

Abstract
Background
5-Fluorouracil (5-FU) is a widely used chemotherapeutic agent; however, its clinical application is often limited by systemic toxicity and the development of drug resistance. To enhance its therapeutic efficacy, novel drug delivery strategies are under investigation. This study evaluated the use of platinum nanoparticles (PtNPs) as a nanocarrier system for 5-FU delivery to glioblastoma cells, focusing on their effects on apoptosis-related proteins.
Methods
The binding affinity and interactions of 5-FU with key apoptotic proteins (BAX, Bcl2, and Caspase-3) were assessed using molecular docking and validated through molecular dynamics (MD) simulations. PtNPs were synthesized and characterized via scanning electron microscopy (SEM), X-ray diffraction (XRD), and dynamic light scattering (DLS). Drug loading and encapsulation efficiency were determined, and cytotoxicity assays were conducted in U87 glioblastoma cells. The expression levels of apoptosis-related genes and proteins were evaluated to determine the biological impact of the formulations.
Results
Docking results confirmed effective binding of 5-FU to Bcl2, Caspase-3, and BAX, with MD simulations supporting stable complex formation, particularly with Bcl2 and Caspase-3. The synthesized PtNPs exhibited favorable physicochemical properties, including uniform morphology and high drug loading efficiency. In vitro release studies revealed a sustained release profile for the PtNPs/5-FU formulation. Furthermore, PtNPs/5-FU significantly downregulated the expression of EMT- and proliferation-related genes (cyclin D1, ZEB1, and Twist) and suppressed Bcl2 protein levels, resulting in enhanced apoptosis in U87 cells.
Conclusion
PtNPs effectively functioned as a delivery platform for 5-FU, improving its release kinetics and promoting apoptotic responses while potentially minimizing systemic toxicity. These findings support further exploration of PtNP-based drug delivery systems as a promising strategy for glioblastoma treatment.
Keywords: 5-Fluorouracil (5-FU), Platinum nanoparticles (PtNPs), Glioblastoma, Apoptosis, Drug delivery, Molecular docking, Molecular dynamics simulation
Introduction
Glioblastoma multiforme (GBM), classified as a World Health Organization (WHO) grade IV glioma, is an extremely aggressive and lethal form of brain cancer. It represents the most common malignant primary tumor of the central nervous system (CNS), accounting for approximately 50% of all malignant CNS tumors. Despite advances in treatment, the 5-year survival rate remains dismally low at only 7.2% [1–3]. GBM is notoriously difficult to manage due to its invasive nature and resistance to conventional therapies. The current standard of care, known as the Stupp protocol, introduced in 2005, involves maximal surgical resection followed by radiotherapy and chemotherapy with temozolomide [4]. However, therapeutic outcomes remain suboptimal, primarily because GBM cells infiltrate the surrounding healthy brain tissue, making complete surgical removal virtually impossible. Moreover, the presence of the blood–brain barrier (BBB) and the blood–brain tumor barrier (BBTB), which regulate the entry of molecules into the brain, poses a significant obstacle to drug delivery. These barriers limit the transport of hydrophilic drugs into tumor tissue by restricting passive diffusion and necessitating the use of specialized transport mechanisms such as absorption-based transcytosis, carrier-mediated transport, or receptor-mediated endocytosis [5].
Epithelial–mesenchymal transition (EMT) is a critical biological process in which epithelial cells lose their cell–cell adhesion and acquire mesenchymal characteristics, enhancing their migratory and invasive properties [6]. In GBM and other solid tumors, dysregulation of interconnected signaling pathways and transcriptional regulators has been shown to promote EMT, contributing to tumor progression and metastasis [7, 8]. Notably, nuclear localization of β-catenin, a hallmark of active Wnt signaling, has been frequently observed in the mesenchymal subtype of GBM. A study involving 30 GBM patients identified heightened Wnt/β-catenin pathway activation in this subtype [9]. Activation of this pathway in primary glioblastoma cells leads to upregulation of EMT-promoting factors such as SLUG, TWIST1, and ZEB1, which, in turn, enhance tumor cell migration and invasion both in vitro and in vivo [9–12]. Importantly, patients with elevated Wnt/β-catenin signaling have poorer overall survival, highlighting the clinical relevance of targeting this pathway in GBM. This underscores the urgent need for further research to clarify the mechanistic role of Wnt/β-catenin signaling in the initiation, progression, and recurrence of GBM [13].
5-Fluorouracil, an anti-metabolite drug approved by the U.S. Food and Drug Administration (FDA), shares structural similarities with the nucleic acid base uracil and is commonly prescribed for various types of cancer. Due to its rapid metabolism, it has a short half-life. This prodrug is converted intracellularly, where it inhibits thymidylate synthase and consequently interferes with the synthesis of RNA and DNA [14]. However, the clinical use of 5-FU is significantly limited by the need for high drug dosages, as well as by issues such as reduced bioavailability, short duration of therapeutic effect, and limited absorption. Additionally, according to the FDA, 5-FU has been associated with a wide range of serious side effects, including neurological toxicity, encephalopathy, hyperammonemia, mucositis, cardiotoxicity, and myelosuppression [15]. The administration of this chemotherapeutic agent—whether alone or in combination—requires substantially elevated doses, which place a heavy burden on the liver, immune system, and other organs, ultimately diminishing overall treatment efficacy.
Because of its hydrophilic nature, 5-FU has poor permeability across the protective BBB [16], making it an unsuitable treatment option for gliomas. Nevertheless, several strategies have been investigated to overcome this limitation, such as the use of 5-fluorocytosine (5-FC) as a prodrug, stereotactic intratumoral delivery, and nanoparticle-mediated drug delivery [17–19]. These approaches have shown promise in enhancing the anticancer efficacy of 5-FU in glioma treatment. Currently, monotherapies such as single-agent chemotherapy have shown limited effectiveness in cancer management. In contrast, combination therapies hold greater promise for improving therapeutic outcomes and minimizing adverse effects. Co-administration of drugs can lead to enhanced synergistic effects and help to overcome drug resistance mechanisms [20–22].
In recent years, nanomedicine has made significant strides in personalizing cancer treatment and addressing key limitations of traditional systemic therapies. The co-delivery of chemotherapeutic agents with nanoparticles has emerged as a highly effective strategy, often achieving superior therapeutic outcomes compared to monotherapy. The integration of diverse nanoparticles with anticancer drugs has evolved into a sophisticated and successful therapeutic approach [23]. Nanomedicine, a rapidly progressing field within nanotechnology, continues to expand due to its wide-ranging applications in biomedical science [10, 11]. Studies have demonstrated that nanoparticles used as nanotherapeutics offer distinct advantages over conventional drugs, including improved bioavailability, targeted delivery, and reduced toxicity [24, 25]. Among various nanocarrier platforms explored in oncology, polymeric nanoparticles, liposomes, and dendrimers have demonstrated considerable success in enhancing drug solubility, targeting, and circulation time. Polymeric systems (e.g., PLGA-based) offer controlled release properties but often face challenges with scalability and drug leakage [26]. Liposomes, such as those used in Doxil®, provide biocompatibility and have achieved clinical translation; however, they can suffer from instability and rapid clearance [27]. Dendrimers allow precise molecular architecture and multivalent interactions but may exhibit toxicity at higher generations [28]. In comparison, metallic nanoparticles, especially PtNPs, offer high drug loading, structural rigidity, ease of surface modification, and inherent therapeutic activity due to their catalytic and oxidative properties [29]. These unique features position PtNPs not merely as passive carriers but as multifunctional therapeutic agents capable of amplifying cytotoxic responses, justifying their evaluation in this study as a dual-functional delivery system for 5-FU [30].
Platinum nanoparticles (PtNPs) have been extensively investigated across a variety of fields, including the chemical and automotive industries, as well as in biomedical and therapeutic applications, owing to their versatile and valuable properties [31]. In the biomedical domain, PtNPs have garnered considerable attention due to their outstanding biocompatibility, high surface-to-mass ratio, nanoscale dimensions, strong reactivity, and excellent electrocatalytic activity [32]. Moreover, their unique optical properties, particularly surface plasmon resonance (SPR), make PtNPs promising candidates for use as radiosensitizing agents in radiotherapy [33]. Research has demonstrated that PtNPs exhibit significantly higher catalytic activity than other metal nanoparticles, such as palladium-based ones. This enhances their therapeutic potential, as platinum atoms within the particles can interact with DNA, thereby inhibiting its replication, a mechanism that underpins their potential anticancer effects [34].
In biomedicine, PtNPs have demonstrated substantial promise as diagnostic tools, components of medical implants, drug delivery vehicles, and sensitizers in photothermal therapy. Unlike other metal nanoparticles such as silver, PtNPs are generally associated with lower cytotoxicity [35–38]. Notably, they exhibit good biocompatibility with a variety of human cells, though cytotoxic effects may still occur at higher concentrations [39–42]. PtNPs are also capable of scavenging reactive oxygen species (ROS), reducing oxidative stress [43, 44]. However, they have also been associated with developmental alterations and elevated heart rate in zebrafish models [45], as well as inducing cellular damage, stress, genotoxicity, and DNA fragmentation in human monocytic cells and in vivo animal studies [46]. Similar cytotoxic effects have been observed in U2OS [42], and LNCaP cell lines [41]. Kutwin, Sawosz [47] revealed that when PtNPs were added to U87 glioma cells, they caused significant changes to their physical appearance, DNA structure, and metabolic functioning. Additionally, harmful effects on the cells' genetic material were detected [47].
The primary objective of this study was to evaluate the properties and biological activity of platinum nanoparticles (PtNPs) as a delivery system for the anticancer drug 5-fluorouracil (5-FU), with a focus on enhancing its pro-apoptotic effects in cancer cells. Specifically, the study aimed to assess the potential synergistic effects of combining PtNPs with 5-FU. The PtNPs were loaded with 5-FU, and their size, dispersion, surface characteristics, and drug encapsulation efficiency were thoroughly characterized. In vitro drug release experiments were conducted to evaluate the release profile of 5-FU from the nanoparticles. The biological effects of free 5-FU, bare PtNPs, and the PtNPs/5-FU complex on U87 glioma cells were examined in terms of cytotoxicity, along with the expression of key genes involved in cancer progression (β-catenin, Cyclin D1, ZEB1, and TWIST1), the anti-apoptotic marker Bcl-2, and the pro-apoptotic markers Bax and caspase-3.
Materials and methods
Materials
The U87 cell line (RRID: CVCL_0022) was obtained from Institut Pasteur. Penicillin–streptomycin, fetal bovine serum (FBS), and Dulbecco’s modified Eagle’s medium (DMEM) were obtained from GIBCO (Burlington, ON, USA). All of the chemicals, including hydrogen hexachloroplatinate (IV) hydrate (H2PtCl6·6H2O, 99.9%), 5-fluorouracil (5-FU), phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), sodium borohydride (NaBH₄), cetyltrimethylammonium bromide (CTAB), SnakeSkin™ dialysis membrane with a 10 kDa molecular weight cut off, annexin V–fluorescein isothiocyanate/propidium iodide (annexin V-FITC/PI) apoptosis kit, and complementary DNA (cDNA) synthesis kit, were acquired from Santa Cruz (NY, USA).
Synthesis of PtNPs
First, a solution was prepared in a beaker containing 10 mL of water with 0.1 M CTAB and 7.5 × 10−4 mM H2PtCl6. The mixture was heated to 60 °C and allowed to sit for a short period. Then, 1.3 mL of 0.01 M NaBH4 was introduced into the hot solution while stirring constantly. The solution quickly turned a deep brown color, indicating the formation of PtNPs. After stirring vigorously for 5 min, the solution was left to cool to room temperature (25 °C) [48].
Characterization of platinum nanoparticles
The synthesized platinum nanoparticles (PtNPs) were characterized to assess their morphology, structure, size distribution, and surface charge. The shape and surface morphology of the PtNPs were analyzed using scanning electron microscopy (SEM; VEGA3, TESCAN, Czech Republic). To evaluate the functional groups present on the NPs surfaces, Fourier-transform infrared spectroscopy (FTIR; Spectrum Two, PerkinElmer, USA) was performed in the spectral range of 400–4000 cm⁻1. The crystalline nature of the PtNPs was confirmed by X-ray diffraction (XRD; X'Pert PRO MPD, PANalytical, Netherlands) within a 2θ range of 10° to 80°. Additionally, dynamic light scattering (DLS) and zeta potential measurements were conducted using a particle size analyzer (Malvern Panalytical, UK) at 25 °C to determine the hydrodynamic diameter, polydispersity index (PDI), and surface charge of the NPs.
Preparation of 5-Fluorouracil loaded PtNPs
Initially, a weighed sample of PtNPs was suspended in pure water and then suitably diluted. The concentration of 5-FU was determined by constructing standard curves through the use of a ultraviolet (UV) spectrophotometer (NanoDrop, Thermo 2000) and measured at 260 nm wavelength. The amount of the drug contained in the nanoparticles was established by a calibration curve. To create PtNPs containing 5-FU, varying concentrations (ranging from 0.1 to 1 mg/mL) of 5-FU were dissolved in distilled water and mixed with PtNPs for 24 h at ambient temperature. The resulting solution was centrifuged at 15,000 rpm for 20 min, then the precipitate was washed with distilled water and the PtNPs with incorporated 5-FU were collected,
Determination of drug encapsulation efficiency (DEE) and drug loading efficiency (DLE)
The 5-FU that was not bound to anything was measured in the supernatant obtained after spinning. The amount of drug present was measured using Nanodrop spectrophotometry. Subsequently, the effectiveness of the loading process and the amount of 5-FU encapsulated were determined using the formulas in Eqs. (1) and (2) respectively. The experiments were carried out three times, and the results are expressed as mean ± SD.
| 1 |
| 2 |
In vitro release studies
To examine the drug release we used a solution containing 3 mg of 5-FU-loaded nanoparticles in 5 mL of 0.1 M PBS buffer solution at a pH of 7.4. The resulting solution was then inserted into a dialysis bag with a molecular weight cut-off range of 12–15 kDa. This bag was placed into 100 mL of phosphate buffer with varying pH levels (7.4 and 5.4). and placed inside a Microbial Shaker Incubator (model JTSL-40, Refrigerated) at 37 °C, with a stirring rate of 100 rpm. To maintain the same volume of the release medium, 5 mL of phosphate buffer was removed and replaced regularly. The absorption at 260 nm was monitored to determine the concentration of 5-FU released over time. The experiment was conducted three times.
In vitro cytotoxicity of 5-FU-loaded nanoparticles against U87 cells
The cytotoxic effects of free 5-FU, PtNPs, and 5-FU-loaded PtNPs (PtNPs/5-FU) were evaluated in vitro using the U87 human glioblastoma cell line. U87 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin, and maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cells were seeded into 96-well plates at a density of 1 × 104 cells per well and incubated for 24 h to allow for cell attachment. Subsequently, cells were treated with various concentrations of PtNPs, PtNPs/5-FU, or free 5-FU. Following a 72-h incubation period, the culture medium was removed and replaced with 200 µL of fresh medium containing 20 µL of MTT solution (5 mg/mL in sterile PBS solution). After 4 h of incubation at 37 °C, the supernatant was carefully discarded, and 200 µL of DMSO was added to each well to solubilize the formazan crystals formed by viable cells. The absorbance of the resulting solution was measured at 570 nm using a microplate reader (STAT FAX 2100, Awareness Technology, USA). Cell viability was calculated as a percentage using the formula:
All experiments were conducted in triplicate, and data were expressed as mean ± standard deviation [49].
Quantitative analysis of EMT- and proliferation-related gene expression
The expression levels of EMT-related and proliferation-associated genes (β-catenin, ZEB1, Cyclin D1, and Twist1) in U87 glioblastoma cells were evaluated at the mRNA level using quantitative real-time PCR (qRT-PCR). Cells were treated with platinum nanoparticles (PtNPs), PtNPs loaded with 5-fluorouracil (PtNPs/5-FU), or free 5-FU. Total RNA was extracted using the SimEX VNA kit and quantified with a NanoDrop spectrophotometer. Subsequently, 200 ng of total RNA from each sample was reverse transcribed into cDNA using the Parstous cDNA Synthesis Kit. Amplification was performed using SYBR Green I dye and the LightCycler® 480 system (SinaSYBR Green HS-qPCRMix, 2x). The PCR protocol included an initial denaturation at 95 °C for 5 min, followed by 40 cycles consisting of denaturation at 95 °C for 10 s, annealing at 60–62 °C for 10 s, and extension at 72 °C for 9 s. All reactions were performed in triplicate. Relative gene expression levels were calculated using the ΔΔCt method, with GAPDH serving as the internal reference gene [49]. The sequences of primers used in the experiment are listed in Table 1.
Table 1.
Sequence of the used primers
| Gene | Sequence (5′ → 3′) |
|---|---|
| ZEB1-F | GCACCTGAAGAGGACCAGAG |
| ZEB1-R | TGCATCTGGTGTTCCATTTT |
| β-catenin -F | CTGAAGAGGACCAGA |
| β-catenin -R | TCCTTCTGCACACATTTGA |
| Twist-F | GACGGTTCGCCATCCAGAC |
| Twist-R | TGTCCATTTTCTCCTTCTCTGGA |
| cyclin D1-F | GCTGCGAAGTGGAAACCATC |
| cyclinD1-R | GCTGCGAAGTGGAAACCATC |
Apoptosis assay
To evaluate apoptosis, the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Life Technologies) was employed, using both Alexa Fluor 488–conjugated Annexin V and propidium iodide (PI). U87 glioblastoma (GBM) cells were seeded at a density of 1 × 104 cells per well and incubated for 24 h. After removing the culture medium, cells were treated with either PtNPs, PtNPs/5-FU, or free 5-FU in complete medium and incubated for an additional 24 h. Following treatment, the cells were harvested and washed with cold phosphate-buffered saline (PBS), then stained using the Annexin V/PI staining protocol provided by the manufacturer. Flow cytometry analysis was performed using a Partec flow cytometer equipped with a xenon/argon laser. Fluorescence emissions were measured at 530 nm and 575 nm, following excitation at 488 nm. Cells exhibiting Annexin V-Alexa Fluor 488 fluorescence were considered to be in the early stages of apoptosis, while those with elevated PI fluorescence were identified as being in the late stages of apoptosis or undergoing necrosis. Data analysis was conducted using FlowJo v10 software, with gating and quadrant settings established based on both positive and negative control samples.
Western blotting
Cells were subjected to different treatments including Pt-NPs, Pt-NPs/5-FU and free 5-FU. The proteins from these cells were extracted by lysing them in radioimmuno precipitation assay buffer on ice for 20 min. Subsequently, they were centrifuged at 13,000 rpm for 1 h at a temperature of 4 °C. The concentration of proteins were determined by the Bradford method, (BioRad Laboratories, Hercules, CA, USA). A specific amount of 100 µg of protein underwent separation on 10% polyacrylamide gel from Biorad Laboratories. Subsequently, the proteins were transferred to a nitrocellulose membrane from Amersham Biosciences in Piscataway, NJ, USA. The membranes underwent a blocking process using 5% nonfat milk for 1 h, followed by exposure to a primary antibody (mouse monoclonal antibody) overnight. The experiment utilized primary antibodies recognizing Bcl2, BAX, and Caspase3. Post-incubation, the membranes were washed and then exposed to a secondary antibody (goat anti-rabbit antibody from Abcam) for 1 h. Luminescent signals were generated upon the addition of enhanced chemiluminescence (ECL) substrate (Pierce Biotechnology in Rockford, IL, USA) and were detected with the use of a fluorescent imager Li-Cor C-Digital Blot Scanner [50].
Protein–ligand docking
The 3D structure of 5-FU was retrieved from the PubChem database and energy-minimized using the MMFF94 force field. Crystal structures of target apoptosis-related proteins, Bcl2 (PDB ID: 1YSW), BAX (PDB ID: 6E6B), and Caspase-3 (PDB ID: 5IBP), were downloaded from the RCSB (Research Collaboratory for Structural Bioinformatics PDB) Protein Data Bank. These proteins were preprocessed by removing water molecules, adding polar hydrogens, and assigning appropriate Gasteiger charges. The binding sites for docking were defined based on biologically relevant ligand-binding regions reported in prior literature and annotated in UniProt. The selected binding pockets correspond to functional domains associated with apoptosis regulation, particularly the BH3 domain of Bcl2 and BAX, and the catalytic pocket of Caspase-3. Docking was performed using Chimera v1.16.1 in combination with AutoDock tools [51]. A grid box was centered over the binding site residues, and side-chain flexibility was allowed where structurally justified. Multiple docking runs were performed for each protein–ligand pair to ensure consistency. The docking protocol was validated via a redocking test, where known co-crystallized ligands (when available) were removed and re-docked to ensure RMSD ≤ 2.0 Å between predicted and experimental poses, ensuring docking reliability. The best binding conformations were selected based on lowest binding free energy and number of hydrogen bonds, and the results were further analyzed using LigPlot + for visualization of key interactions. No non-specific interactions were included, and binding orientations were assessed to confirm placement in catalytically or functionally relevant regions.
Molecular dynamics (MD) simulations
To assess the structural stability of the protein–ligand complexes over time, molecular dynamics (MD) simulations were conducted using GROMACS v4.6.5 with the CHARMM36 all-atom force field. Ligand topologies for 5-FU were generated using the CGenFF server. Each protein–ligand complex was solvated in a dodecahedral water box using TIP3P water molecules and neutralized with counter ions. The systems were energy-minimized using the steepest descent algorithm to eliminate steric clashes. Following minimization, Number–Volume–Temperature (NVT) and constant Number–Pressure–Temperature (NPT) equilibration steps were carried out at 300 K and 1 bar for 100 ps each, using the Berendsen thermostat and Parrinello–Rahman barostat. Position restraints of 1000 kJ/mol were applied to heavy atoms during equilibration. Subsequently, 100 ns production MD simulations were performed for each complex, with a time step of 2 fs. All covalent bonds involving hydrogen were constrained using the linear constraint solver (LINCS) algorithm. Electrostatics were calculated using the Particle Mesh Ewald (PME) method with a cutoff of 1.0 nm. Simulation stability was monitored through RMSD (root mean square deviation) and RMSF (root mean square fluctuation) plots. All three complexes (Bcl2/5-FU, BAX/5-FU, Caspase-3/5-FU) demonstrated stable RMSD values after 20 ns, indicating convergence of the system and reliable protein–ligand interaction dynamics. Grace software was used for trajectory visualization and data plotting. These results confirm that the docked poses remain stable within the biological binding pockets throughout the simulation, supporting the relevance of 5-FU interaction with apoptosis-regulatory targets.
Statistical analysis
The software program GraphPad Prism 8.4.3 was utilized to examine the statistical data collected from the experiment. The data was assessed using analysis of variance (ANOVA), and any variances in the mean were compared using the Tukey method. The findings were displayed as a mean with corresponding standard deviation. A significance level of P < 0.05 was used to determine the significance of any observed differences.
Results
Synthesis and characterization of PtNPs
X-ray diffraction (XRD) analysis
XRD analysis was performed to investigate the crystalline structure of the synthesized PtNPs. The diffraction pattern (Fig. 1) exhibited prominent peaks at 2θ values of approximately 40°, 46.5°, and 67.8°, which correspond to the (111), (200), and (220) planes of a face-centered cubic (fcc) crystal structure of platinum (JCPDS card no. 04–0802) [52]. These peaks confirm the crystalline nature of the PtNPs and their consistency with bulk platinum structure. To estimate the average crystallite size, the Debye–Scherrer equation was applied to the full width at half maximum (FWHM) of the (111) peak:
where D is the crystallite size, K is the shape factor (0.9), λ is the X-ray wavelength (1.5406 Å for Cu Kα), β is the FWHM (in radians), and θ is the Bragg angle corresponding to the (111) plane. Based on this calculation, the crystallite size was found to be approximately 10.6 nm, confirming that the synthesized PtNPs are within the nanoscale regime. This nanoscale crystallite size supports the suitability of PtNPs for biomedical applications, as it allows for high surface area and enhanced biological interaction. Furthermore, the similarity between the observed diffraction pattern and that of bulk platinum suggests that the nanoparticles retain the characteristic fcc structure of platinum even at the nanoscale.
Fig. 1.
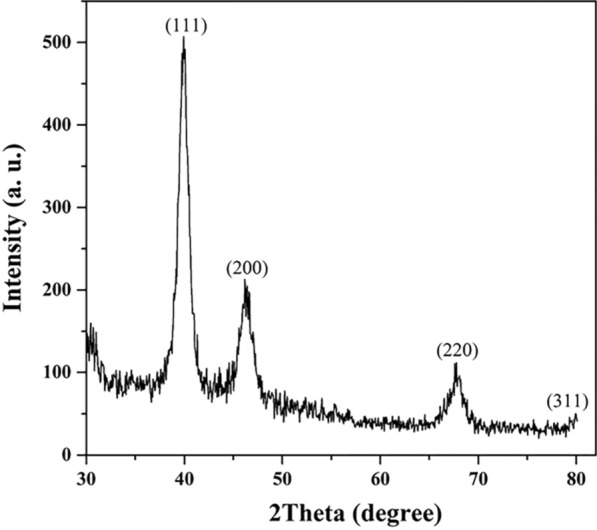
XRD pattern of Pt nanoparticles showing characteristic peaks at (111), (200), and (220) planes, confirming their face-centered cubic (fcc) structure
FTIR analysis
To confirm the successful synthesis and surface functionalization of PtNPs, FTIR spectroscopy was performed. The FTIR spectrum of the synthesized PtNPs (Fig. 2) reveals several key absorption bands indicative of both the nanoparticle surface chemistry and the presence of stabilizing agents. Prominent peaks at 2922.60 cm⁻1 and 2853.77 cm⁻1 correspond to C–H stretching vibrations from the methylene groups of CTAB, which was used as a capping and stabilizing agent during synthesis. These peaks confirm the adsorption of CTAB onto the nanoparticle surface, enhancing colloidal stability and preventing aggregation [53].
Fig. 2.

FTIR spectrum of Pt nanoparticles showing key absorption peaks at 2922 and 2853 cm⁻1 (C–H stretching), 1640 cm⁻1 (C = O stretching), 1024 cm⁻1 (C–O stretching), and additional fingerprint region peaks indicative of surface functional groups involved in stabilization
A strong peak observed at 1640.08 cm⁻1 is attributed to the bending vibration of O–H bonds, likely originating from adsorbed water molecules or residual hydroxyl groups in the synthesis medium [54]. The band at 1491.07 cm⁻1 may be assigned to N–H bending or CH₂ scissoring vibrations, further supporting the presence of CTAB or similar nitrogen-containing ligands on the PtNP surface [53]. Additional peaks at 1215.79 cm⁻1 and 1026.53 cm⁻1 correspond to C–N and C–O stretching modes, respectively, providing further evidence of organic moieties interacting with or stabilizing the NP surface [55]. Of particular interest is the low-frequency band observed at 436.27 cm⁻1, which is characteristic of Pt–O or Pt–N bond stretching. This peak strongly supports the successful reduction of H₂PtCl₆ to elemental Pt and the subsequent formation of metal–ligand bonds between Pt atoms and the stabilizing molecules in the reaction mixture. The presence of additional bands in the fingerprint region (900–600 cm⁻1) further suggests complex surface interactions likely resulting from residual surfactants and reducing agents.
Collectively, the FTIR data confirm not only the formation of PtNPs but also their surface modification by CTAB and other functional groups derived from the synthesis medium. These surface characteristics play a critical role in drug binding and nanoparticle stability in biological environments.
Scanning electron microscopy (SEM) analysis
The morphology and surface structure of the synthesized platinum nanoparticles (PtNPs) were examined using SEM at 75,000 × magnification (Fig. 3). The SEM image revealed that the PtNPs were predominantly spherical and well-dispersed, without evidence of significant aggregation. Based on scale bar analysis and visual inspection, the majority of NPs ranged approximately between 100 and 150 nm in diameter. This size range is consistent with typical nanomaterial classifications and supports their suitability for biomedical applications. The relatively uniform spherical shape and size distribution observed in the SEM images indicate that the synthesis protocol successfully produced NPs within a narrow nanoscale range, with morphological features conducive to cellular uptake and drug delivery.
Fig. 3.

SEM image of synthesized platinum nanoparticles (PtNPs), showing predominantly spherical morphology with size distribution primarily in the 100–120 nm range, consistent with nanoscale features suitable for biomedical applications
Dynamic light scattering (DLS) and Zeta potential analysis
DLS was employed to evaluate the hydrodynamic diameter and colloidal behavior of the synthesized PtNPs in aqueous dispersion. As shown in Fig. 4, the PtNPs exhibited a monomodal and relatively narrow size distribution, with an average hydrodynamic diameter of approximately 234.6 nm and a PDI of 1.0. The hydrodynamic size observed by DLS reflects not only the metallic core but also the surrounding solvation layer and any surface-adsorbed surfactants (e.g., CTAB), as well as potential minor aggregation under fluidic conditions. In comparison with the dry-state morphology seen under SEM, where particle sizes predominantly ranged from 100 to 150 nm, these results confirm the nanoscale characteristics of the synthesized PtNPs.
Fig. 4.

Dynamic light scattering (DLS) analysis of PtNPs showing a narrow size distribution centered around 234.6 nm with a polydispersity index (PDI) of 1.0. The measured zeta potential was – 1.319 mV, indicating low surface charge and moderate colloidal stability
Zeta potential measurements indicated a surface charge of approximately –1.319 mV. It is acknowledged that this value lies near the electrostatic neutrality zone, and thus may be suboptimal for ensuring long-term colloidal stability under certain conditions. Typically, nanoparticles with zeta potentials greater than ± 30 mV exhibit enhanced electrostatic repulsion and reduced aggregation. However, it is important to note that the use of surfactants such as CTAB, as well as steric stabilization mechanisms, may contribute to the transient stability of the nanoparticle suspension in the absence of strong electrostatic repulsion. Moreover, no visible signs of aggregation or precipitation were observed during synthesis, drug loading, or biological assays, indicating adequate short-term dispersion stability within the timeframe of these experiments. Future formulation optimization may consider surface modification strategies to enhance zeta potential and improve long-term stability for in vivo applications.
Drug encapsulation and loading efficiency
Efficient drug loading and entrapment are essential for enhancing the therapeutic performance of nanoparticle-based drug delivery systems. In this study, PtNPs were incubated with varying concentrations of 5-FU, ranging from 0.1 to 1 mg/mL, while maintaining a fixed nanoparticle mass of 3 mg. Following incubation, unencapsulated 5-FU was quantified from the supernatant using NanoDrop® spectrophotometry at 260 nm. DEE and DLE were then calculated using established formulas (Eqs. 1 and 2). The results demonstrated a maximum encapsulation efficiency (DEE) of 70%, indicating that a substantial proportion of the drug was successfully retained by the nanoparticles. Furthermore, the DLE reached up to 41%, reflecting a favorable drug-to-carrier ratio. These findings confirm the high loading capacity of the synthesized PtNPs and support their potential for use in targeted and sustained drug delivery applications in cancer therapy.
In vitro drug release
To assess the release profile of 5-FU from the synthesized PtNPs, in vitro release studies were conducted over a 72-h period at both physiological (pH 7.4) and acidic (pH 5.4) conditions, simulating the systemic and tumor microenvironments, respectively. The drug-loaded PtNPs were enclosed in dialysis membranes and immersed in PBS at 37 °C with regular medium replacement. The release profiles of free 5-FU and PtNPs-encapsulated 5-FU were compared under identical conditions. As shown in Fig. 5, free 5-FU exhibited a typical burst release, with approximately 60–65% of the drug released within the first 6 h at both pH levels. In contrast, the PtNPs/5-FU system demonstrated a substantially slower release, with only 25–30% of the drug released in the same timeframe. By the end of the 72-h period, cumulative release from PtNPs/5-FU reached approximately 40% (pH 5.4) and 35% (pH 7.4), while free 5-FU showed plateaued release within the first 10 h. These results support the capacity of PtNPs to mitigate premature release and prolong 5-FU availability, a desirable feature for reducing systemic toxicity and enhancing therapeutic efficiency.
Fig. 5.

Comparison of actual and predicted drug release profiles fitted to the biexponential kinetic model for free 5-FU and PtNPs-loaded 5-FU at pH 5.5 and 7.4. Each subplot displays the experimental cumulative release data (black diamonds) and the fitted biexponential model curve for: a free 5-FU at pH 5.5, b PtNPs-5FU at pH 5.5, c free 5-FU at pH 7.4, and d PtNPs-5FU at pH 7.4. The model captures both the initial burst and sustained release phases across all conditions
To quantitatively interpret the underlying release mechanisms, the drug release data were fitted to several conventional kinetic models, including zero-order, first-order, Higuchi, Korsmeyer–Peppas, and Hixson–Crowell. However, none of these standard models yielded an acceptable fit across all conditions, indicating a more complex, non-linear release behavior. Subsequently, a biexponential kinetic model was applied, which better described the biphasic release pattern involving an initial burst phase followed by a slower sustained release. The model is defined as:
where C(t) is the cumulative release at time t, A and B are the contributions of the burst and sustained phases, and k₁ and k₂ are the corresponding release rate constants. The fitted parameters and correlation coefficients (R2) are summarized in Table 2 and visualized in Fig. 5.
Table 2.
Fitting parameters of the biexponential kinetic model for 5-FU release from free drug and PtNPs formulations at pH 7.4 and 5.5
| Group | A (initial burst) | k₁ (h⁻1) | B (sustained phase) | k₂ (h⁻1) | R2 |
|---|---|---|---|---|---|
| PtNPs-5FU at pH 7.4 | 36.26 | 3.46 | 0 | 0.11 | 0.948 |
| Free 5FU at pH 7.4 | 60.13 | 2.80 | 1.64 | 0.03 | 0.986 |
| PtNPs-5FU at pH 5.5 | 24.95 | 1.31 | 3.29 | 0.05 | 0.928 |
| Free 5FU at pH 5.5 | 63.57 | 4.91 | 0.34 | 0.02 | 0.995 |
Parameters A and B represent the contributions of the burst and sustained phases, respectively, while k₁ and k₂ are the corresponding release rate constants
R2 indicates the goodness of fit
The strong correlation coefficients (R2 > 0.92) indicate that the biexponential model accurately captures the release kinetics across all conditions. In particular, PtNPs/5-FU demonstrated smaller burst release constants (k₁) and more prominent sustained phases compared to free 5-FU, validating the nanoparticles’ role in modulating release dynamics. The differences in behavior between pH 5.4 and 7.4 also suggest the pH-responsiveness of the system, which may facilitate selective release in the acidic tumor microenvironment. These findings confirm that the PtNPs/5-FU formulation offers a more controlled and extended drug delivery profile than the free drug. While some early-phase release remains, likely due to surface-adsorbed drug, the overall sustained release behavior supports the potential of PtNPs as effective nanocarriers. Future formulations employing surface modification strategies such as PEGylation or smart pH-sensitive coatings could further improve release control and reduce burst effects.
In vitro cytotoxicity activity
The cytotoxic potential of free 5-FU, PtNPs, and 5-FU-loaded PtNPs (PtNPs/5-FU) was evaluated against U87 glioblastoma cells using the MTT assay after 72 h of treatment. As shown in Fig. 6A, treatment with free 5-FU led to a dose-dependent reduction in cell viability, with marked cytotoxic effects observed at concentrations as low as 14 μg/mL. In contrast, Fig. 6B demonstrates that PtNPs alone exhibited significantly lower cytotoxicity across the tested concentration range, indicating minimal intrinsic toxicity of the carrier at lower doses. However, at elevated concentrations, a sharp decrease in cell viability was observed, suggesting dose-dependent effects potentially linked to oxidative stress or membrane disruption. Figure 6C highlights the cytotoxic profile of PtNPs/5-FU. The nanoparticle formulation induced cytotoxicity in a concentration-dependent manner, but its potency lagged behind that of free 5-FU at equivalent drug doses. A comparable reduction in cell viability to that observed with 14 μg/mL of free 5-FU required approximately 103 μg/mL of the nanoparticle formulation. This delayed cytotoxicity likely reflects the controlled and sustained release kinetics of 5-FU from the nanoparticle matrix, aligning with the design goals of nanoparticle-mediated drug delivery systems.
Fig. 6.

Cytotoxic effects of A free 5-fluorouracil (5-FU), B platinum nanoparticles (PtNPs), and C PtNPs loaded with 5-FU (PtNPs/5-FU) on U87 glioblastoma cells after 72 h, as measured by MTT assay. Dose-dependent cell viability is plotted against different concentrations of each treatment. Trendline equations and R2 values indicate the goodness of fit for each dose–response curve
Collectively, these findings confirm that while free 5-FU exhibits rapid and potent cytotoxic effects, PtNPs/5-FU offers a more gradual but sustained cytotoxic profile, which may be advantageous for reducing systemic toxicity and achieving prolonged therapeutic action in vivo.
Apoptosis assay
The Annexin V/PI assay was employed to evaluate the mode of cell death induced by different treatments in U87 glioblastoma cells. This technique distinguishes between viable, apoptotic, and necrotic cells based on phosphatidylserine exposure and membrane integrity. As shown in Fig. 7, untreated control cells displayed minimal signs of apoptosis or necrosis. Treatment with free 5-FU resulted in 14.64% late apoptotic cells and negligible necrosis, indicating its known apoptotic action. Interestingly, PtNPs alone led to significant necrotic cell death (55.83%) and only modest late apoptosis (13.04%), a pattern consistent with findings in other studies where unmodified metal nanoparticles induced oxidative stress, reactive oxygen species (ROS) generation, or disruption of plasma membrane integrity.
Fig. 7.

Flow cytometric analysis of apoptosis in U87 glioblastoma cells after 24-h treatment. Cells were stained with Annexin V-FITC and propidium iodide (PI) to distinguish between live, early apoptotic, late apoptotic, and necrotic populations. A Untreated control cells, B free 5-FU-treated cells, C PtNP-treated cells, and D PtNPs loaded with 5-FU (PtNPs/5-FU). The percentages in each quadrant indicate: Q1 (necrotic), Q2 (late apoptotic), Q3 (viable), and Q4 (early apoptotic) cells. PtNPs/5-FU notably increased apoptotic cell populations compared to control and individual treatments
However, when PtNPs were used to deliver 5-FU (PtNPs/5-FU), a marked shift in the cell death profile was observed. The combination treatment resulted in 7.59% late apoptosis and 33.40% necrosis, indicating a more balanced and regulated cytotoxic effect compared to PtNPs alone. While necrosis was present to some extent, particularly with the carrier alone, it is noteworthy that the PtNPs/5-FU formulation still promoted apoptosis as a primary mechanism of action. This apoptotic trend is crucial, as programmed cell death is a desired therapeutic outcome in cancer treatment due to its non-inflammatory nature. The reduced necrosis in the drug-loaded system compared to bare PtNPs suggests that 5-FU modulates the cytotoxic mechanism toward a more controlled pathway. These findings highlight that while necrosis remains detectable, particularly with unmodified PtNPs at higher concentrations, the data highlight the potential for further improvement in the nanoparticle design, such as surface modification or targeting strategies, to minimize off-target effects and favor apoptosis. These findings underscore the relevance of nanoparticle formulation in determining therapeutic outcomes and safety profiles.
Effects of treatments on gene expression by RT-qPCR
To investigate the molecular response of glioblastoma cells to treatment, the expression levels of key genes involved in proliferation and epithelial-mesenchymal transition (EMT), including β-catenin, ZEB1, Twist1, and Cyclin D1, were quantified using RT-qPCR in U87 cells following treatment with 5-FU, PtNPs, and PtNPs/5-FU. As illustrated in Fig. 8, all treatment groups led to a downregulation of these oncogenic and EMT-related transcripts compared to untreated controls. Notably, the PtNPs/5-FU combination produced the most significant suppression across all four genes, indicating a synergistic or enhanced effect of the nanoparticle-mediated delivery system. Free 5-FU also reduced gene expression levels, but to a lesser extent than the nanoparticle formulation, suggesting that sustained release and improved intracellular delivery may enhance therapeutic efficacy. PtNPs alone exhibited mild gene-suppressive effects, possibly due to intrinsic cytotoxic or stress-inducing properties of the metal nanoparticles. Among the analyzed genes, β-catenin and Cyclin D1, both critical regulators of cell proliferation and survival, showed the sharpest decline with PtNPs/5-FU treatment, emphasizing the potential of this formulation to interrupt Wnt/β-catenin signaling and cell cycle progression. These findings reinforce the therapeutic value of PtNP-mediated 5-FU delivery in targeting multiple molecular pathways involved in glioblastoma progression.
Fig. 8.

Quantitative real-time PCR analysis of gene expression levels in U87 glioblastoma cells following treatment. mRNA expression levels of A β-Catenin, B ZEB1, C Twist1, and D Cyclin D1 were evaluated in untreated cells, cells treated with free 5-FU, PtNPs, and the combination of PtNPs loaded with 5-FU (PtNPs/5-FU). Data are shown as log2 fold changes relative to the untreated control group, normalized to GAPDH. Combination treatment resulted in a significantly greater downregulation of all target genes compared to monotherapies. Statistical significance was determined using ANOVA with p-values indicated
Effects of treatments on protein levels by Western blotting
Western blot analysis was conducted to evaluate the expression levels of key apoptosis-related proteins, Bcl2, BAX, and Caspase-3, in U87 glioblastoma cells following treatment with free 5-FU, PtNPs alone, and the PtNPs/5-FU complex. β-actin was used as a loading control (Fig. 9). The results demonstrated a consistent downregulation of Bcl2, an anti-apoptotic protein, across all treatment groups, with the most pronounced reduction observed in the PtNP-treated cells. This significant suppression of Bcl2 may indicate an intrinsic pro-apoptotic or cytotoxic effect associated with unmodified platinum nanoparticles. In contrast, treatment with PtNPs/5-FU resulted in moderate Bcl2 downregulation, reflecting a more controlled apoptotic response. Both Caspase-3 and BAX, which are pro-apoptotic markers, showed relatively stable or increased expression in response to 5-FU and PtNPs/5-FU, suggesting activation of the intrinsic apoptotic pathway. Notably, the combination treatment (PtNPs/5-FU) maintained BAX and Caspase-3 expression while reducing Bcl2 levels, creating a favorable pro-apoptotic ratio (BAX/Bcl2), which is critical in tipping the balance toward programmed cell death. These findings provide molecular confirmation of apoptosis induction and support the notion that PtNP-mediated 5-FU delivery promotes a more desirable apoptotic mechanism compared to PtNPs alone, which may trigger necrotic or unregulated cell death pathways at higher doses.
Fig. 9.

Western blot analysis of apoptosis-related proteins in U87 glioblastoma cells treated with free 5-FU, platinum nanoparticles (PtNPs), and 5-FU-loaded PtNPs (Complex). Expression levels of Bcl2 (anti-apoptotic), Bax and Caspase 3 (pro-apoptotic) were evaluated. β-actin served as the internal loading control. Combination treatment (Complex) notably reduced Bcl2 expression while enhancing Bax and Caspase 3 levels, indicating a stronger apoptotic response compared to single-agent treatments
Protein–ligand docking
In this research, the crystal structure data for Bcl2 (1YSW), BAX (6E6B) and Caspase3 (5IBP) was utilized to demonstrate how 5-FU binds to these proteins. Binding pockets were defined based on published active site residues and UniProt annotations, followed by grid box generation to constrain ligand placement. Molecular docking was carried out using AutoDock Vina, and the top-ranked binding poses were selected based on the lowest binding energies and strongest hydrogen bonding patterns. Docking scores indicated that 5-FU exhibited favorable binding energies with all three targets: − 6.1 kcal/mol with Bcl2, − 5.4 kcal/mol with BAX, and − 6.3 kcal/mol with Caspase-3. Detailed analysis of the interaction networks using LigPlot + confirmed the presence of key hydrogen bonds and van der Waals contacts. Specifically, 5-FU formed hydrogen bonds with His91 and Lys15 in Bcl2 (Fig. 10), Lys57 and Gln32 in BAX (Fig. 11), and Arg64, Arg207, and Gln161 in Caspase-3 (Fig. 12), suggesting strong and specific ligand–receptor interactions.
Fig. 10.

Molecular docking analysis of 5-fluorouracil (5-FU) with Bcl2 (PDB ID: 1YSW). The left panel shows the 3D structure of Bcl2 with 5-FU bound in the active site, and the inset highlights key interactions with residues Lys15 and His91. The right panel presents a 2D interaction map generated by LigPlot + , showing hydrogen bonds and hydrophobic contacts stabilizing the 5-FU–Bcl2 complex
Fig. 11.

Molecular docking visualization of 5-fluorouracil (5-FU) bound to the pro-apoptotic protein BAX (PDB ID: 6E6B). The left panel illustrates the 3D binding pocket, highlighting interactions between 5-FU and key residues Lys57 and Gln32. The inset provides a magnified view of the interaction interface. The right panel shows a 2D interaction diagram generated by LigPlot + , indicating hydrogen bonds and hydrophobic contacts stabilizing the 5-FU–BAX complex
Fig. 12.

Molecular docking visualization of 5-fluorouracil (5-FU) bound to caspase-3 (PDB ID: 5IBP). The left panel shows the 3D binding conformation of 5-FU within the active site of caspase-3, highlighting interactions with key amino acid residues Arg64, Arg207, and Gln161. The inset provides an enlarged view of hydrogen bonding between 5-FU and the active site residues. The right panel presents a 2D LigPlot + interaction map, showing hydrogen bonds and hydrophobic interactions stabilizing the 5-FU–caspase-3 complex
Docking validation was performed by redocking co-crystallized ligands into the binding sites of reference proteins, yielding RMSD values below 2.0 Å, thereby confirming the reliability and accuracy of the docking protocol. Moreover, the convergence of multiple top-ranked poses around the same residues further validated the stability of the binding configurations. These findings underscore the biological relevance of the selected binding sites, all of which are involved in critical apoptotic regulation. The ability of 5-FU to target Bcl2 (anti-apoptotic), BAX (pro-apoptotic), and Caspase-3 (executioner caspase) provides mechanistic support for its role in apoptosis induction. The docking results thus form a theoretical basis for the enhanced apoptotic activity observed in U87 glioblastoma cells upon delivery of 5-FU via PtNPs.
Molecular dynamics simulation
The MD simulations were carried out for the Bcl2/5-FU, BAX/5-FU, and Caspase-3/5-FU complexes over a 100 ns trajectory to investigate their structural stability and dynamic behavior. The backbone root-mean-square deviation (RMSD) and residue-level root-mean-square fluctuation (RMSF) were analyzed to assess global and local conformational stability, respectively.
As shown in Fig. 13A, the RMSD plot revealed that the Bcl2/5-FU and Caspase-3/5-FU complexes exhibited relatively stable trajectories throughout the simulation, with average RMSD values fluctuating between 0.25 and 0.35 nm. This stability suggests that 5-FU forms stable interactions with both Bcl2 and Caspase-3, maintaining conformational integrity over time. In contrast, the BAX/5-FU complex showed greater fluctuations with RMSD values approaching ~ 0.45 nm, indicating a comparatively less stable interaction between BAX and 5-FU.
Fig. 13.

Molecular dynamics simulation results of 5-FU complexes with BAX, Bcl2, and Caspase-3. A RMSD (Root Mean Square Deviation) plot representing backbone stability of each complex over 100 ns simulation, indicating higher structural stability for Bcl2–5-FU and Caspase-3–5-FU compared to BAX–5-FU, B RMSF (Root Mean Square Fluctuation) plot showing atomic flexibility of protein residues upon 5-FU binding
The RMSF plot (Fig. 13B) provides further insight into the flexibility of specific amino acid residues. The BAX complex exhibited higher fluctuations across several residues, particularly in loop regions, suggesting greater structural mobility and reduced local stability. In contrast, the Bcl2 and Caspase-3 complexes displayed lower RMSF values, particularly at the binding site residues, confirming that ligand binding helped stabilize these protein regions. Therefore, these findings indicate that 5-FU establishes more stable and rigid interactions with Bcl2 and Caspase-3 compared to BAX. This supports its potential role in modulating apoptotic signaling pathways through preferential stabilization of key regulatory proteins.
Discussion
In this study, we investigated the effects of 5-FU alone and 5-FU delivered by PtNPs on apoptosis related proteins, Bcl2, BAX, and Caspase3. Firstly, we conducted protein–ligand docking studies, and further evaluation using molecular dynamics simulations. Next we synthesized and characterized PtNPs as vehicles for transporting 5-FU, evaluating their potential cytotoxicity, ability to induce apoptosis, and effects on gene and protein expression in U87 glioblastoma cells.
5-FU is a commonly utilized chemotherapy drug with a strong track record in treating various forms of cancer. This anti-tumor drug acts as an analog of naturally occurring pyrimidine bases, inhibiting the synthesis of DNA and hindering the growth of cancer cells, ultimately causing their death [14]. Despite being clinically effective, the widespread harm it causes in the body and its ability to induce resistance are still significant problems [56]. As a result, there is a growing interest in discovering drug delivery methods to improve its therapeutic ratio while reducing its harmful side effects [57]. This study has investigated the binding between 5-FU and proteins involved in apoptosis, and the potential application of PtNPs for transporting 5-FU to glioblastoma cells.
For protein–ligand docking, we utilized the established protein structures of Bcl2, BAX, and Caspase3, and showed that 5-FU has a strong affinity for these specific proteins. By analyzing the specific components of the binding site for each protein, we gained a useful understanding of the dynamics of 5-FU interactions. Our findings further support previous research indicating the vital role of hydrogen bonds in the specificity and stability of drug-target complexes [58].
By using the crystal structures of Bcl2, BAX, and Caspase3, the docking experiments demonstrated that 5-FU can successfully bind to these proteins, supporting earlier evidence of its involvement in apoptosis mechanisms [59]. The identification of key binding sites, including Tyr7 and Asp8 for Bcl2 and Pro49 and Arg37 for BAX, as well as the interaction with Caspase3, is essential in comprehending the mechanistic processes underlying 5-FU's ability to promote apoptosis (Chou et al., 2013).
The stability of Bcl2 and Caspase3 complexes with 5-FU was also verified through molecular dynamics simulations, which showed a reduction in RMSD throughout the simulation. The lower RMSF in these complexes suggests minimal changes in structure, indicating a strong bond with 5-FU in comparison to BAX. These findings are consistent with prior research indicating a high affinity between 5-FU and regulators of apoptosis [60].
The structures of the PtNPs were confirmed to be face-centered cubic crystals using XRD, demonstrating their stability as a carrier for drug delivery. The nanoscale and narrow size distribution of the PtNPs were verified through SEM and dynamic light scattering analysis, making them well-suited for use in biological contexts. Additionally, measurements of the zeta potential revealed that the PtNPs had appropriate suspension stability, making them effective for drug delivery purposes [61].
Analysis of the effectiveness of drug encapsulation revealed notable success in both capturing a large proportion of the drug (DEE) and loading it efficiently into the nanoparticles (DLE), making them promising for fighting tumors. The steady and prolonged release rate of 5-FU from PtNPs at various pH levels is consistent with controlled-release drug vehicles, which improve treatment outcomes while reducing negative effects [62]. The findings from the in vitro experiments comparing cytotoxicity indicate that although unbound 5-FU displayed greater initial toxicity, the formulation of PtNP-loaded 5-FU offers a more balanced cytotoxic effect which could potentially extend the duration of its therapeutic effects.
Experiments measuring the toxicity of the agents on U87 glioblastoma cells showed that initially, unbound 5-FU caused greater toxicity. However, the use of PtNP as a delivery system resulted in a more moderate level of cell death. These findings imply that PtNPs may have the ability to specifically attack cancer cells for a longer duration, thereby reducing the risk of harmful effects on the entire body [63]. While PtNPs demonstrated promising drug loading and delivery capacity, the observation of pronounced necrosis in cells treated with PtNPs alone raises important considerations for their clinical use. Metal NPs, including platinum-based ones, are known to induce necrosis through several mechanisms such as disruption of cellular membranes, generation of ROS, mitochondrial dysfunction, and inflammatory signaling [64]. Such uncontrolled cell death can lead to inflammatory responses, potentially damaging adjacent healthy tissues. This is especially concerning in non-target tissues where PtNPs might accumulate [65, 66]. However, our findings indicate that conjugation with 5-FU mitigates this effect, shifting the cell death mechanism toward apoptosis, a more controlled and favorable mode for cancer therapy. This suggests that while bare PtNPs may elicit undesired necrotic responses, their role as carriers can be harnessed safely when combined with therapeutic agents. Further optimization, such as PEGylation or ligand-mediated targeting, may enhance biocompatibility and minimize off-target toxicity, strengthening the translational potential of PtNP-based systems.
The findings of this study reinforce the strategic selection of PtNPs as a drug delivery system for 5-FU. While polymeric nanoparticles and liposomes are well-established nanocarriers with notable advantages in terms of biocompatibility and controlled release, their limitations, such as premature drug leakage or instability, remain significant hurdles [67]. In contrast, PtNPs demonstrated both effective encapsulation and improved sustained release, along with intrinsic cytotoxic activity that synergized with 5-FU to induce apoptosis and suppress EMT markers in glioblastoma cells. This dual functionality differentiates PtNPs from more conventional carriers, positioning them as a promising alternative or complement to current nanocarrier technologies. The observed biological effects further substantiate the unique therapeutic potential of PtNPs, especially in targeting aggressive tumors such as glioblastoma.
Prior studies have indicated that 5-FU displays high levels of toxicity in comparison to nanoparticles containing drugs. For instance, combining Dox with CS-Au-5-FU NPs showed great potential in inhibiting tumor cell growth when coupled with an aptamer, surpassing the effectiveness of using anticancer drugs alone [68]. Gurunathan et al., determined the IC50 value of PtNPs against THP-1 cells to be 75 µg/mL after a duration of 24 h [69]. Bendale et al. [39] demonstrated that PtNPs can cause harm to cancerous cell lines, without affecting normal cells, even at high levels. Studies have shown that PtNPs possess both anti-cancer and cytotoxic properties towards different types of cancerous and non-cancerous cells. These nanoparticles are able to enter cells through diffusion or endocytosis and ultimately form clusters within the cells [70, 71].
Confirmation of apoptosis initiation was established through the Annexin V/PI assay, which exhibited a higher occurrence of cell death caused by the 5-FU/PtNP complexes. Further investigation into gene expression using RT-qPCR showed a decrease in critical oncogenes including cyclin D1, ZEB1, and Twist, further reinforcing the ability of 5-FU to regulate pathways involved in cancer progression [72]. The Wnt/β-catenin pathway has a significant impact on cell growth and the formation of tumors, and was significantly downregulated by the use of the 5-FU nanoparticle delivery system, highlighting its important amplifying effect [73].
By utilizing apoptosis assays, it was verified that both 5-FU and PtNP-loaded 5-FU were effective in causing cell death. Furthermore, the effect on Bcl2 family proteins served as further evidence of the effectiveness of targeting apoptosis pathways in cancer treatment [74]. The more pronounced decrease of Bcl2 levels caused by PtNPs and PtNP/5-FU combinations is directly related to the observed increase in markers of apoptosis.
Mechanistically, integrated molecular docking and molecular dynamics (MD) simulation results offer critical insights into the apoptotic effects observed experimentally. The docking studies demonstrated that 5-FU exhibits strong binding affinities with apoptosis-related proteins, Bcl2, BAX, and Caspase-3, at biologically relevant regulatory sites. Specifically, 5-FU interacted with the BH4 domain of Bcl2, a region essential for its anti-apoptotic function, via key residues such as Tyr7, Asp8, and Lys15. Inhibition of this domain may impair Bcl2’s ability to neutralize pro-apoptotic factors, thereby facilitating mitochondrial outer membrane permeabilization (MOMP) and subsequent cytochrome c release [75]. In the case of BAX, 5-FU binding near residues Pro49 and Arg37, components of the core dimerization interface, could stabilize the active conformation of BAX, promoting oligomerization and membrane insertion, which are pivotal events in apoptotic pore formation [75]. Furthermore, binding of 5-FU to Caspase-3 near catalytic residues such as Arg64 and Cys163 may enhance its proteolytic activity or stabilize the active enzyme conformation [76]. These interactions correlate with the observed upregulation of cleaved Caspase-3 protein levels in vitro. RMSD and RMSF analyses from molecular dynamics simulations further confirmed that 5-FU forms more stable complexes with Bcl2 and Caspase-3 than with BAX, supporting the differential expression levels and apoptotic induction seen experimentally. These docking sites are functionally critical regions of the proteins, and the ability of 5-FU to occupy them reinforces its mechanism of action in disrupting the apoptotic balance toward cell death.
Additionally, the significant downregulation of EMT-related genes (β-catenin, Twist, ZEB1, and Cyclin D1) in the PtNPs/5-FU group indicates suppression of mesenchymal transition and proliferative signaling. Given that Wnt/β-catenin signaling is tightly linked to EMT induction and cell invasiveness in glioblastoma, the suppression of β-catenin observed at both gene and protein levels may stem from indirect inhibition of this pathway following 5-FU’s interaction with transcriptional regulators or through apoptosis-induced gene silencing [77, 78]. Overall, the combined use of PtNPs and 5-FU creates a synergistic effect, enhancing drug-protein binding, promoting apoptosis, and concurrently repressing EMT, thereby disrupting both survival and migration programs in glioma cells. This multi-target action underscores the therapeutic advantage of nanoparticle-assisted delivery platforms in precision oncology. The research has shown that PtNPs can serve as an effective vehicle observation for transporting 5-FU. This aligns with recent developments in nanomedicine. With improved drug stability, targeted precision, and effectiveness against tumors, using PtNPs as a vehicle for delivering 5-FU shows great potential for managing glioblastoma. Further progress and clinical trials are necessary to fully realize its benefits in treating cancer.
Conclusion
This study presents a comprehensive investigation into the therapeutic potential of 5-fluorouracil (5-FU) delivered via platinum nanoparticles (PtNPs) in the treatment of glioblastoma. The synthesized PtNPs demonstrated favorable physicochemical properties for drug delivery, including nanoscale size, structural uniformity, and appropriate surface charge. In vitro cytotoxicity assays revealed that while bare PtNPs alone exerted necrotic effects, the PtNP/5-FU complex predominantly induced apoptosis, suggesting a safer and more targeted mechanism of action.
Molecular docking and molecular dynamics (MD) simulations confirmed strong and stable interactions between 5-FU and key apoptotic regulators, including Bcl2, BAX, and Caspase-3. These in silico findings were corroborated by gene and protein expression analyses, which showed upregulation of pro-apoptotic markers and downregulation of anti-apoptotic and EMT-associated genes such as β-catenin, Twist1, ZEB1, and cyclin D1. Furthermore, in vitro drug release studies demonstrated that the PtNPs enabled a more controlled and sustained release of 5-FU compared to the free drug, particularly under tumor-mimicking acidic conditions. While initial burst release was partially observed, fitting the data to a biexponential kinetic model provided a more accurate representation of the release mechanism than traditional models.
The comparative discussion of PtNPs with other nanocarriers highlights their unique advantage as both delivery vehicles and cytotoxic agents, offering dual functionality not present in conventional platforms such as liposomes or polymeric systems. Collectively, these results underscore the translational potential of PtNPs/5-FU as a multifunctional nanotherapeutic approach for glioblastoma, capable of enhancing drug stability, promoting apoptosis, and suppressing tumor progression pathways. Further in vivo studies and surface modifications are warranted to optimize therapeutic efficacy and minimize off-target effects.
Acknowledgements
We would like to thank The Advocate Center for Clinical Research, Ayatollah Yasrebi Hospital and Shahrekord University of Medical Sciences for their nice support in this project.
Author contributions
The authors confirm contributions to this paper as follows: Investigation and Data collection, writing, and draft preparation: Atena Abed, Merat Karimi, Majid Nejati, Review and editing and validation: Atena Abed, Michael R Hamblin, Seyed Abbas Mirzaei, Mostafa Sarvizadeh, Hamed Mirzaei. Supervision and project administration: Seyed Abbas Mirzaei, Hamed Mirzaei.
Funding
This study was financially supported by Shahrekord University of Medical Sciences (Grant No. SKUMS-5673), and Kashan University of Medical Sciences and Ayatollah Yasrebi Hospital (Grant No. 99197).
Availability of data and materials
The data that support the findings of this study are available on request from the corresponding author.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mostafa Sarvizadeh, Email: sarvizadeh83@yahoo.com.
Hamed Mirzaei, Email: mirzaei-h@kaums.ac.ir, Email: h.mirzaei2002@gmail.com.
References
- 1.Koshy M, et al. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol. 2012;107:207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(1):Supplement_1-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu W, et al. Glioblastoma multiforme (GBM): an overview of current therapies and mechanisms of resistance. Pharmacol Res. 2021;171: 105780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- 5.Dong X. Current strategies for brain drug delivery. Theranostics. 2018;8(6):1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alizadeh J, et al. Simultaneous detection of autophagy and epithelial to mesenchymal transition in the non-small cell lung cancer cells. Autoph Diff Tissue Maint Methods Prot. 2019;1854:87–103. [DOI] [PubMed] [Google Scholar]
- 7.Mehta S, Lo Cascio C. Developmentally regulated signaling pathways in glioma invasion. Cell Mol Life Sci. 2018;75:385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madan B, Virshup DM. Targeting Wnts at the source—new mechanisms, new biomarkers, new drugs. Mol Cancer Ther. 2015;14(5):1087–94. [DOI] [PubMed] [Google Scholar]
- 9.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahlert UD, et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012;325(1):42–53. [DOI] [PubMed] [Google Scholar]
- 11.Foltz G, et al. Epigenetic regulation of Wnt pathway antagonists in human glioblastoma multiforme. Genes Cancer. 2010;1(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atkinson JM, et al. Activating the Wnt/β-catenin pathway for the treatment of melanoma–application of LY2090314, a novel selective inhibitor of glycogen synthase kinase-3. PLoS ONE. 2015;10(4): e0125028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, et al. Wnt/beta-Catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clin Exp Med. 2011;11:105–12. [DOI] [PubMed] [Google Scholar]
- 14.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8. [DOI] [PubMed] [Google Scholar]
- 15.Jha SK, et al. Enhanced antitumor efficacy of bile acid-lipid complex-anchored docetaxel nanoemulsion via oral metronomic scheduling. J Control Release. 2020;328:368–94. [DOI] [PubMed] [Google Scholar]
- 16.Beale JM, Block J, Hill R. Organic medicinal and pharmaceutical chemistry. Philadelphia: Lippincott Williams & Wilkins; 2010. [Google Scholar]
- 17.Menei P, et al. Stereotaxic implantation of 5-fluorouracil-releasing microspheres in malignant glioma: a phase I study. Cancer. 2004;100(2):405–10. [DOI] [PubMed] [Google Scholar]
- 18.Shinde G, et al. Enhanced brain targeting efficiency using 5-FU (fluorouracil) lipid–drug conjugated nanoparticles in brain cancer therapy. Prog Biomater. 2020;9:259–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cloughesy TF, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra75-341ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGuire WP, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, et al. Dairy milk fat augments paclitaxel therapy to suppress tumour metastasis in mice, and protects against the side-effects of chemotherapy. Clin Exp Metas. 2011;28:675–88. [DOI] [PubMed] [Google Scholar]
- 22.Lane D, et al. Acquired resistance to TRAIL-induced apoptosis in human ovarian cancer cells is conferred by increased turnover of mature caspase-3. Mol Cancer Ther. 2006;5(3):509–21. [DOI] [PubMed] [Google Scholar]
- 23.Mignani S, et al. Advances in combination therapies based on nanoparticles for efficacious cancer treatment: an analytical report. Biomacromol. 2015;16(1):1–27. [DOI] [PubMed] [Google Scholar]
- 24.Asharani P, et al. DNA damage and p53-mediated growth arrest in human cells treated with platinum nanoparticles. Nanomedicine. 2010;5(1):51–64. [DOI] [PubMed] [Google Scholar]
- 25.Conde J, Doria G, Baptista P. Noble metal nanoparticles applications in cancer. J Drug Deliv. 2012;2012(1): 751075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Operti MC, et al. PLGA-based nanomedicines manufacturing: technologies overview and challenges in industrial scale-up. Int J Pharm. 2021;605: 120807. [DOI] [PubMed] [Google Scholar]
- 27.Kraft JC, et al. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci. 2014;103(1):29–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain K, et al. Dendrimer toxicity: let’s meet the challenge. Int J Pharm. 2010;394(1):122–42. [DOI] [PubMed] [Google Scholar]
- 29.Yang Q, et al. Porous Au@Pt nanoparticles: therapeutic platform for tumor chemo-photothermal co-therapy and alleviating doxorubicin-induced oxidative damage. ACS Appl Mater Interfaces. 2018;10(1):150–64. [DOI] [PubMed] [Google Scholar]
- 30.Abed A, et al. Platinum nanoparticles in biomedicine: preparation, anti-cancer activity, and drug delivery vehicles. Front Pharmacol. 2022;13:797804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeyaraj M, et al. A comprehensive review on the synthesis, characterization, and biomedical application of platinum nanoparticles. Nanomaterials. 2019;9(12):1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawar AA, et al. Usage of platinum nanoparticles for anticancer therapy over last decade: a review. Part Part Syst Charact. 2021;38(10):2100115. [Google Scholar]
- 33.Testa G, et al. Functionalized platinum nanoparticles with surface charge trigged by pH: synthesis, characterization and stability studies. Beilstein J Nanotechnol. 2016;7(1):1822–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohammadi H, et al. Evaluation of synthesized platinum nanoparticles on the MCF-7 and HepG-2 cancer cell lines. Int Nano Lett. 2013;3:1–5. [Google Scholar]
- 35.Gurunathan S, et al. Cytotoxicity of biologically synthesized silver nanoparticles in MDA-MB-231 human breast cancer cells. Biomed Res Int. 2013;2013(1): 535796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt (II) agents, nanoparticle delivery, and Pt (IV) prodrugs. Chem Rev. 2016;116(5):3436–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, et al. Osteotropic peptide-mediated bone targeting for photothermal treatment of bone tumors. Biomaterials. 2017;114:97–105. [DOI] [PubMed] [Google Scholar]
- 38.Doherty RE, et al. Photodynamic killing of cancer cells by a platinum (II) complex with cyclometallating ligand. Sci Rep. 2016;6(1):22668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bendale Y, Bendale V, Paul S. Evaluation of cytotoxic activity of platinum nanoparticles against normal and cancer cells and its anticancer potential through induction of apoptosis. Integr Med Res. 2017;6(2):141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown AL, et al. Biodistribution and toxicity of micellar platinum nanoparticles in mice via intravenous administration. Nanomaterials. 2018;8(6):410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gurunathan S, et al. Graphene oxide–platinum nanoparticle nanocomposites: a suitable biocompatible therapeutic agent for prostate cancer. Polymers. 2019;11(4):733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurunathan S, et al. Tangeretin-assisted platinum nanoparticles enhance the apoptotic properties of doxorubicin: combination therapy for osteosarcoma treatment. Nanomaterials. 2019;9(8):1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gatto F, et al. Platinum nanoparticles decrease reactive oxygen species and modulate gene expression without alteration of immune responses in THP-1 monocytes. Nanomaterials. 2018;8(6):392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsumi H, et al. Pharmacokinetics and preventive effects of platinum nanoparticles as reactive oxygen species scavengers on hepatic ischemia/reperfusion injury in mice. Metallomics. 2014;6(5):1050–6. [DOI] [PubMed] [Google Scholar]
- 45.Asharani P, et al. Comparison of the toxicity of silver, gold and platinum nanoparticles in developing zebrafish embryos. Nanotoxicology. 2011;5(1):43–54. [DOI] [PubMed] [Google Scholar]
- 46.Gurunathan S, et al. The effects of apigenin-biosynthesized ultra-small platinum nanoparticles on the human monocytic THP-1 cell line. Cells. 2019;8(5):444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kutwin M, et al. Investigation of platinum nanoparticle properties against U87 glioblastoma multiforme. Arch Med Sci. 2017;13(6):1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu X, Wang T, Dong S. Rapid synthesis of cubic Pt nanoparticles and their use for the preparation of Pt nanoagglomerates. J Nanosci Nanotechnol. 2006;6(7):2056–61. [DOI] [PubMed] [Google Scholar]
- 49.Gholamian Dehkordi N, et al. Intelligent TAT-coupled anti-HER2 immunoliposomes knock downed MDR1 to produce chemosensitize phenotype of multidrug resistant carcinoma. J Cell Physiol. 2019;234(11):20769–78. [DOI] [PubMed] [Google Scholar]
- 50.Hosseini SA, et al. Epigenetic disruption of histone deacetylase-2 accelerated apoptotic signaling and retarded malignancy in gastric cells. Epigenomics. 2024;16(5):277–92. [DOI] [PubMed] [Google Scholar]
- 51.Chen S, et al. Uncovering the mechanism of resveratrol in the treatment of diabetic kidney disease based on network pharmacology, molecular docking, and experimental validation. J Transl Med. 2023;21(1):380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Şen F, Gökaǧaç G. Different sized platinum nanoparticles supported on carbon: an XPS study on these methanol oxidation catalysts. J Phys Chem C. 2007;111(15):5715–20. [Google Scholar]
- 53.Borodko Y, et al. Spectroscopic study of tetradecyltrimethylammonium bromide Pt−C14TAB nanoparticles: structure and stability. Langmuir. 2009;25(12):6665–71. [DOI] [PubMed] [Google Scholar]
- 54.Nayak S, McPherson IJ, Vincent KA. Adsorbed intermediates in oxygen reduction on platinum nanoparticles observed by in situ IR spectroscopy. Angew Chem Int Ed. 2018;57(39):12855–8. [DOI] [PubMed] [Google Scholar]
- 55.Dablemont C, et al. FTIR and XPS study of Pt nanoparticle functionalization and interaction with alumina. Langmuir. 2008;24(11):5832–41. [DOI] [PubMed] [Google Scholar]
- 56.Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Exp Opin Drug Saf. 2009;8(2):191–202. [DOI] [PubMed] [Google Scholar]
- 57.Chandran SP, et al. Nano drug delivery strategy of 5-fluorouracil for the treatment of colorectal cancer. J Cancer Res Pract. 2017;4(2):45–8. [Google Scholar]
- 58.Morris GM, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garcia-Mayea Y, et al. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol. 2020;60:166. [DOI] [PubMed] [Google Scholar]
- 61.Narayanan R, El-Sayed MA. Catalysis with transition metal nanoparticles in colloidal solution: nanoparticle shape dependence and stability. J Phys Chem B. 2005;109:12663–76. [DOI] [PubMed] [Google Scholar]
- 62.Khan MI, et al. Recent progress in nanostructured smart drug delivery systems for cancer therapy: a review. ACS Appl Bio Mater. 2022;5(3):971–1012. [DOI] [PubMed] [Google Scholar]
- 63.Kumari A, et al. Biodegradable polymeric nanoparticles based drug delivery systems. Coll Surf B Biointerf. 2010;75(1):1–18. [DOI] [PubMed] [Google Scholar]
- 64.Summer M, et al. Inflammatory response of nanoparticles: mechanisms, consequences, and strategies for mitigation. Chemosphere. 2024;363: 142826. [DOI] [PubMed] [Google Scholar]
- 65.Pedone D, et al. Platinum nanoparticles in nanobiomedicine. Chem Soc Rev. 2017;46(16):4951–75. [DOI] [PubMed] [Google Scholar]
- 66.Schrand AM, et al. Metal-based nanoparticles and their toxicity assessment. WIREs Nanomed Nanobiotechnol. 2010;2(5):544–68. [DOI] [PubMed] [Google Scholar]
- 67.De Leo V, et al. Recent advancements in polymer/liposome assembly for drug delivery: from surface modifications to hybrid vesicles. Polymers (Basel). 2021;13(7):1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sathiyaseelan A, et al. pH-controlled nucleolin targeted release of dual drug from chitosan-gold based aptamer functionalized nano drug delivery system for improved glioblastoma treatment. Carbohyd Polym. 2021;262: 117907. [DOI] [PubMed] [Google Scholar]
- 69.Gurunathan S, et al. Anisotropic platinum nanoparticle-induced cytotoxicity, apoptosis, inflammatory response, and transcriptomic and molecular pathways in human acute monocytic leukemia cells. Int J Mol Sci. 2020;21(2):440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gehrke H, et al. Platinum nanoparticles and their cellular uptake and DNA platination at non-cytotoxic concentrations. Arch Toxicol. 2011;85:799–812. [DOI] [PubMed] [Google Scholar]
- 71.Prasek M, et al. Influence of nanoparticles of platinum on chicken embryo development and brain morphology. Nanoscale Res Lett. 2013;8(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. [DOI] [PubMed] [Google Scholar]
- 73.Clevers HJC. Wnt/β-catenin signaling in development and disease. Cell. 2006;127(3):469–80. [DOI] [PubMed] [Google Scholar]
- 74.Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17(9):2941–2941. [DOI] [PubMed] [Google Scholar]
- 75.Gillies LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014;115(4):632–40. [DOI] [PubMed] [Google Scholar]
- 76.Flanagan L, et al. Low levels of caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016;7(2):e2087–e2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim KH, et al. Wnt/β-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013;15(2):161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He L, et al. Wnt/β-catenin signaling cascade: a promising target for glioma therapy. J Cell Physiol. 2019;234(3):2217–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.