

Abstract
Peptide synthesis is a highly optimized process that has led to the production of new classes of therapeutics and materials. The process of peptide synthesis is straightforward: commercially available, orthogonally protected amino acids can be linked on the solid phase using highly efficient coupling agents. However, the simplicity of peptide synthesis masks a significant drawback of the current method: it is highly wasteful and utilizes a solvent that is facing restrictions on its use. A catalyst that allows solid phase synthesis of peptides in benign solvents without requirement for excess reagents and protected amino acids would have a significant impact. Here, we describe the development of a small molecule catalyst for peptide synthesis. The catalyst design incorporates redox recycling of diselenide and phosphine with air as the ultimate oxidant and phenylsilane as the ultimate reductant. The catalyst affords efficient coupling of amino acids in the solution and solid phase. Significantly, the catalyst functions with acetonitrile, bypassing the need for DMF. The current effort builds on mechanistic analysis of reaction rates and intermediates in our prior work which led to a hydrogen bonding catalyst: [ Handoko; PanigrahiN. R., ; AroraP. S., . J. Am. Chem. Soc. 2022, 144, 3637–3643 ]. Here, we significantly simplified earlier designs to afford an easily accessible small molecule catalyst.


Introduction
Peptides are gaining momentum as therapeutics with over 100 candidates currently in clinical trials and dozens approved as drugs. Chemical synthesis remains a mainstay method for the production of these therapeutics. Merrifield’s seminal efforts to develop solid phase peptide synthesis (SPPS), along with the advent of orthogonal protecting groups and highly efficient coupling reagents have allowed automation of peptide synthesis. The success of SPPS relies on the use of excess reagents to drive reactions to completion. However, this inherent advantage of SPPS leads to a resource intensive and wasteful process that consumes excess reagents and solvents. Conventional SPPS methods typically require 3–5 equiv of both protected amino acids and coupling agents for each amide bond formed. ,, The requirement for excess reagents is even higher in the emerging flow peptide chemistry technology. Additionally, the preferred solvent for amino acid coupling in peptide synthesis, N,N-dimethylformamide (DMF), possesses reprotoxic properties, leading to restrictions to its use. , While SPPS remains a vital method, addressing its nonsustainability has become a critical challenge to sustain the success of peptides as therapeutics.
Numerous strategies to achieve catalytic amide bond formation from carboxylic acids and amines have been discussed. − Lewis acid catalysts, particularly boron-based reagents, reported by Yamamoto in 1996, have garnered significant attention. Borate esters , and multiboron − catalysts enable condensation of a range of amines and carboxylic acids. Progress has also been made in the development of transition metal catalysts, with ZrCl4 and Ti(Oi-Pr)4 emerging as promising options. However, the use of these reagents in SPPS has remained limited because these catalysts require prolonged reaction times, and organometallic catalysts may chelate with oligoamide backbones. An ideal catalyst for peptide synthesis would possess the following attributes: (i) short coupling times, preferably 1–2 h, (ii) compatibility with less toxic solvents, and (iii) compatibility with solid phase conditions.
We recently reported a biomimetic organocatalyst for peptide synthesis. − Our approach, inspired by the work of Mukaiyama and others, centers on a redox condensation procedure to activate carboxylic acids as selenoesters in the presence of the diselenide organocatalyst and exogenous phosphine. − The catalytic system required four components: the urea–diselenide catalyst 1a, substoichiometric amount of strained phosphorus reagent 2-[O], silane for recycling of phosphorus(V) to phosphorus(III), and oxygen to reoxidize selenol to diselenide. The system was designed to have an amino acid reversibly attach as a selenoester to the organocatalyst, which features a urea scaffold designed to stabilize the tetrahedral intermediate between amino acids and facilitate amide bond formation (Figure A). ,− The catalyst enabled coupling of Fmoc-amino acids, without detectable epimerization, and showed promise for oligopeptide synthesis on a solid support. The dual catalytic system leverages silanes as the terminal reductant and oxygen as the terminal oxidant (Figure A). Significantly, the reaction proceeded in acetonitrile, allowing us to bypass DMF as a solvent.
1.

(A) Our previous work on sustainable peptide synthesis showed that a combination of urea diselenide catalyst 1a and phosphetane oxide 2-[O] can lead to catalytic formation of the amide bond. This dual catalytic system utilizes phenylsilane as the terminal reductant and molecular oxygen as the terminal oxidant to drive the catalytic cycle. (B) 31P NMR analysis of key intermediates reveals that the intermolecular reaction between phosphetane 2 and diselenide 1b to generate the key selenophosphonium intermediate 3 is sluggish, requiring approximately 4 h for completion. (C) In the prior work, the selenophosphonium intermediate was generated via an intermolecular reaction between phosphetane 2 and a diaryldiselenide. (D) In the current effort, we developed a small molecule organocatalyst for peptide synthesis wherein the selenophosphonium intermediate is generated through an intramolecular reaction.
Here, we build on this previous effort to introduce a simpler and more efficient catalyst for amide bond formation. The new design results from a rigorous analysis of the reaction mechanism and the intermediates. The redesigned catalyst can be synthesized on a multigram scale and enables rapid peptide synthesis, in solution or solid phase, from conventionally protected amino acids with acetonitrile as the solvent.
Results
Preliminary Studies Support the Intramolecular Catalyst Design
The goal of this project is to reduce the requirement for superstoichiometric amounts of protected amino acids and coupling agents in solid phase peptide synthesis. Our strategy to create an efficient catalyst for amide bond formation builds on a four-component redox recycling mechanism (Figure A). − We rigorously investigated the mechanism of the catalytic cycle by 1H, 19F, 31P, and 77Se nuclear magnetic resonance (NMR) to identify the critical intermediates and enhance its overall efficiency. The mechanistic analysis with a model diselenide 1b revealed that the intermolecular reaction of sterically hindered phosphetane 2 with 1b to afford selenophosphonium 3 is slow (Figure B). The selenophosphonium adduct constitutes a key intermediate in the catalytic cycle, whose condensation with carboxylic acid affords a selenoester. Based on this insight, we designed a small molecule catalyst in which the selenophosphonium intermediate is obtained from an intramolecular reaction (Figure C,D).
The reenvisioned catalytic cycle to access the selenophosphonium intermediate via an intramolecular reaction is shown in Figure A. Reduction of phosphetane oxide-linked diselenide (I) with silane is expected to provide intermediate II, which is predicted to undergo an intramolecular reaction to yield the desired selenophosphonium adduct III. Reaction of carboxylic acid with III would yield selenoester V, after an acyl transfer step involving intermediate IV. Condensation of the selenoester with an amine provides an amide bond and liberates selenol, which is oxidized under aerobic conditions to yield diselenide I for the next cycle. The small molecule catalyst design draws inspiration from recent efforts to promote redox organocatalytic reactions. ,−
2.

(A) Proposed catalytic cycle for the redesigned organocatalyst for amide bond synthesis. The proposed cycle begins with the reduction of phosphetane oxide to generate II which is predicted to undergo an intramolecular reaction to generate selenophosphonium intermediate III. The subsequent reaction of carboxylic acid with III would lead to the formation of selenoester V via an acyl transfer step involving intermediate IV. Condensation of the selenoester with an amine facilitates amide bond formation, releasing selenol VI which is then oxidized under aerobic conditions to regenerate catalyst I, completing the catalytic cycle. (B) Preliminary comparison of the catalytic efficiency of the intermolecular catalytic system (1c + 2-[O]) and intramolecular catalytic system 3a and 3b for catalytic amide bond formation.
To determine if the switch to an intramolecular reaction to obtain the selenophosphonium adduct leads to an overall increase in the efficiency of amide bond formation, we compared the rates of formation of a model amide product (6) from toluic acid (4) and benzylamine (5). Treatment of the carboxylic acid and amine with 20 mol % diphenyl-diselenide (1c) and 20 mol % phosphetane oxide (2-[O]), along with phenylsilane (2 equiv) in acetronitrile at 80 °C affords the amide product 6 in 15% yield after 30 min. [Two equiv of phenylsilane are required in the catalytic cycle because one equivalent is needed for absorption of water produced during the reaction.] In contrast, the benzylphosphetane oxide diphenyl-diselenide 3a, which would enable intramolecular formation of a 5-membered selenophosphonium intermediate, exhibited a substantial increase in the yield of the amide product under identical conditions within 30 min. We next compared the impact of the ring sizes on the reaction progress. Catalyst 3b features an ethylphosphetane oxide-tethered to diphenyl-diselenide and is designed to generate a 6-membered selenophosphonium intermediate. As expected, the 5-membered ring intermediate leads to the amide product in higher yields as compared to the 6-membered ring intermediate, highlighting the kinetics of the 5-membered ring formation.
The above results support our hypothesis that intramolecular access to the selenophosphonium intermediate would result in a more efficient catalyst. We next focused on optimizing the electronics of the aryl ring to optimize the catalyst. In our proposed catalytic cycle (Figure ), selenium acts as an electrophile in the formation of selenophosphonium intermediate III from II but as a nucleophile in the formation of selenoester (IV → V). A selenide is also a leaving group in step II to III. Our foray into catalyst optimization began with the synthesis and screening of electronically diverse analogs of 3a (Figure ).
3.

(A) Overview of two distinct synthetic routes developed for phosphetane oxide-tethered aryldiselenide catalysts. (B) Structures of catalysts 3a–3k. These analogs were compared for their efficiency for catalytic amidation for a model reaction between toluic acid 4 and benzylamine 5 to yield 6. Reaction conditions: 4 (50 μmol), 5 (65 μmol), 3a–3k (20 mol %, 10 μmol), PhSiH3 (2 equiv, 100 μmol) in CH3CN (1 mL) at 80 °C under air. The %product formation was assessed by HPLC using an internal control (Supporting Information).
Synthesis of Phosphetane Oxide-Tethered Diselenides
We developed two strategies for the synthesis of phosphetane oxide-tethered diaryl diselenides (Figure A). In Method 1, bromination of bis(benzyl alcohol) diselenide 7 afforded bis(bromobenzyl) diselenide 8 in good yields. Subsequent treatment of 8 with bromine, followed by a double displacement reaction with phosphetane 2-H, led to the formation of selenophosphonium salt 9. Finally, aerobic hydrolysis of 9 with aqueous potassium carbonate furnished the bis(benzylphosphetane oxide) diselenides 3 in near quantitative yields. As an alternative, we explored modification of substituted bromobenzyl bromides to obtain the desired bis(benzylphosphetane oxide) diselenides. In Method 2, phosphetane 2-H is reacted with benzyl bromide 10 to first afford a phosphonium salt, which is oxidized to phosphetane oxide 11 upon treatment with aqueous sodium hydroxide and exposure to air. We found that 11 can be efficiently converted to aryl diselenides via boronate 12. Borylation of arylhalide with B2(Pin)2 followed by the treatment of the boronate with elemental selenium and silver nitrate furnishes bis(benzylphosphetane oxide) diselenides 3 in good-to-excellent yields.
Evaluation of Phosphetane Oxide-Tethered Diselenides as Catalysts for Amide Bond Formation
Using these methods, we synthesized several analogs of 3a as illustrated in Figure B. These analogs contain electron donating or withdrawing groups at the meta (R 1 and R 3 substituents on the ring) or para (R 2 substituent) positions relative to the diselenide. Method 1 is suitable for the synthesis of majority of catalysts due to its cost-effectiveness and higher overall yields. However, for some derivatives (such as 3c), method 2 afforded higher yields, as attempts to isolate the corresponding selenophosphonium salt 9 in method 1 were unsuccessful. Method 2 is a viable alternative for synthesizing all derivatives, but the step to obtain boronate esters 12 from 11 proved to be low yielding for several analogs. We evaluated the effectiveness of catalysts 3a–3k in facilitating amide bond formation between toluic acid 4 and benzylamine 5. Reactions were conducted at 20 mol % catalyst loading with phenylsilane (2 equiv) at 80 °C in CH3CN. Catalyst 3c bearing an electron donating OMe group at the R 2 position (para to the diselenide group) was initially tested but proved ineffective for amide bond formation. We postulated that the electron donating group in 3c likely disfavors the nucleophilic attack by the phosphine onto the diselenide, thereby reducing the rate of formation of the selenophosphonium intermediate III. Interestingly, the introduction of electron withdrawing groups at the R 3 (3d) or R 2 position (3e–3f) also did not enhance the catalytic efficiency compared to the parent 3a. In contrast, we found that substitution of electron withdrawing substituents at the R 1 position (3g–3j) provided a significant improvement in the catalyst performance. Notably, 20 mol % of 3g afforded near quantitative yields of product 6 within 30 min of reaction time (Figure B).
Based on the survey of derivatives in Figure B, catalyst 3g was selected for further exploration. We next assessed reaction parameters including solvents, catalyst loading, additives, and temperature to further optimize reaction yields (Supporting Information, Table S1). Counterions have been shown to impact the stability and reactivity of the selenophosphonium intermediate; as part of these optimization studies, we evaluated the impact of counterions on the reaction progress. The initial reaction conditions (20 mol % catalyst, acetonitrile as solvent, and high temperature of 80 °C) were chosen based on an extensive optimization with the intermolecular system discussed in Figure . For example, the high reaction temperature is required for efficient reduction of the phosphetane oxide. We found that the initial conditions remain optimal for intramolecular catalyst 3g.
Mechanistic Analysis of the Catalytic Cycle by NMR and DFT
To gain insight into the reaction mechanism, we performed single turnover studies with NMR spectroscopy to identify the intermediates. Fluorobenzoic acid 13 was chosen to allow monitoring of the reaction progress by 19F NMR. Single turnover studies were designed to assess the involvement of both phosphorus and selenium components in the catalytic cycle for amide bond formation (Figure ). For these studies, we designed two derivatives of the catalyst, where the diselenide is substituted with selenobromide (3l) or selenoether (3m). The selenobromide derivative would be expected to yield the same cyclic selenophosphonium intermediate as 3g, whereas 3m would not be expected to react. We tracked the formation of amide 15 by reacting equimolar amounts of p-fluorobenzoic acid 13, benzylamine 5, and the selenium adduct 3m or 3l, along with 2 equiv PhSiH3 (Supporting Information ). Under these single turnover conditions, amide 15 is formed rapidly in the presence of 3l (Figure A). As expected, selenoether 3m did not lead to product formation.
4.

We used a suite of NMR active isotopes (1H, 19F, 31P, and 77Se) to assess the proposed mechanism. Carboxylic acid (p-fluorobenzoic acid) was chosen to follow product formation by 19F NMR for these studies. (A) Analogs 3l and 3m were used to confirm that a leaving group on selenium is required for the initiation of the catalytic cycle. Comparison of the performance of 3g under O2 versus N2 environments supports the hypothesis that air reoxidation of selenol is needed for the reaction cycle. (B) We simultaneously monitored the 19F (left) and 31P (right) NMRs to identify the selenophosphonium intermediates 3g-III and 3g-VI. (C) Intermediates 14, 3g-III, 3g-VI, and product 15 were identified by NMR.
A combination of 1H, 19F, 31P, and 77Se NMRs allowed us to map the presence of other important mechanistic intermediates in catalytic reaction conditions with 3g (Supporting Information). We were particularly interested in identifying the critical cyclic selenophosphonium adduct 3g-III (Figure C, and the proposed catalytic cycle Figure A). Prior to PhSiH3 addition, the 19F NMR spectrum exhibited a broad signal at 111.65 ppm indicative of salt formation between carboxylic acid 13 and amine 5 (Figure B). The corresponding 31P NMR spectrum (Figure B) displayed two signals at 56.90 (major) and 62.30 (minor), corresponding to diastereomeric catalyst 3g.
Upon introduction of PhSiH3, gas evolution (H2) was observed and within 5 min, the 19F NMR revealed complex signals between −106 ppm and –107 ppm (Figure B left), consistent with the formation of silyl ester intermediates 14 along with a signal at −110.91 ppm corresponding to amide product 15. The 31P NMR spectrum, obtained in tandem, showed two new signals at 107.81 (major) and 100.28 ppm (minor), indicative of the formation of selenophosphonium intermediate 3g-III (Figure B right). After the reaction is completed, the amounts of 3g-III diminish relative to 3g (Figure B, 30 min plot). Over the course of 30 min, the condensation of 13 and 5 to give 14 was completed as evidenced by the disappearance of the silyl ester signals in the 19F NMR spectrum. The 31P spectrum displayed a gradual decrease in the intensity of the 3g-III signals, along with the reappearance of the 3g signal, confirming the catalyst regeneration. We tentatively assigned 3g-VI to the peak at 55.96 ppm, but this peak could represent various forms of phosphine oxide intermediates that may form in the catalytic cycle.
In these studies, we also confirmed the involvement of oxygen in the catalytic cycle for amide synthesis. When the model reaction with 3g (20 mol %) was conducted under a nitrogen environment, product formation was terminated after one cycle (Figure A).
We pursued density functional theory (DFT) calculations to understand how the electronics of the aryl group may impact the catalytic cycle and contribute to the enhanced reactivity of 3g relative to those of other catalysts (Figure B). DFT calculations were carried out using the ORCA 6 software package and visualized with CYLview. An initial conformer search was performed at the GFN2-xTB level to identify global minimum structures. All geometries were optimized at the r2SCAN-3c-SMD(MeCN) level, − and single-point energies were subsequently evaluated at the DLPNO–CCSD(T)/def2-TZVPP level. Frequency and thermochemistry calculations were performed at 80 °C to align with the experimental conditions. Additional methodological details and benchmarking are provided in the Supporting Information.
We generated model systems for two key steps in the catalytic cycle, involving selenophosphonium generation and selenoester formation (Figure ). Our computational results aligned with experimental trends, specifically indicating that the meta-fluorine substituted catalyst (3g) displays the lowest energy barriers across both steps compared to those of the unsubstituted 3a and para-substituted 3e. We further analyzed the intermediates using natural bond order (NBO) analysis. This analysis suggests that the inductive effect of the fluorine substituent is greatest when it is meta to the selenium center, thereby lowering the energy barriers for each step (Supporting Information). The natural charge on selenium is most negative for 3g in species IV, which facilitates the nucleophilic attack leading to selenoester formation (Figure B and Supporting Information). The NBO analysis showed consistent trends across a variety of functionals and basis sets (Supporting Information). ,
5.
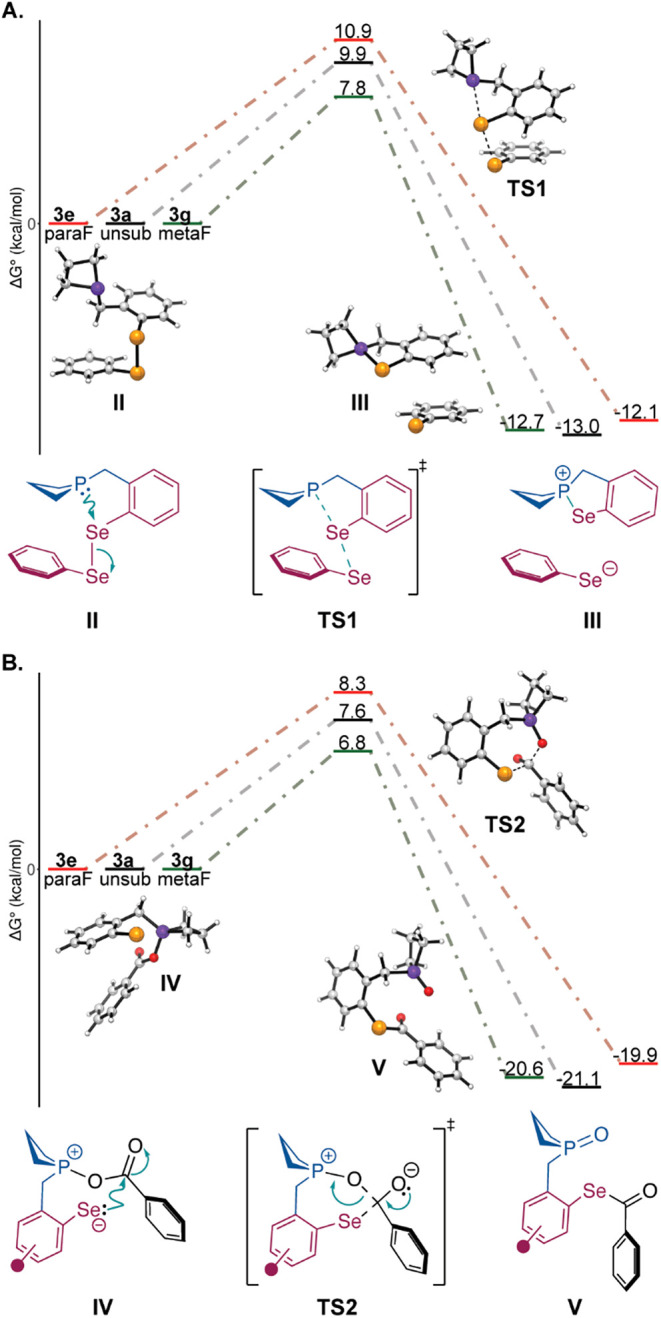
Computational analysis of the impact of ring substituents on the catalyst performance. (A, B) Transition states leading to selenophosphonium adduct III and selenoester V were calculated. Free energies were calculated at the DLPNO–CCSD(T)/def2-TZVPP//r2SCAN-3c-SMD(MeCN) level.
Application of Phosphetane Oxide-Tethered Diselenide 3g to Dipeptide Synthesis
We next evaluated the efficiency of catalyst 3g in promoting the coupling of amino acids. Our investigation focused on a diverse set of commercially available Boc-protected amino acids and amino acid esters with results summarized in Scheme . Condensation of Boc-Ala-OH with benzylamine and alanine tert-butyl ester in acetonitrile provided >90% yields of dipeptides 16a and 16b in <1.5 h. Encouraged by these results, we expanded our investigation to include phenylalanine and tryptophan, valine, proline, lysine, aminoisobutyric acid (Boc-Aib), histidine, aspartic acid, and arginine amino acids (16c–16s). In all cases, we were pleased to observe high yields of dipeptide products within 1–2 h, particularly for the β-branched valine, secondary amine proline, and sterically hindered Boc-Aib. No amide product 16u was obtained with aniline due to the low nucleophilicity of the aromatic amine. We have previously demonstrated that little to no epimerization is observed with carbamate-protected amino acids activated as arylselenoesters. ,
1. Catalytic Synthesis of Dipeptides .

a Reaction condition: R1COOH (0.5 mmol), R2R3NH (0.55 mmol), 3g (20 mol %), and PhSiH3 (2 × 1 equiv every 30 min) in CH3CN (5 mL) at 80 °C under air. Progress of the reaction was analyzed by thin layer chromatography and isolated yields are reported. We analyzed the potential epimerization of amino acids in the catalyst-mediated coupling by NMR or HPLC; the diastereomeric ratio (d.r.) is highlighted in gray. Detailed reaction procedures and epimerization analysis are included in Supporting Information.
To determine if diselenide 3g catalyzed reactions lead to epimerization-free coupling of amino acids, we analyzed crude NMRs or HPLC spectra of dipeptides Boc-l-Val-l-Val-O t Bu (16l), Boc-l-Phe-l-Phe-O t Bu (16g), Boc-l-Pro-l-Ala-O t Bu (16o), Boc-l-His-l-Ala-O t Bu (16q), and Boc-l-Phg-l-Ala-O t Bu (16t). Crude spectra used to determine the diastereomeric ratios are included in the Supporting Information. Spectra of the L,L dipeptides were compared to those of the D,L diastereomer as standards. We chose histidine, valine, and phenylglycine because they are known to be sensitive to epimerization as carboxylic acids in peptide synthesis. − The catalyzed synthesis of dipeptides resulted in undetectable levels of epimerization for all dipeptides, with the exception of phenylglycine (Scheme ). The results are in agreement with our previous analyses and show that negligible epimerization of carbamate-protected natural amino acids results in the selenoester-mediated amide bond formation.
Solid Phase Peptide Synthesis with Catalyst 3g
The overarching goal of this catalyst design is to improve the sustainability of solid phase peptide synthesis by replacing stoichiometric coupling reagents with a catalytic system while employing an environmentally benign solvent. Fmoc solid phase peptide synthesis (SPPS) is the method of choice for peptide synthesis. For any catalyst to have a practical impact, it would need to be operational under the Fmoc SPPS conditions. The advantage of SPPS is that reactions can be driven to completion by the use of excess reagents. This key attribute of SPPS has led to waste in contemporary peptide synthesis. We sought to determine if catalyst 3g would provide efficient synthesis of oligopeptides on solid phase with a slight excess of Fmoc-amino acids (1.5 equiv), 20 mol % 3g, and 2.5 equiv of PhSiH3 in acetonitrile. We used Tentagel-S-RAM resin due to its superior swelling properties in acetonitrile.
We tested three different sequences with a range of amino acid residues and side chain protecting groups (Figure ). We began by synthesizing pentamer Fmoc-KCGFG-NH2 (17a) using standard Fmoc-amino acids in acetonitrile. Peptide elongation was carried out at 80 °C using 20 mol % of 3g, 1.5 equiv of Fmoc-amino acids, and 2.5 equiv of PhSiH3 in acetonitrile for each coupling step. Kaiser test was used to monitor reaction progress. Cycles of coupling and deprotection (see Supporting Information ) were repeated with different amino acids until the complete peptide was obtained. HPLC and mass spectrometry analyses of the crude peptide confirmed the formation of a high purity product (Figure A). We next synthesized 5-mer Ac-LWFGA-NH2 (17b) and 11-mer Ac-QCFVAYKCFG-NH2(17c). While 17b was obtained with satisfactory purity, 17c required two coupling cycles per residue beyond the sixth amino acid for complete conversion. Nevertheless, HPLC analysis demonstrated high purity for the final product, as compared to the peptide prepared using standard Fmoc solid phase synthesis procedure with 5 equiv each of HBTU and Fmoc-amino acids (Figure D).
6.

Catalytic solid phase peptide synthesis using 3g. HPLC chromatograms (λ 220 nm) of crude peptides (A) Fmoc-KCGFG-NH2 (17a), (B) Ac-LWFGA-NH2 (17b), (C) Ac-QCFVAYKCGFG-NH2 (17c) obtained from catalytic SPPS, and (D) 17c synthesized using standard Fmoc solid phase peptide synthesis (SPPS) using 5 equiv each of HBTU and Fmoc-amino acid per coupling. Detailed reaction conditions and HPLC purification protocols are included in the Supporting Information.
Conclusions
In this study, we have developed a small molecule organocatalyst for peptide synthesis. The catalyst design incorporates redox recycling of aryldiselenide and phosphetane oxide with air as the ultimate oxidant and phenylsilane as the ultimate reductant. The catalyst can be readily accessed from simple starting materials and provides high yields for a range of dipeptide synthesis and, notably, solid phase synthesis of oligopeptides. Two salient features of the catalyst are that the reaction can be run in air and is compatible with acetonitrile, which is an attractive solvent to DMF both for its lower toxicity and damaging environmental impact. Despite the current progress, challenges remain, which may limit the practical utility of 3g. In particular, the sensitivity of catalyst 3g to water necessitates a need for a higher mol % of the catalyst and phenylsilane. Acetonitrile proved to be a suitable solvent for the solid phase synthesis of short peptides tested herein but its use as a general replacement for DMF remains to be explored. Our current efforts focus on reducing the amount of catalyst needed for efficient amide bond formation and exploring the effectiveness of the catalyst in automated synthesizers.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-2108150) for financial support of this work.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.5c07242.
Synthesis and characterization of compounds, description of DFT analysis (PDF)
The authors declare no competing financial interest.
References
- Al Musaimi O.. FDA’s stamp of approval: Unveiling peptide breakthroughs in cardiovascular diseases, ACE, HIV, CNS, and beyond. J. Pept. Sci. 2024:e3627. doi: 10.1002/psc.3627. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B.. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963;85:2149–2154. doi: 10.1021/ja00897a025. [DOI] [Google Scholar]
- Carpino L. A., Han G. Y.. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972;37:3404–3409. doi: 10.1021/jo00795a005. [DOI] [Google Scholar]
- Albericio F., Bofill J. M., El-Faham A., Kates S. A.. Use of Onium Salt-Based Coupling Reagents in Peptide Synthesis1. J. Org. Chem. 1998;63:9678–9683. doi: 10.1021/jo980807y. [DOI] [Google Scholar]
- Coin I., Beyermann M., Bienert M.. Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007;2:3247–3256. doi: 10.1038/nprot.2007.454. [DOI] [PubMed] [Google Scholar]
- Behrendt R., White P., Offer J.. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016;22:4–27. doi: 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartrampf N., Saebi A., Poskus M., Gates Z. P., Callahan A. J., Cowfer A. E., Hanna S., Antilla S., Schissel C. K., Quartararo A. J., Ye X., Mijalis A. J., Simon M. D., Loas A., Liu S., Jessen C., Nielsen T. E., Pentelute B. L.. Synthesis of proteins by automated flow chemistry. Science. 2020;368:980–987. doi: 10.1126/science.abb2491. [DOI] [PubMed] [Google Scholar]
- Hong S.-J., Zhang X.-N., Sun Z., Zeng T.. The potential health risks of N,N-dimethylformamide: An updated review. J. Appl. Toxicol. 2024;44:1637–1646. doi: 10.1002/jat.4590. [DOI] [PubMed] [Google Scholar]
- Sherwood J., Albericio F., de la Torre B. G.. N,N-Dimethyl Formamide European Restriction Demands Solvent Substitution in Research and Development. ChemSusChem. 2024;17:e202301639. doi: 10.1002/cssc.202301639. [DOI] [PubMed] [Google Scholar]
- Sabatini M. T., Boulton L. T., Sneddon H. F., Sheppard T. D.. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019;2:10–17. doi: 10.1038/s41929-018-0211-5. [DOI] [Google Scholar]
- Todorovic M., Perrin D. M.. Recent developments in catalytic amide bond formation. Pept. Sci. 2020;112:e24210. doi: 10.1002/pep2.24210. [DOI] [Google Scholar]
- Massolo E., Pirola M., Benaglia M.. Amide Bond Formation Strategies: Latest Advances on a Dateless Transformation. Eur. J. Org. Chem. 2020;2020:4641–4651. doi: 10.1002/ejoc.202000080. [DOI] [Google Scholar]
- Sabatini M. T., Boulton L. T., Sneddon H. F., Sheppard T. D.. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019;2:10–17. doi: 10.1038/s41929-018-0211-5. [DOI] [Google Scholar]
- Shen B., Makley D. M., Johnston J. N.. Umpolung reactivity in amide and peptide synthesis. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieter K. E., Shen B., Shackleford J. P., Leighty M. W., Johnston J. N.. Umpolung Amide Synthesis Using Substoichiometric N-Iodosuccinimide (NIS) and Oxygen as a Terminal Oxidant. Org. Lett. 2014;16:4714–4717. doi: 10.1021/ol502089v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo R. M., Suppo J.-S., Campagne J.-M.. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016;116:12029–12122. doi: 10.1021/acs.chemrev.6b00237. [DOI] [PubMed] [Google Scholar]
- Lecomte M., Lipshultz J. M., Kim-Lee S.-H., Li G., Radosevich A. T.. Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C–N and C–C Bond Formation. J. Am. Chem. Soc. 2019;141:12507–12512. doi: 10.1021/jacs.9b06277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara S., Okada Y., Kitano Y., Chiba K.. Biphasic electrochemical peptide synthesis. Chem. Sci. 2021;12:12911–12917. doi: 10.1039/D1SC03023J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K., Ohara S., Yamamoto H.. 3,4,5-Trifluorobenzeneboronic Acid as an Extremely Active Amidation Catalyst. J. Org. Chem. 1996;61:4196–4197. doi: 10.1021/jo9606564. [DOI] [PubMed] [Google Scholar]
- Sabatini M. T., Boulton L. T., Sheppard T. D.. Borate esters: Simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 2017;3:e1701028. doi: 10.1126/sciadv.1701028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomber C. E., Laserna V., Martin L. T., Smith P. D., Hailes H.C., Porter M. J., Sheppard T. D.. Catalytic directamidations in tert-butyl acetate using B (OCH 2 CF 3) 3. Org. Biomol. Chem. 2019;17:6465–6469. doi: 10.1039/C9OB01012B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda H., Furutachi M., Asada Y., Shibasaki M., Kumagai N.. Unique physicochemical and catalytic properties dictated by the B3NO2 ring system. Nat. Chem. 2017;9:571–577. doi: 10.1038/nchem.2708. [DOI] [PubMed] [Google Scholar]
- Shimada N., Hirata M., Koshizuka M., Ohse N., Kaito R., Makino K.. Diboronic acid anhydrides as effective catalysts for the hydroxy-directed dehydrative amidation of carboxylic acids. Org. Lett. 2019;21:4303–4308. doi: 10.1021/acs.orglett.9b01484. [DOI] [PubMed] [Google Scholar]
- Koshizuka M., Makino K., Shimada N.. Diboronic acid anhydride-catalyzed direct peptide bond formation enabled by hydroxy-directed dehydrative condensation. Org. Lett. 2020;22:8658–8664. doi: 10.1021/acs.orglett.0c03252. [DOI] [PubMed] [Google Scholar]
- Sawant D. N., Bagal D. B., Ogawa S., Selvam K., Saito S.. Diboron-catalyzed dehydrative amidation of aromatic carboxylic acids with amines. Org. Lett. 2018;20:4397–4400. doi: 10.1021/acs.orglett.8b01480. [DOI] [PubMed] [Google Scholar]
- Michigami K., Sakaguchi T., Takemoto Y.. Catalytic dehydrative peptide synthesis with gem-diboronic acids. ACS Catal. 2020;10:683–688. doi: 10.1021/acscatal.9b03894. [DOI] [Google Scholar]
- Lundberg H., Tinnis F., Adolfsson H.. Direct amide coupling of non-activated carboxylic acids and amines catalysed by zirconium (IV) chloride. Chem.- Eur. J. 2012;18:3822–3826. doi: 10.1002/chem.201104055. [DOI] [PubMed] [Google Scholar]
- Lundberg H., Tinnis F., Adolfsson H.. Titanium (IV) isopropoxide as an efficient catalyst for direct amidation of nonactivated carboxylic acids. Synlett. 2012;23:2201–2204. doi: 10.1055/s-0032-1316993. [DOI] [Google Scholar]
- Handoko, Panigrahi N. R., Arora P. S.. Two-Component Redox Organocatalyst for Peptide Bond Formation. J. Am. Chem. Soc. 2022;144:3637–3643. doi: 10.1021/jacs.1c12798. [DOI] [PubMed] [Google Scholar]
- Handoko, Satishkumar S., Panigrahi N. R., Arora P. S.. Rational Design of an Organocatalyst for Peptide Bond Formation. J. Am. Chem. Soc. 2019;141:15977–15985. doi: 10.1021/jacs.9b07742. [DOI] [PubMed] [Google Scholar]
- Wu H., Handoko, Raj M., Arora P. S.. Iterative design of a biomimetic catalyst for amino acid thioester condensation. Org. Lett. 2017;19:5122–5125. doi: 10.1021/acs.orglett.7b02412. [DOI] [PubMed] [Google Scholar]
- Endo T., Ikenaga S., Mukaiyama T.. A convenient method for the preparation of thiolesters. Bull. Chem. Soc. Jpn. 1970;43:2632–2633. doi: 10.1246/bcsj.43.2632. [DOI] [Google Scholar]
- Ghosh S. K., Singh U., Mamdapur V. R.. One pot amide/peptide synthesis via two redox reactions. Tetrahedron Lett. 1992;33:805–808. doi: 10.1016/S0040-4039(00)77720-3. [DOI] [Google Scholar]
- Liebeskind L. S., Gangireddy P., Lindale M. G.. Benzoisothiazolone organo/copper-cocatalyzed redox dehydrative construction of amides and peptides from carboxylic acids using (EtO) 3P as the reductant and O2 in air as the terminal oxidant. J. Am. Chem. Soc. 2016;138:6715–6718. doi: 10.1021/jacs.6b03168. [DOI] [PubMed] [Google Scholar]
- Zhao W., Yan P. K., Radosevich A. T.. A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PVO Redox Cycling. J. Am. Chem. Soc. 2015;137:616–619. doi: 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]
- Hondal R. J., Nilsson B. L., Raines R. T.. Selenocysteine in native chemical ligation and expressed protein ligation. J. Am. Chem. Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- Reich H. J., Hondal R. J.. Why Nature Chose Selenium. ACS Chem. Biol. 2016;11:821–841. doi: 10.1021/acschembio.6b00031. [DOI] [PubMed] [Google Scholar]
- Raj M., Wu H., Blosser S. L., Vittoria M. A., Arora P. S.. Aldehyde capture ligation for synthesis of native peptide bonds. J. Am. Chem. Soc. 2015;137:6932–6940. doi: 10.1021/jacs.5b03538. [DOI] [PubMed] [Google Scholar]
- Acosta G. A., del Fresno M., Paradis-Bas M., Rigau-DeLlobet M., Côté S., Royo M., Albericio F.. Solid-phase peptide synthesis using acetonitrile as a solvent in combination with PEG-based resins. J. Pept. Sci. 2009;15:629–633. doi: 10.1002/psc.1158. [DOI] [PubMed] [Google Scholar]
- Beddoe R. H., Andrews K. G., Magné V., Cuthbertson J. D., Saska J., Shannon-Little A. L., Shanahan S. E., Sneddon H. F., Denton R. M.. Redox-neutral organocatalytic Mitsunobu reactions. Science. 2019;365:910–914. doi: 10.1126/science.aax3353. [DOI] [PubMed] [Google Scholar]
- Pagire S. K., Shu C., Reich D., Noble A., Aggarwal V. K.. Convergent Deboronative and Decarboxylative Phosphonylation Enabled by the Phosphite Radical Trap “BecaP”. J. Am. Chem. Soc. 2023;145:18649–18657. doi: 10.1021/jacs.3c06524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- But T. Y. S., Toy P. H.. Organocatalytic Mitsunobu Reactions. J. Am. Chem. Soc. 2006;128:9636–9637. doi: 10.1021/ja063141v. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Liu Y., Wang Y.. Weak Interaction Activates Esters: Reconciling Catalytic Activity and Turnover Contradiction by Tailored Chalcogen Bonding. J. Am. Chem. Soc. 2024;146:13296–13305. doi: 10.1021/jacs.4c01541. [DOI] [PubMed] [Google Scholar]
- Neese F.. Software update: The ORCA program systemVersion 5.0. WIREs Comput. Mol. Sci. 2022;12:e1606. doi: 10.1002/wcms.1606. [DOI] [Google Scholar]
- Legault, C. Y. CYLview20 Université de Sherbrooke 2020. http://www.cylview.org.
- Bannwarth C., Ehlert S., Grimme S.. GFN2-xTBAn Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019;15:1652–1671. doi: 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]
- Grimme S., Hansen A., Ehlert S., Mewes J.-M.. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021;154:064103. doi: 10.1063/5.0040021. [DOI] [PubMed] [Google Scholar]
- Marenich A. V., Cramer C. J., Truhlar D. G.. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Garcia-Ratés M., Neese F.. Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 2020;41:922–939. doi: 10.1002/jcc.26139. [DOI] [PubMed] [Google Scholar]
- Riplinger C., Sandhoefer B., Hansen A., Neese F.. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013;139:134101. doi: 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]
- Glendening E. D., Landis C. R., Weinhold F.. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019;40:2234–2241. doi: 10.1002/jcc.25873. [DOI] [PubMed] [Google Scholar]
- Cohen L. A., Takahashi S.. Reexamination of electronic effects in ring-substituted phenyl esters. Correlation of spectral and kinetic properties with sigma0. J. Am. Chem. Soc. 1973;95:443–448. doi: 10.1021/ja00783a022. [DOI] [Google Scholar]
- Müller M., Hansen A., Grimme S.. ωB97X-3c: A composite range-separated hybrid DFT method with a molecule-optimized polarized valence double-ζ basis set. J. Chem. Phys. 2023;158:014103. doi: 10.1063/5.0133026. [DOI] [PubMed] [Google Scholar]
- Caldeweyher E., Mewes J.-M., Ehlert S., Grimme S.. Extension and evaluation of the D4 London-dispersion model for periodic systems. Phys. Chem. Chem. Phys. 2020;22:8499–8512. doi: 10.1039/D0CP00502A. [DOI] [PubMed] [Google Scholar]
- Liang C., Behnam M. A. M., Sundermann T. R., Klein C. D.. Phenylglycine racemization in Fmoc-based solid-phase peptide synthesis: Stereochemical stability is achieved by choice of reaction conditions. Tetrahedron Lett. 2017;58:2325–2329. doi: 10.1016/j.tetlet.2017.04.047. [DOI] [Google Scholar]
- Sakakibara S.. Chemical synthesis of proteins in solution. Pept. Sci. 1999;51:279–296. doi: 10.1002/(SICI)1097-0282(1999)51:4<279::AID-BIP4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Yang Y., Hansen L., Baldi A.. Suppression of Simultaneous Fmoc-His(Trt)-OH Racemization and Nα-DIC-Endcapping in Solid-Phase Peptide Synthesis through Design of Experiments and Its Implication for an Amino Acid Activation Strategy in Peptide Synthesis. Org. Process Res. Dev. 2022;26:2464–2474. doi: 10.1021/acs.oprd.2c00144. [DOI] [Google Scholar]
- Duengo S., Muhajir M. I., Hidayat A. T., Musa W. J. A., Maharani R.. Epimerisation in Peptide Synthesis. Molecules. 2023;28:8017. doi: 10.3390/molecules28248017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.