

Abstract
Background
Hao–Fountain syndrome (HAFOUS) is a rare neurodevelopmental disorder manifesting as several known symptoms, including speech and language delay, behavioral abnormalities, and intellectual disability. This rare condition is usually diagnosed by heterozygous deletion or mutation in the ubiquitin-specific protease 7 gene in conjunction with phenotype features.
Case presentation
We report the case of a 5-year-old Persian girl with this rare syndrome. The process of diagnosis, from perinatal examinations to the latest laboratory and clinical tests, is described for the first time in Iran.
Conclusion
Reporting all the symptoms of such a rare genetic case in detail emphasizes the importance of interdisciplinary teamwork and the necessity of raising awareness among therapists about probable upcoming problems; sharing such evident information with parents would help them manage the complexity of raising children with rare syndromes.
Keywords: Hao–Fountain syndrome, USP7, Developmental delay, Speech delay, Speech impairment, Occupational therapy, Case report
Background
Hao–Fountain syndrome (HAFOUS) is a rare neurodevelopmental disorder with a 1/1,000,000 prevalence worldwide and was first described in 2015 by Fountain et al., who reported six children with variable neurodevelopmental disorders. Global developmental delay, variably impaired intellectual development with significant speech delay, behavioral abnormalities, and mild dysmorphic faces were among the first described clinical characteristics of the syndrome, and other significant clinical observations are being added to further published case reports [1]. Since the first representation of this rare condition, several reports describing Hao–Fountain syndrome from genetic and clinical aspects have been published, and a recent cohort study described 32 novel cases to provide a clearer phenotype. The behavioral and physical characteristics of 16 patients, including prominent speech delays, intellectual disability ranging from borderline intellectual functioning to mild–moderate intellectual disability, and autism spectrum disorder (ASD) diagnosis or manifestations of autistic features in some children, were described in detail in an article by Fountain et al. in 2019. In addition, impulsivity, compulsivity, stubbornness, manipulative behaviors, and a propensity for temper tantrums and aggressive behaviors and attention deficit hyperactivity disorder (ADHD) were also noted. Among the physical characteristics, facial dysmorphisms, hypogonadism, abnormal pain thresholds, gastroesophageal reflux disease (GERD), a history of feeding problems, seizures, and abnormal brain magnetic resonance imaging (MRI) results have also been reported [2, 3]. Difficulties in coordination tests and the management of a sequence of movements and decreased sensitivity to pain were also described in an 18-year-old girl with HAFOUS whose rare condition of isolated tubal torsion was the main point of her case report. The Fallopian tube was necrotic in this patient, with adhesions to the bladder, bowel, and iliac vessels, all of which were managed [4]. Some HAFOUS phenotypes, such as visual deficits, hypotonia, impaired cognition and aggressive behavior, were revealed when USP7 was conditionally deleted in glutamatergic neurons in the mouse forebrain by Chen et al. [5]. Sun et al. reported developmental delay, specifically speech delay and mild-to-moderate autism symptoms, as well as abnormal magnetic resonance imaging (MRI) findings, in two cases [6].
A rare condition affecting several body functions and structures needs to be managed by a multidisciplinary team consisting of physicians, occupational therapists, speech pathologists, physiotherapists, psychologists, social workers, administrative staff, specialist medical services, and allied health services [7]. Occupational therapy, as a client-centered health profession concerned with promoting health and well-being through occupation [8], could be considered one of the key team members for managing multidimensional symptoms of Hao–Fountain syndrome; thus, this report aims to shed light on this syndrome’s effect on patients’ daily living functions and caregivers, as well as the importance of a multidisciplinary treatment approach.
Case presentation
The patient is a 5-year-old Persian girl, the second child of consanguineous and healthy parents, who was diagnosed with Hao–Fountain syndrome based on whole-exome sequencing. She was born at 38 weeks of pregnancy, with a birth weight of 3100 g and height of 51 cm and head circumference of 37 cm, and the growth parameters at the time of the report were height of 120 cm, weight of 25 kg, and head circumference of 52 cm. Gestational sonography (anomaly scan) was performed three times, and an echogenic itra cardiac focus (2–3 mm) was identified. At birth, her hypotonia and difficulty in breastfeeding were remarkable. She was admitted for 5 days to the neonatal intensive care unit (NICU) owing to low blood sugar and patent ductus arteriosus (PDA).
Frequent urinary infections led to the diagnosis of vesicoureteral reflux (VUR) when she was 2 months old. Sonographic assessment of the urinary system revealed a normal size, shape, and echotexture of the kidney, but the distal portion of the uterus was mildly dilated and showed increased peristalsis. Bilateral high-grade vesicoureteral reflux was diagnosed after the instillation of 37 megabecquerel (MBq) technetium-99m (Tc-99m) pretechnetate into the bladder via a catheter, and then dynamic imaging was performed via radionuclide cystography. Gel injection (hyaluronic acid/dextranome) into the bladder wall for VUR treatment at the age of 6 months was not successful; therefore, she underwent surgery (ureteroocystostomy) at the age of 2 years.
At the age of 1 year, a metabolic test (organic acids and acylglycines profile) was performed because of the developmental delay. The urine organic acid test, analyzing 112 analytes, revealed no abnormality.
Some facial dysmorphisms similar to Down syndrome and hypotonia led the medical team to rule out this diagnosis by karyotyping. A total of 20 G-banded metaphases at 450–550 band resolution was analyzed. In accordance with the International System for Human Cytogenomic Nomenclature (ISCN, 2016), the karyotype of the patient was reported as 46, XX (normal female). Whole-exome sequencing (WES) was performed on the basis of the phenotype, which led to the discovery of a likely pathogenic variant in the ubiquitin-specific peptidase 7 (USP7) gene at the age of 2.5 years. The variant was confirmed via Sanger sequencing to be heterozygous in our patient, and the parents and her older brother were normal.
The USP7 gene regulates many target proteins and interactors through its deubiquitinating activity and has been shown to play an essential role in regulating the stability of proteins that are implicated in crucial cellular processes such as mitosis, apoptosis, the cell cycle, DNA replication, and neuronal development.
The brain MRI report at the age of 2 years showed mild dilation of sulci of the occipital lobe and mild increased signals of the white matter adjacent to trigone of lateral ventricle (Fig. 1). Dysrhythmic discharges in both hemispheres were noted by electroencephalogram (EEG) findings when she was 1 year old, and pirimidon was prescribed by a pediatric neurologist. The MRI report at the age of 2 years was normal but somewhat smoggy, addressed by a pediatric neurologist. EEGs were repeated at least twice a year to track the abnormal discharges over time; the background was normal with dominant beta activity, but frequent generalized epileptical discharges (spike) was seen. Two generalized tonic colonic and some focal seizures occurred at the ages of 3 and 4 years, and since then, sodium valproate and diazepam have been added to her medications. After 2 years of medical management, epileptic discharges have decreased and seem to be under control.
Fig. 1.
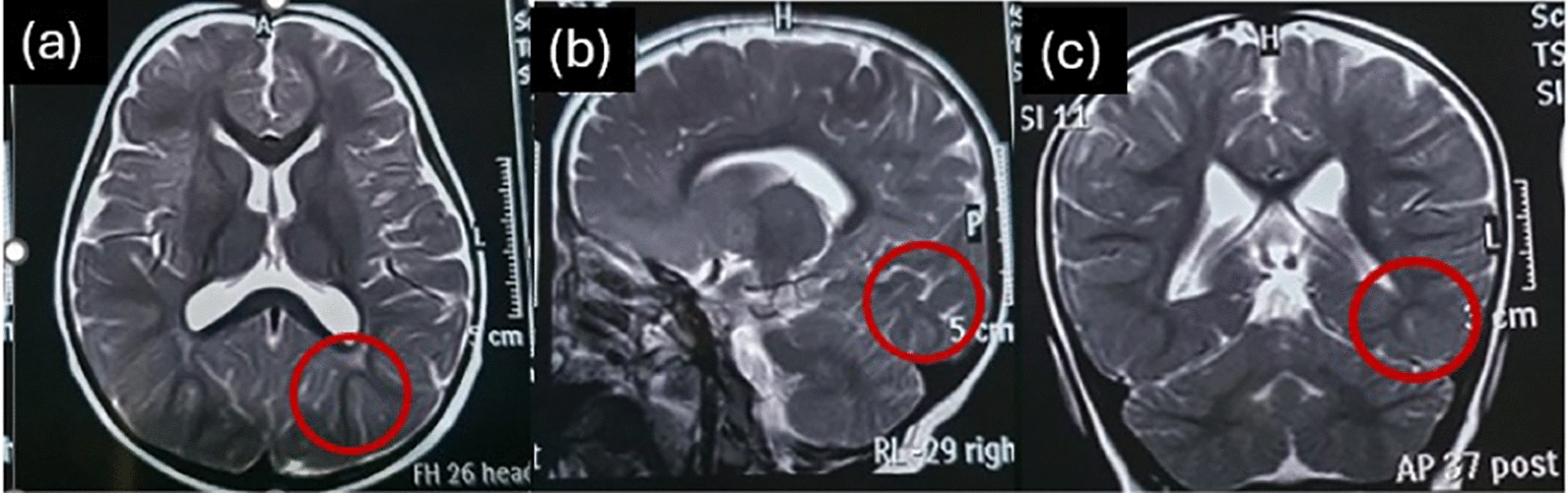
Mild dilation of sulci of the occipital lobe and mild increased signal of white matter adjacent to the trigone of the lateral ventricle (shown by red circles) in (a) axial, (b) sagittal, and (c) coronal view of brain magnetic resonance image
Her sleep disturbance had also been present for almost 2 years, but recently, her sleep pattern has tended to be normal. Recurrent respiratory infections have always led to increased antibiotic intake, and the recovery phase has lasted longer than that in children of the same age. Hypotonia, delays in milestone achievement, speech delays, significant behavioral disorders such as refusal to follow rules, and poor gross motor control and ataxic gait are the main clinical symptoms of this condition now, as determined by occupational therapy and speech pathology assessments. At the age of 3 years, she uttered her first word and still spoke single words; she also had some stuttering for approximately 6 months. Walking milestones were achieved by receiving 6 months of consistent neurodevelopmental occupational therapy at the age of 2 years. Dynamic balance is still challenging for her. Postural problems such as back knee deformity and forefoot pronation are also present, leading to imbalance while standing or walking, which are our current objectives in line with her independent walking and running in playground, goals set by the occupational therapist and the family.
She has not been formally evaluated by a standardized intelligence quotient (IQ) test, but special education is needed for age-related learning. She had never been formally diagnosed with autism spectrum disorder (ASD), but she exhibited symptoms such as poor eye contact. Hyperactivity and problems in attending to age-appropriate play and activities have always been a matter of family complaint, but no pharmacological treatment has been prescribed to manage these symptoms.
The main concern of her family has always been her attendance at school and being ready for successful participation in common peer-group activities and engagement in activities of daily life. The team general management approach for this client is to bring as much independence as possible, considering the extent of symptoms in need of attention.
The preterm aneuploidy, the result of karyotype and whole-exome sequencing in line with the main symptoms of our case: hypotonia, speech impairment, intellectual and behavioral problems, led her medical team to confirm the diagnosis of Hao–Fountain syndrome.
Discussion and conclusion
We report the case of a 5-year-old girl with HAFOUS with a background of developmental delay, urinary tract disease, intellectual disability, and speech delay who was the first patient diagnosed with this syndrome in Iran and who has been receiving occupational therapy services under supervision of the first author since she was 2 years old. Previous studies noted sleep disturbances, seizures, and postural deformities, which were also present in our patient [2]. Neuroradiology examination via brain MRI analysis and/or chart review revealed neuroradiology anomalies in 11 of 15 individuals in the Fountain et al. report, while some abnormality was reported in our case [2]. The high prevalence of developmental delay/intellectual disability, ASD, and hypotonia was confirmed in more than 50% of cases, and seizures were relatively less common; however, this is the main health concern in our case [2].
The characteristic facial features, including almond-shaped eyes, a small-appearing mouth with down-turned corners and a thin upper lip, and dolichocephaly in infancy, were reported, and almond-shaped eyes, and small mouth and nose were present in our case [9]. Recurrent respiratory infections were another symptom in common with our report, which was noted in a case report of a patient with USP7 with a description of fetal complications during pregnancy and cardiac involvement [9].
A recent questionnaire-based cohort study characterizing the phenotype of Hao–Fountain syndrome investigated 32 new cases, hyperphagia and increased body weight, an increased rate of birth complications, congenital anomalies, and abnormal pain thresholds. Speech impairment, the most delayed developmental milestone in our case, was found to be a potential hallmark of Hao–Fountain syndrome [3].
In a study published in 2023, researchers mapped a sensitive and specific DNAm episignature for pathogenic variants in USP7, and their main clinical findings were intellectual disability (100%) and developmental delay (100%). Most patients had speech delay (88%), behavioral abnormalities (93%), and hypotonia (53%). Facial features revealed that most of the patients had bulbous nasal tips (60%) or full lips (47%). Microcephaly (6%) and epilepsy (12%) were present in only a small number of patients [10].
In another report, abdominal “café au lait” spots were described in addition to other common characteristics [11]. This study described three novel patients with pathogenic/likely pathogenic missense and truncating variants in the USP7 gene. All three male patients manifested developmental delay/intellectual disability and brain abnormalities, which is in line with our findings and other reports [12]. Chen et al. conditionally deleted USP7 in glutamatergic neurons in the mouse forebrain, which revealed some HAFOUS phenotypes, such as visual deficits, hypotonia and abnormal gait, impaired cognition specifically in associated learning, and aggressive behavior [5]. In another report of two cases by Sun et al., developmental delay, specifically speech ,delay and mild-to-moderate autism symptoms and also abnormal MRI findings were noted [6].
This is the first description of a rare genetic disorder affecting several body functions and structures that leads to impairments in the daily functioning of individuals and involved families in Iran. Although no cure has yet been identified for this rare genetic syndrome, mapping the clinical features of such conditions may facilitate health management and help medical teams approach functional achievements. More studies are needed to determine the specific rehabilitation perspective toward this syndrome.
Acknowledgements
We acknowledge the KaryoGen private genetic laboratory in Isfahan Province, Iran, which confirmed the genetic analysis and proposed the diagnosis of Hao–Fountain syndrome. We also thank the family, who provided clinical data.
Abbreviations
- ASD
Autism spectrum disorder
- ADHD
Attention deficit hyperactivity disorder
- MRI
Magnetic resonance imaging
- VUR
Vesicoureteral reflux
- USP7
Ubiquitin-specific-processing protease 7
- PDA
Patent ductus arteriosus
- WES
Whole-exome sequencing
Author contributions
Search and data collection: Fatemeh Rafeienejad; analysis and interpretation of results: Nazila Akbarfahimi, Elahe Keyhani, and Fatemeh Rafeienejad; draft manuscript preparation: Fatemeh Rafeienejad; reviewed and edited paper: Nazila Akbarfahimi, Elahe Keyhani, Fatemeh Rafeienejad, and Narges Nouri. Genetic analysis: Narges Nouri. All the authors read and approved the final manuscript.
Funding
None.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Declarations
Ethics approval and consent to participate
IR.USWR.REC.1403.087.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests in this section.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hao Y-H, Fountain Michael D Jr, Fon Tacer K, Xia F, Bi W, Kang S-Hae L,, et al. USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol Cell. 2015;59(6):956–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fountain MD, Oleson DS, Rech ME, Segebrecht L, Hunter JV, McCarthy JM, et al. Pathogenic variants in <em>USP7</em> cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies. Genet Med. 2019;21(8):1797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimmer MC, Brennenstuhl H, Hirsch S, Dötsch L, Unser S, Caro P, et al. Hao-Fountain syndrome: 32 novel patients reveal new insights into the clinical spectrum. Clin Genet. 2024;105(5):499–509. [DOI] [PubMed] [Google Scholar]
- 4.Zampieri N, Pulvirenti R, Pedrazzoli E, Camoglio FS. Hao-Fountain syndrome and genital disorders: report of a new possible association. Ital J Pediatr. 2022;48(1):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Ferguson CJ, Mitchell DC, Risch I, Titus A, Paulo JA, et al. The Hao-Fountain syndrome protein USP7 regulates neuronal connectivity in the brain via a novel p53-independent ubiquitin signaling pathway. Cell Rep. 2025;44(2):115231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun M, Li Q, Zhang Y, Cai Y, Dong Y, Shu J, et al. Identification of two variants c.2697A > C and c.3305A > C in USP7 by analysis of whole-exome sequencing in Chinese patients with Hao-Fountain syndrome. Glob Med Genet. 2024;11(1):13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dall’Alba L, Gray M, Williams G, Lowe S. Early intervention in children (0–6 Years) with a rare developmental disability: the occupational therapy role. Hong Kong J Occup Ther. 2014;24(2):72–80. [Google Scholar]
- 8.Therapists WFoO. DEFINITIONS OF OCCUPATIONAL THERAPY from MEMBER ORGANISATIONS: World Federation of Occupational Therapists 2013 [Available from: https://wfot.org/resources/definitions-of-occupational-therapy-from-member-organisations.
- 9.Capra AP, Agolini E, La Rosa MA, Novelli A, Briuglia S. Correspondence on “Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies” by Fountain et al. Genet Med. 2021;23(2):421–2. [DOI] [PubMed] [Google Scholar]
- 10.van der Laan L, Karimi K, Rooney K, Lauffer P, McConkey H, Caro P, et al. DNA methylation episignature, extension of the clinical features and comparative epigenomic profiling of Hao-Fountain syndrome caused by variants in USP7. Genet Med. 2023. 10.1016/j.gim.2023.101050. [DOI] [PubMed] [Google Scholar]
- 11.Itani KN, Elfaki S. A rare case of Hao-Fountain syndrome mimicking fragile X syndrome. Cureus. 2023;15(9):e45332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng H, Mei S, Li F, Wei L, Wang Y, Huang J, et al. Expansion of the mutation spectrum and phenotype of USP7-related neurodevelopmental disorder. Front Mol Neurosci. 2022;15:970649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.