroma crosstalk (Fig. S1C). These findings delineate a rewired interaction landscape in tumors, driven by fibroblast-epithelial synergy and pathway-specific activation.
Metabolite-mediated fibroblast-epithelial crosstalk and spatial architecture
Metabolite-mediated crosstalk between fibroblast and epithelial subpopulations was markedly amplified in tumors. Comparative analysis revealed a significant increase in metabolite-driven interactions involving ASCL1 + Epi, MKI67 + Epi, TFF2 + Epi, and Fb_COL11A1 in tumor tissues, whereas normal tissues showed the main metabolite-driven interactions in MKI67 + Epi, C11orf97 + Epi, ASCL1 + Epi, and Fb_MFAP5 (Fig. 5A). Network visualization further demonstrated stronger and more complicated interaction intensities in tumors (Fig. S2A). These findings suggest a tumor-specific reliance on metabolic crosstalk to fuel epithelial-stromal crosstalk.
Fig. 5.

Metabolic crosstalk and spatial architecture. A Bar plots comparing metabolite-mediated interactions between tumor (left) and normal (right) tissues. B–D UMAP clustering (B), spatial mapping (C), and co-localization (D) in LUAD tissues. E–G Normal tissue counterparts (E UMAP; F spatial mapping; G co-localization)
Deconvolution mapping highlighted heterogeneous cellular distributions in tumors, with green spots representing raw spatial transcriptomic data and red overlays aligning single-cell reference profiles (Fig. S2B). UMAP visualization and co-localization analysis of spatial transcriptomes further identified dense interactions between epithelial cell subsets (mainly MKI67 + Epi and MUC21 + Epi) and fibroblasts in LUAD tumor tissues (Fig. 5B–D). In contrast, normal lung tissues exhibited uniform UMAP clustering (Fig. 5E), homogeneous spatial mappings (Fig. 5F), and minimal stromal-epithelial overlap (Fig. 5G), underscoring tumor-specific rewiring of cellular ecosystems.
Prognostic value of MCI score and molecular signatures in LUAD
To confirm that the three key subgroups of MUC21 + Epi, Fb_COL11A1, and Fb_IGFBP4 are not only associated with LUAD, but also closely related to the survival of patients with other types of NSCLC as well, we collected data from TCGA LUAD and LUSC patients to construct prognostic models. Prioritizing highly expressed genes from three critical subpopulations (MUC21 + Epi, Fb_COL11A1, and Fb_IGFBP4), we identified six survival-associated candidates. Multi-variable Cox regression found six survival-associated genes (ADAM10, MARVELD1, IER5L, MYLIP, PELI1, ANKRD65), with ADAM10 (HR = 1.716, p = 0.0044) and MARVELD1 (HR = 1.348, p = 0.0415) showing the strongest prognostic relevance (Fig. 6A). The MCI score, constructed from the expression profiles of these six genes, was statistically correlated with T stage, N stage and clinical stage, among which higher MCI score predicted aggressive clinicopathological features in general (Fig. 6B).
Fig. 6.

Prognostic value of the MCI score. A Forest plot of six survival-associated genes derived from critical epithelial and fibroblast subpopulations using multivariable Cox regression. B Violin plots linking MCI score to TNM stage and clinical stage. C–D Univariate/multivariate Cox analysis of prognostic factors. E Kaplan-Meier curves stratified by MCI score (high vs. low, p < 0.0001) in lung adenocarcinoma/lung squamous cell carcinoma. F Nomogram integrating MCI score and clinical variables for survival prediction. G Time-dependent ROC curves (1-/3-/5-years)
Univariate and multivariate analyses confirmed MCI score as an independent prognostic factor (HR > 1, p < 0.001), outperforming traditional variables like TNM stage (Fig. 6C–D). To verify whether MCI scores accurately stratify patients, we performed a survival analysis, using 0.95 as a threshold to divide patients into groups with high MCI scores and low MCI scores. Patients with high MCI score exhibited markedly worse overall survival compared to the low-score group in LUAD and LUSC (p < 0.0001; Fig. 6E). A nomogram integrating MCI score, TNM stage, gender, and age provided robust 1-/3-/5-year survival predictions (Fig. 6F), with time-dependent AUC values all more than 0.6, further validating its clinical utility (Fig. 6G). These results position MCI score as a potent biomarker for NSCLC risk stratification and prognosis.
Discussion
Our integrated single-cell and spatial transcriptomic analysis of LUAD revealed profound cellular heterogeneity and dynamic stromal-epithelial crosstalk. We identified eight epithelial subpopulations, including tumor-enriched subsets such as MUC21 + Epi (extracellular matrix remodeling) and ASCL1 + Epi (neuroendocrine-like), alongside nine fibroblast subclusters, with Fb_IGFBP4 emerging as key prognostic drivers. Tumor-specific interactions, mediated by COL1A1/SDC4 and MK signaling pathways, were amplified between these subpopulations. Spatial deconvolution further demonstrated colocalization of epithelial cells with fibroblasts in tumor niches. Finally, a six-gene MCI score derived from critical subpopulations robustly predicted patient survival, highlighting its clinical relevance.
Our clustering analysis uncovered several previously unrecognized epithelial subpopulations in LUAD, including the TMEM45A + Epi and RTKN2 + Epi subsets, which exhibit distinct functional specializations within the tumor microenvironment. These findings significantly expand the known heterogeneity of LUAD epithelial cells and provide new cellular targets for understanding tumor progression. Previous studies have linked MUC21 and ASCL1 expression to lung cancer progression [22, 23], but our study is the first to identify these markers as defining specific tumor-associated epithelial subpopulations (MUC21 + Epi and ASCL1 + Epi) at single-cell resolution. While the association between CNVs and extracellular matrix has been reported in colorectal cancer, our identification of this feature in MUC21 + Epi cells represents its first demonstration in LUAD epithelial subpopulations [24]. The differentiation trajectory from SFTPC + Epi to RTKN2 + Epi mirrors findings by Venkataramani et al. [25], who described neural-progenitor-like tumor cell transitioning to invasive states.
The nine fibroblast subsets identified here expand upon the traditional iCAF/myCAF classification method [26, 27]. Notably, Fb_IGFBP4, linked to extracellular matrix organization or Golgi vesicle transport, emerged as a potent prognostic marker, contrasting with prior studies that focused on FAP + or PDGFRα + fibroblasts [28, 29]. This discrepancy may reflect tissue-specific fibroblast adaptations in LUAD. The poor survival associated with Fb_IGFBP4 aligns with emerging evidence that IGFBP4 promotes epithelial-mesenchymal transition in lung cancer [30]. Moreover, we identified Fb_MFAP5 as a normal tissue-enriched subpopulation that may represent a homeostatic fibroblast state, distinct from tumor-associated subsets.
Our spatial analysis provides direct evidence of colocalization between epithelial cells with fibroblasts, a phenomenon previously inferred but not visualized in lung cancer [31]. While collagen-mediated stromal-epithelial crosstalk has been described in breast cancer [32], the LUAD-specific COL1A1/SDC4 interaction network localized to these niches represents a novel mechanism. This contrasts with prior lung cancer spatial studies emphasizing immune-stromal interactions [33], underscoring the need to reevaluate epithelial-stromal units as central regulators of tumor ecosystems.
This study has several limitations. First, the cohort’s limited ethnic diversity, along with underrepresentation of more lung cancer subtypes, may restrict the generalizability of our findings. Since this study only focused on LUAD, we will focus on a wider range of lung cancer subtypes, including LUSC and small cell lung cancer. Second, while we identified novel epithelial and fibroblast subpopulations through multi-omics integration, their functional roles in tumor progression remain to be validated through organoid co-culture or genetic perturbation experiments. Third, although the MCI score independently predicted survival outcomes, its modest accuracy (AUC < 0.7) highlights the need for more robust prognostic models incorporating dynamic multi-omics data (e.g., proteomics, methylation) or longitudinal monitoring. These limitations emphasize the necessity for large-scale, multicenter validation and mechanistic studies to advance the clinical translation of targeting these newly defined cellular subpopulations.
Conclusion
This study delineates NSCLC-specific epithelial and fibroblast subpopulations driving tumor progression through collagen-mediated crosstalk and metabolic symbiosis. Spatial mapping reveals their coordinated niche formation, while a prognostic model derived from these interactions enhances clinical risk stratification. These findings prioritize stromal-epithelial networks as therapeutic targets in LUAD.
Author contributions
Conceptualization, Jiajin Yang, Yanjun Lu; Investigation, Qiuping Xu; Software, Qiuping Xu; Formal Analysis, Jiajin Yang, Yanjun Lu; Writing—original draft, Jiajin Yang, Yanjun Lu; Writing—review and editing, Jiajin Yang, Yanjun Lu; Supervision, Jiajin Yang, Yanjun Lu; Visualization, Qiuping Xu; Funding acquisition, Jiajin Yang, Yanjun Lu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Not applicable.
Author contributions
YJJ, LYJ and XQP wrote the main manuscript text. All authors reviewed the manuscript.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval and consent to participate
The data utilized in this study were sourced from publicly accessible databases and were managed under approved ethical exemptions.
Consent for publication
The authors agreed to publication in the journal.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I et al (2024) Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin 74(3):229–263 [DOI] [PubMed] [Google Scholar]
- 2.Wu F, Fan J, He Y, Xiong A, Yu J, Li Y et al (2021) Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun 12(1):2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW et al (2020) Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11(1):2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Yu Q, Song T, Wang Z, Song L, Yang Y et al (2022) The heterogeneous immune landscape between lung adenocarcinoma and squamous carcinoma revealed by single-cell RNA sequencing. Signal Transduct Target Therapy 7(1):289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin G, Gao Z, Wu S, Zheng J, Guo X, Zheng X et al (2024) scRNA-seq revealed high stemness epithelial malignant cell clusters and prognostic models of lung adenocarcinoma. Sci Rep 14(1):3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li PH, Zhang X, Yan H, Xia X, Deng Y, Miao Q et al (2024) Contribution of crosstalk of mesothelial and tumoral epithelial cells in pleural metastasis of lung cancer. Transl Lung Cancer Res 13(5):965–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanley CJ, Waise S, Ellis MJ, Lopez MA, Pun WY, Taylor J et al (2023) Single-cell analysis reveals prognostic fibroblast subpopulations linked to molecular and immunological subtypes of lung cancer. Nat Commun 14(1):387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim D, Kim JS, Cheon I, Kim SR, Chun SH, Kim JJ et al (2022) Identification and characterization of cancer-associated fibroblast subpopulations in lung adenocarcinoma. Cancers 14(14) [DOI] [PMC free article] [PubMed]
- 9.Chen C, Guo Q, Liu Y, Hou Q, Liao M, Guo Y et al (2023) Single-cell and Spatial transcriptomics reveal POSTN(+) cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin Transl Med 13(12):e1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng C, Zhu G, Li Y, Wang H, Wang S, Li C et al (2023) Single-cell transcriptome analysis reveals cellular and molecular alterations in small cell lung cancer tumors following chemotherapy. Int J Cancer 153(6):1273–1286 [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Fan G, Li L, He Y, Lou N, Xie T et al (2023) Integrative analyses of bulk and single-cell RNA-seq identified cancer-associated fibroblasts-related signature as a prognostic factor for immunotherapy in NSCLC. Cancer Immunol Immunother CII 72(7):2423–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satija R, Farrell JA, Gennert D, Schier AF, Regev A (2015) Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33(5):495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K et al (2019) Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 16(12):1289–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao M, Huang J, Yang B, Liu Q, Luo M, Yang B et al (2023) Identification of efferocytosis-related subtypes in gliomas and elucidating their characteristics and clinical significance. Front Cell Dev Biol 11:1295891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang M, Armenteros JJA, Gulati GS, Gleyzer R, Avagyan S, Brown EL et al (2024) Mapping single-cell developmental potential in health and disease with interpretable deep learning. bioRxiv 2024:2024.2003.2019.585637.
- 16.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N et al (2018) Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19(1):477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen K, Wang Y, Hou Y, Wang Q, Long D, Liu X et al (2022) Single cell RNA-seq reveals the CCL5/SDC1 receptor-ligand interaction between T cells and tumor cells in pancreatic cancer. Cancer Lett 545:215834 [DOI] [PubMed] [Google Scholar]
- 18.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z et al (2021) ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. innovation (Cambridge (Mass) 2(3):100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH et al (2021) Inference and analysis of cell-cell communication using cellchat. Nat Commun 12(1):1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui K, Gao X, Wang B, Wu H, Arulsamy K, Dong Y et al (2023) Epsin nanotherapy regulates cholesterol transport to fortify atheroma regression. Circul Res 132(1):e22–e42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei R, He S, Bai S, Sei E, Hu M, Thompson A et al (2022) Spatial charting of single-cell transcriptomes in tissues. Nat Biotechnol 40(8):1190–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshimoto T, Matsubara D, Soda M, Ueno T, Amano Y, Kihara A et al (2019) Mucin 21 is a key molecule involved in the incohesive growth pattern in lung adenocarcinoma. Cancer Sci 110(9):3006–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meder L, König K, Ozretić L, Schultheis AM, Ueckeroth F, Ade CP et al (2016) NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int J Cancer 138(4):927–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heiser CN, Simmons AJ, Revetta F, McKinley ET, Ramirez-Solano MA, Wang J et al (2023) Molecular cartography uncovers evolutionary and microenvironmental dynamics in sporadic colorectal tumors. Cell 186(25):5620–5637e5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wißmann N et al (2022) Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185(16):2899–2917e2831 [DOI] [PubMed] [Google Scholar]
- 26.Chen B, Chan WN, Xie F, Mui CW, Liu X, Cheung AHK et al (2023) The molecular classification of cancer-associated fibroblasts on a pan-cancer single-cell transcriptional atlas. Clin Translat Med 13(12):e1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kennel KB, Bozlar M, De Valk AF, Greten FR (2023) Cancer-associated fibroblasts in inflammation and antitumor immunity. Clin Cancer Res Off J Am Assoc Cancer Res 29(6):1009–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z et al (2022) Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun 13(1):1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng H, An M, Luo Y, Diao X, Zhong W, Pang M et al (2024) PDGFRα(+)ITGA11(+) fibroblasts foster early-stage cancer lymphovascular invasion and lymphatic metastasis via ITGA11-SELE interplay. Cancer Cell 42(4):682–700e612 [DOI] [PubMed] [Google Scholar]
- 30.Xiao Y, Zhu S, Yin W, Liu X, Hu Y (2017) IGFBP-4 expression is adversely associated with lung cancer prognosis. Oncol Lett 14(6):6876–6880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen D, Shao M, Song Y, Ren G, Guo F, Fan X et al (2023) Single-cell RNA-seq with Spatial transcriptomics to create an atlas of human diabetic kidney disease. FASEB J Off Publ Feder Am Soc Exp Biol 37(6):e22938 [DOI] [PubMed] [Google Scholar]
- 32.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C et al (2015) Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol Quant Biosci Nano Macro 7(10):1120–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiang H, Pan Y, Sze MA, Wlodarska M, Li L, van de Mark KA et al (2024) Single-Cell analysis identifies NOTCH3-Mediated interactions between stromal cells that promote microenvironment remodeling and invasion in lung adenocarcinoma. Cancer Res 84(9):1410–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.