

Abstract
Rhodopsin is the G protein-coupled receptor that is activated by light and initiates the transduction cascade leading to night (rod) vision. Naturally occurring pathogenic rhodopsin (RHO) mutations have been previously identified only in humans and are a common cause of dominantly inherited blindness from retinal degeneration. We identified English Mastiff dogs with a naturally occurring dominant retinal degeneration and determined the cause to be a point mutation in the RHO gene (Thr4Arg). Dogs with this mutant allele manifest a retinal phenotype that closely mimics that in humans with RHO mutations. The phenotypic features shared by dog and man include a dramatically slowed time course of recovery of rod photoreceptor function after light exposure and a distinctive topographic pattern to the retinal degeneration. The canine disease offers opportunities to explore the basis of prolonged photoreceptor recovery after light in RHO mutations and determine whether there are links between the dysfunction and apoptotic retinal cell death. The RHO mutant dog also becomes the large animal needed for preclinical trials of therapies for a major subset of human retinopathies.
Rhodopsin, the visual pigment of rod photoreceptors, is one of many G protein-coupled receptors involved in human disease (1–3). Numerous rhodopsin gene (RHO) mutations (mostly point mutations) cause retinitis pigmentosa (RP), a blinding human retinal degeneration (see RetNet, http://www.sph.uth.tmc.edu/Retnet/; refs. 4 and 5). RP caused by RHO mutations exhibits two different phenotypes (4, 5). One is an early-onset disorder with rapid loss of rods uniformly across the retina. The second has a protracted natural history of vision loss and puzzling features: rod vision can be normal early in life, and degeneration slowly spreads from a disease focus in one retinal region. Furthermore, and independent of disease stage, there is abnormally slow recovery of rod vision after bright light exposure. Variation in severity of this second phenotype may indicate that epigenetic factors play an important role in its progression (5–7) and suggests that it may be particularly amenable to preventive or ameliorative therapies.
Other than in humans, naturally occurring disease-causing RHO mutations have not been identified previously in mammals. Genetically engineered animals and mutagenized flies have been the mainstay for in vivo research and treatment attempts in the past decade (reviewed in ref. 8).
Naturally occurring hereditary retinal degenerations in dogs, termed progressive retinal atrophies (PRAs), are widespread and have provided several models of autosomal recessive and X-linked RP (9–13). We have now identified an autosomal dominant form of PRA, one that closely resembles the second human phenotype described above. This model should advance understanding of the pathophysiology of the disease and therapies for this major subset of dominant RP.
Materials and Methods
Rhodopsin Gene Sequence Analysis.
Mutation analysis by comparative sequencing of the five canine RHO exons was performed by using published primers (14). Detection of the C to G transversion at nucleotide 11 followed PCR, using primers OPIAF (5′-GCA GCA CTC TTG GGA CTG AG) and OPIAR (5′-TGT AGT TGA GAG GTG TAC GC). Digestion of the 275-bp product with BsmFI results in fragments of 202, 47, and 26 bp (wild type) or 249 and 26 bp (nt11C to G). For Northern analysis, wild-type cDNA was amplified with primers D849 and D855 (14) to generate a 701-bp probe. RNA isolation, electrophoresis, transfer, and hybridization were conducted with standard procedures, and the blot was hybridized with β-ACTIN to confirm equal amounts of RNA loading.
Clinical Electroretinograms (ERGs).
Clinical ERGs were recorded from halothane-anesthetized, dark-adapted dogs as described (15).
ERG Photoresponses.
Dogs were dark-adapted overnight, premedicated, and anesthetized as described (16). Pupils were dilated with cyclopentolate (1%) and phenylephrine (10%). Pulse rate, oxygen saturation, and temperature were monitored. Full-field ERGs were recorded with Burian–Allen (Hansen Ophthalmics, Iowa City, IA) contact lens electrodes and a computer-based system. High-energy flashes [1-ms duration; maximum luminance of unattenuated white flash = 3.66 log scotopic (scot)-cd⋅s⋅m−2] were attenuated and spectrally shaped with Wratten (Eastman Kodak) filters. Dark-adapted photoresponses were elicited by a series of five stimuli presented with 2-min interstimulus intervals: one white, two blue (Wratten 47A), and two red (Wratten 26) flashes (3.66, 2.87, 2.27, 1.27, and 0.5 log scot-cd⋅s⋅m−2, respectively). White flash bleaches of 3.66, 5.0, and 5.8 log scot-cd⋅s⋅m−2 were used in bleaching adaptation experiments and white backgrounds in the range of −0.9 to 2.3 log scot-cd⋅m−2 were used in background adaptation experiments. The rod-isolated component of bleaching and background adaptation functions was estimated with the highest energy blue flash (2.87 log scot-cd⋅s⋅m−2).
ERG Analyses.
Rod and cone components of dark-adapted photoresponse series were analyzed with a model of rod and cone phototransduction activation (5, 17) fitted as an ensemble to the leading edges of the five waveforms. Two parameters, maximum amplitude and sensitivity, were allowed to vary during the fitting process; all other parameters were fixed to the same values in normal and mutant dogs (18). Maximum amplitude represents the circulating current, and sensitivity represents the amplification factor of each photoreceptor type averaged across the retina (19). Cone components estimated from dark-adapted data were used to isolate the rod component in bleaching and background adaptation functions performed with a single blue flash. Photoresponse parameters as a function of background were fitted to the hyperbolic function F = I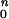 /[I
/[I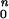 + I
+ I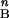 ], where F is the maximum amplitude or sensitivity parameter specified as a fraction of dark-adapted value, I0 is the background luminance that halves the parameter of interest, IB is the background luminance, and n is the slope of the function.
], where F is the maximum amplitude or sensitivity parameter specified as a fraction of dark-adapted value, I0 is the background luminance that halves the parameter of interest, IB is the background luminance, and n is the slope of the function.
Ocular axial length and pupil diameter were measured after each ERG experiment. The ratio of pupil diameter squared to axial length squared averaged 0.33 in normal and mutant dogs; this was approximately 3 times the typical value for a normal adult human. Assuming anterior focal length of the dog and human eyes to be a constant fraction of axial length (20) and stereotypy of mammalian rod photoreceptors (19), we estimate 1 scot-cd⋅s⋅m−2 to cause ≈1,200 isomerizations per rod in the dog. Estimates of percent bleach assume similar photosensitivity at the level of the retina in humans and dogs.
Clinical Retinal Examination and Definition of Disease Progression.
Animals were examined ophthalmoscopically for areas of retinal thinning as evidenced by hyperreflectivity of the fundus (first observed in RHO T4R/+ animals 6–12 mo old). Observation of retinal vascular attenuation (first observed after 12 mo) and pallor of the optic nerve head (ONH) (before 36 mo) define advancing stages of the disease.
Optical Coherence Tomography (OCT).
Cross-sectional retinal reflectivity profiles were obtained with OCT from two normal and three RHO T4R/+ dogs. Principles of the technique (21) and our methods (22, 23) have been described. Multiple scans (4.5- or 8.0-mm-long, at various orientations) were performed to give maximal coverage of an area 9–12 mm in diameter, approximately centered on the ONH. A custom program was used to determine the precise location and orientation of each scan relative to the retinal features (blood vessels and ONH). The individual longitudinal reflectivity profiles (LRPs) that make up each scan were allotted to 0.3 mm × 0.3 mm bins based on their location. The LRPs in each bin were aligned with a dynamic cross-correlation algorithm (22, 23), were median-filtered to remove speckle noise, and were averaged. The difference between the most vitreal maxima and the most scleral minima of the slope in the average LRP was used as a measure of retinal thickness at each location. A full retinal thickness map was generated by interpolation between the nonuniformly spaced retinal thickness estimates.
Histopathology and Immunocytochemistry.
Retinal sections for morphologic studies were prepared with either a triple fixation protocol (15) before embedding in plastic or 4% paraformaldehyde fixation for diethylene glycol distearate (DGD) embedding (24) and immunocytochemistry. For reconstructing the topographic distribution of disease, 1-μm retinal plastic sections, extending from the optic disk to the periphery (ora serrata), were evaluated from continuous overlapping 110-μm fields (25). For immunocytochemical studies, sections from DGD-embedded retinas were labeled with mAb K16-107C, directed at the C-terminal domain of opsin (26).
Human Studies.
RHO mutation analyses in the patients have been reported (5, 27, 28). Bleaching and background adaptation functions were obtained as described (5, 28, 29). Thresholds as a function of background were fitted to the hyperbolic function T = log [(I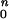 + I
+ I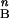 )/I
)/I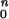 ], where T is log threshold, I0 is the background luminance that raises the threshold by 0.3 log units, IB is the background luminance, and n is the slope of the function. Note that the model used for the human psychophysical thresholds is the inverse of the function used for dog photoresponse parameters. OCT methods have been published (30). Informed consent for all procedures was obtained and the research procedures were in accordance with institutional guidelines and the Declaration of Helsinki.
], where T is log threshold, I0 is the background luminance that raises the threshold by 0.3 log units, IB is the background luminance, and n is the slope of the function. Note that the model used for the human psychophysical thresholds is the inverse of the function used for dog photoresponse parameters. OCT methods have been published (30). Informed consent for all procedures was obtained and the research procedures were in accordance with institutional guidelines and the Declaration of Helsinki.
Results
Identification of a Dominant Canine Progressive Photoreceptor Disease and the Causative Gene Mutation.
PRA in English Mastiff dogs was found to be inherited as an apparently autosomal dominant disease. Confirmation by testmating (Fig. 1a) prompted exon scanning of canine RHO in affected dogs. A single nonsynonymous C → G transversion at nucleotide 11 changes Thr-4 to Arg (T4R) and cosegregates with disease in the test pedigree (Fig. 1 a and b). A mutation of Thr-4 to Lys (T4K) has been associated with human RP (31). Testing for T4R in PRA-affected (n = 26) and related -unaffected (n = 21) Mastiffs showed the association predicted for a mutation causing dominant disease (23/26 affecteds T4R/+; 3/26 affecteds T4R/T4R; 21/21 normals +/+). The absence of T4R from 156 clinically normal dogs from 17 other canine breeds further indicates T4R is not neutral but causes autosomal dominant PRA.
Figure 1.

Autosomal dominant PRA (adPRA) caused by RHO T4R mutation. (a) A normal, male, mixed-breed dog (I:1) was crossbred to an adPRA-affected female Mastiff (I:2). Among the six progeny (II:1–4 shown), two were affected with PRA at 12 mo of age and four were normal. PCR restriction fragment length polymorphism analysis with BsmFI in the normal sire (I:1) yields one fragment of 202 bp; the affected dam (II:2) has an additional 249-bp fragment diagnostic of the T4R mutation. (b) RHO sequence analysis from an affected dog shows the heterozygous C > G transversion at position 11, which predicts substitution of Arg for Thr at residue 4. (c) Northern blot analysis shows no difference in RHO expression comparing an RHO T4R/+ and wild-type animal. (d) ERGs recorded from dark-adapted RHO mutant and control dogs. Each vertical image presents the responses to a blue flash, to 5-Hz low-intensity white light flashes, and to 30-Hz high-intensity white light flicker: these elicit rod b-waves, rod-, and cone-specific responses, respectively. Upper images show recordings from normal (rod b-wave amplitude mean = 234.9 μV; SEM = 20.3 μV and cone flicker mean = 63.9 μV; SEM = 4.7 μV) and T4R/+ dogs (rod mean = 202.5 μV, SEM = 13.5 μV; cone mean = 60 μV, SEM = 2.8 μV) each at 2 mo of age (mo). These were not significantly different (P = 0.2 and 0.5, respectively). Lower images show recordings from a normal dog at 11 mo and a T4R/+ animal at 14 mo where rod and cone ERGs are markedly reduced, although cone responses show relative preservation. (e) ERG photoresponses (thin noisy lines) in a representative normal dog and three RHO mutant dogs evoked by one white (W), two blue (B1, B2), and two red (R1, R2) flash stimuli. Waveforms are fitted with a phototransduction activation model (thick line) that is the sum of rod (dashed lines) and cone (dotted lines) components. T4R/+ dogs at 3–6 mo of age have rod (Max. Amplitude, Rmax = 249 ± 14 μV; sensitivity, σ = 3.46 ± 0.11 log scot-cd−1⋅m2⋅s−3) and cone (Rmax = 26.1 ± 5.0 μV; σ = 3.93 ± 0.22 log phot-cd−1⋅m2⋅s−3) photoresponses within the normal range (rod: Rmax = 242 ± 12 μV, σ = 3.52 ± 0.09 log scot-cd−1⋅m2⋅s−3; cone: Rmax = 26.6 ± 1.3 μV, σ = 3.89 ± 0.11 log phot-cd−1⋅m2⋅s−3). At 13 mo of age there were abnormal rod (Rmax = 147 μV, σ = 3.72 log scot-cd−1⋅m2⋅s−3) and cone (Rmax = 11.7 μV, σ = 4.09 log phot-cd−1⋅m2⋅s−3) photoresponses.
Clinical ERGs defined the disease as a progressive retinal degeneration. ERG rod- and cone-mediated responses were not significantly different between 2-mo-old normal and T4R/+ heterozygous affected dogs (Fig. 1d). By 12–18 mo of age, however, ERG b-wave amplitudes were severely abnormal in heterozygous affected dogs.
Photoreceptor-specific ERG function was evaluated with high-energy stimuli and a model of phototransduction activation (Fig. 1e). RHO T4R/+ dogs at 3–6 mo of age have rod and cone photoresponses within the normal range. Fully dark-adapted rod photoreceptors thus have normal circulating dark currents and a normal gain of phototransduction amplification despite the RHO mutation. Consistent with clinical ERG results in the heterozygous dogs, by 13 mo of age rod and cone photoresponses were abnormal. A third homozygous (T4R/T4R) dog at 3 mo had photoresponses similar to a 13-mo-old heterozygote (data not shown). The normal photoresponse sensitivity in the T4R/T4R dog suggests the mutant allele can produce a normal gain of phototransduction. The natural history of disease, however, seemed to be accelerated because of the increased mutant allele dosage. The reduced maximum amplitude in older T4R/+ and in young T4R/T4R dogs is consistent with loss of rods, shortening of their outer segments (the rhodopsin-containing cell component), or both.
Abnormal Photoreceptor Adaptation in RHO Mutant Dogs Imitates a Human Phenotype.
Our studies of human RHO mutations (5, 27, 28) induced us to ask whether rod photoreceptor function in RHO T4R/+ dogs can recover normally in the dark after adapting light exposure. Rod recovery was probed with ERG photoresponses after different levels of adapting light flashes. Rod-isolated photoresponses of RHO T4R/+ dogs recovered similarly to those of wild-type dogs after the two dimmer adapting flashes (estimated to cause 7% and 78% bleaches, respectively). After the brightest adapting flash (99% bleach; Fig. 2a), however, photoresponses did not recover to baseline. Analysis of recovery with the maximum amplitude parameter of rod photoresponses showed there was an initial period of near-normal recovery kinetics that was interrupted at approximately 15 min; the resulting abnormal plateau did not appear to change for the duration of the experiment (Fig. 2c). A T4R/T4R dog had adaptation kinetics similar to the T4R/+ dogs (not shown). Recovery of rod sensitivity was similar in normal and mutant animals. Dim adapting lights could be used both in wild-type and RHO T4R/+ dogs to simulate the photoresponse properties of mutant dogs during the abnormal plateau of bleach recovery (Fig. 2b). Analysis of photoresponse parameters as a function of increasing levels of background light showed hyperbolic saturation for maximum amplitude and sensitivity both for wild-type and RHO mutant dogs (Fig. 2d). The decay of equivalent background obtained with a Crawford transformation (32) had dominant linear components on semilogarithmic plots supporting exponential decays of bleach byproducts; the rate of decay was −0.1 log min−1 (time constant = 4.3 min) for the maximum amplitude and −0.05 log min−1 (time constant = 8.7 min) for sensitivity (Fig. 2e).
Figure 2.

Bleaching and background adaptation in canine and human RHO mutations. (a and b) A model of rod phototransduction activation (smooth lines) fitted to the leading edges of rod-isolated ERG photoresponses (noisy lines) in representative normal and RHO T4R/+ mutant dogs. The photoresponses shown were evoked either fully dark-adapted (gray lines), or in the dark at specified times after three levels of bleaching flashes (a), or on a background of 1 scot-cd⋅m−2 (b). Ordinates are normalized by the maximum amplitude under fully dark-adapted conditions. (c) Recovery of maximum amplitude and sensitivity parameters as a function of time after bleaching flashes in normal wild-type (empty symbols) and RHO T4R/+ (filled symbols) dogs. Symbols and bars represent mean ± SEM. (d) Change of maximum amplitude and sensitivity parameters as a function of background luminance in wild-type (empty symbols) and RHO T4R mutant (filled symbols) dogs. Symbols and bars represent mean ± SE. Hyperbolic saturation functions (gray lines) with I0 of 0.3 and 0.35 log scot-cd⋅m−2 and n of 1.0 and 0.7 were fitted to maximum amplitude and sensitivity, respectively. (e) Recovery of equivalent background as a function of time for a range of adapting flashes estimated by applying the inverse of the saturation functions shown in d to the data in c. Parallel lines (gray) fitted to the two larger bleaches have slopes of −0.1 min−1 (Max. Amplitude) and −0.05 min−1 (Sensitivity) on semilogarithmic coordinates. (f) Thresholds (psychophysically determined) in representative normal (empty symbols) or RHO mutant (filled symbols) human subjects after an adapting light that bleached >95% of available rhodopsin; prebleach thresholds shown near time 0. (g) Thresholds obtained on a range of background levels. Normal background adaptation could be well described with a hyperbolic saturation function (gray lines) with I0 of −4.5 log scot-cd⋅m−2 and n of 0.85. Approximately equal elevations in absolute threshold and I0 described the data from the patients. (h) Recovery of equivalent background as a function of time after the bright adapting light was estimated by applying the inverse of the saturation functions shown in g to the data in f. The major portion of normal recovery and the portion of recovery before the abnormal interruption in patients could be fitted with a line (gray) of −0.25 min−1 slope on semilogarithmic coordinates. Recovery of patient thresholds continued at an abnormally slow slope of −0.025 min−1.
Defective dark (or bleaching) adaptation in RHO T4R/+ dogs was indeed similar to that in patients with RP with certain RHO mutations (Class B1; ref. 5) (Fig. 2 f–h). In these patients, recovery of psychophysically determined rod thresholds is abnormal after a bright (>95% bleach) adapting exposure (Fig. 2f). Qualitatively, patients with T58R, G106R, and G190D RHO mutations showed an abrupt change of recovery kinetics occurring 15–20 min after light offset (Fig. 2f). The timing and level of the interrupted recovery was consistent within mutations but tended to differ between mutations (5, 27). Adaptation of rod thresholds to dim background lights fits a hyperbolic saturation function in normal subjects; in patients, results could be described by shifting the normal curve up and to the right by approximately equal amounts (Fig. 2g). Similar to the photoresponse data for the dogs, human psychophysical thresholds were converted to equivalent background with the Crawford transformation. The equivalent background corresponding to the major portion of rod recovery in the normal subject decayed at a rate of −0.25 log min−1 on semilogarithmic coordinates (time constant = 1.74 min). In patients with RHO mutations, there was an early component of recovery with the same slope followed by a slower decay of −0.025 min−1 (time constant = 17.4 min). Mutant dogs did not seem to have the slower recovery component seen in patients.
Topography of Photoreceptor Degeneration in the Canine RHO Mutant Retina.
Clinical examination of T4R/+ animals, in the earliest detectable stage of the disease, revealed a variably sized and located area of retinal thinning in the central fundus, clearly demarcated from surrounding clinically normal retina (Fig. 3a). Topographical variation of the retinal degeneration was independently observed in cross-sectional images measured in vivo with OCT. In normal dogs, LRPs indicate a relatively constant retinal thickness (Fig. 3b). LRPs from the RHO T4R/+ retina show localized thinning with a narrow transition zone (≈1.0 mm) separating normal and thin retina. Topographical maps of retinal thickness, based on OCT LRPs, provide further support for the localized distribution of the disease in its early stages (Fig. 3c). Intraretinal variation in severity also occurs in patients with adRP because of RHO mutations (5, 6, 27, 33). A visual field from such a patient shows a scotoma in the superior field (Fig. 3d). Average LRPs from a vertical scan (arrow on the visual field) indicate that the retina becomes progressively thinner as the scan passes into the dysfunctional region (Fig. 3e).
Figure 3.

Regional retinal disease gradient in canine and human RHO mutations. (a) Superimposed drawings of ophthalmoscopically evident lesions in one eye each from four RHO mutant dogs (displayed as left eyes) illustrate the range of topographic variation in disease. Darker grays represent higher frequencies of overlap of lesions among eyes. Lesions are drawn superimposed on a schematic of the dog fundus; green outlines tapetal area; red are blood vessels; white circle is the ONH. (b) OCT LRPs from an 8-mm-long horizontal scan located ≈3.3 mm superior to the ONH for a normal dog and right eye of an RHO T4R mutant dog; arrow is region from which LRPs are derived. For the normal dog, retinal thickness is the same throughout the length of the scan; the mutant dog shows distinct regions of thinning. (c) Topographical maps of retinal thickness in the left eyes of normal and RHO T4R/+ mutant dogs. A very small focus of retinal thinning in the 6-mo-old dog and a larger area of thinning in the 13-mo-old mutant dog are apparent in the superior temporal quadrants. (d) Kinetic visual field showing an altitudinal defect in a patient with adRP caused by an RHO G106R mutation. (e) LRPs derived from scans from the region indicated by the arrow on the visual field. The retina becomes progressively thinner as the scan moves toward the region of dysfunction.
Morphologic examination of RHO T4R/+ retinas confirmed and extended the results of noninvasive studies (Fig. 4). Three observations confirm that RHO T4R/+ retinal photoreceptors develop normally, and the mutation does not impair RHO expression or trafficking. Firstly, rod photoreceptors in 8–9-week-old affected dogs (n = 8) were indistinguishable from wild-type +/+ littermate controls (n = 9) (Fig. 4a). Secondly, rhodopsin localization with Abs directed at the N- (amino acids 3–8, data not shown) and C-terminal (amino acids 340–348; Fig. 4 h1 and h2) domains showed the normal pattern of label intensity and distribution restricted to rod outer segments. Finally, comparison of mRNA expression detected no difference in the intensity or size of the RHO transcript (Fig. 1c). The nonuniform degeneration of photoreceptors detected by both clinical examination and OCT was also observed morphologically (Fig. 4 a–g, j, and k). Photoreceptor disease and degeneration occurs in older animals, with advanced degeneration present at 4.5 and 11 years. In younger affected adult dogs, the disease is expressed with striking topographic variation. In different retinal locations, photoreceptors can be normal or show different gradations of disease (Fig. 4 a–g). The topographic distribution of disease was examined in an 11-mo-old RHO T4R/+ mutant dog by serial reconstruction of retinal sections from the major quadrants of the eye (Fig. 4j). In general, more severe disease (stages 3–6) was present in an area surrounding the ONH but centered in the temporal tapetal region of the fundus. Beyond this severely diseased region, there was an abrupt transition zone (Fig. 4 j and k1-7) beyond which photoreceptors were structurally and quantitatively normal. Thus, at this age and stage of disease, despite the severe disease centrally, most of the retina was comprised of structurally intact photoreceptors.
Figure 4.

Retinal disease morphology and RHO immunocytochemistry. (a–g) Sections from an 11-mo-old RHO T4R/+ retina. (a) Normal section of T4R/+ retina. (b) Early rod loss is associated with drop out of diseased rods and shortened outer segments in remaining rods. (c and d) The degenerative phase is characterized by rod loss with preservation of cones. (e–g) End-stage atrophy results in the progressive and sequential loss of all photoreceptors and retinal pigmented epithelium. (h1 and h2) Immunocytochemistry in a 2-mo-old RHO T4R/+ retina shows a normal pattern of intense opsin labeling limited to the outer segments. (i1 and i2) Examination of an 11-mo-old T4R/+ retina shows RHO label is present in the short, disorganized, outer segments of the few remaining rods adjacent to regions devoid of opsin staining where only cones remain. Calibration marker = 10 μm. (j) Location of 7 1-μm retinal plastic sections, extending from the optic disk to the periphery, taken to reconstruct the topographic distribution of disease. (k) Sections were evaluated in continuous overlapping fields and assigned stage 0–6 to correspond with disease severity as defined in a–g, respectively. All regions are drawn to scale. More severe disease (stages 3–6) was observed surrounding the ONH and centered in the temporal tapetal region of the fundus. Dotted line indicates approximate area of retinal thinning apparent on gross examination of the fixed eyecup; colored line represents inferred margin of degenerate area from evaluation of fixed sections.
Discussion
The phenotype of the RHO mutant dog is distinctly different from other canine retinal degenerations (15, 25), and these differences are precisely the similarities it shares with certain human RHO mutation phenotypes. Normal retinal structure, rhodopsin expression, receptor activation, and postreceptoral signaling in young affected dogs suggest that the pathogenesis does not involve abnormal photoreceptor development. The defect in dark adaptation and the focal initiation of photoreceptor degeneration uniquely characterize both this canine disease and that in human patients with RP with class B1 RHO mutations (5). The compelling question then is how are these features linked together?
Rhodopsin, like other G protein-coupled receptors, has seven transmembrane α-helical segments, and N- (intradiscal or extracellular) and C-terminal (cytoplasmic or intracellular) domains (Fig. 5). The T4R mutation would be expected to affect the extracellular surface, a domain comprised of the N terminal and three interhelical loops (34). The mutation could alter one of two consensus glycosylation sequences in mammalian RHO (35) (sites 2–4 and 15–17). Although glycosylation has been implicated in RHO transportation, the consequences of its deficiency remain unclear. In vitro investigation of the T17M mutant indicated mislocalization (36) whereas analysis of a human postmortem donor retina from a patient with RHO T17M failed to show evidence of missorting (7). The amount and distribution of mature RHO in nondegenerate areas of canine RHO T4R/+ retinas were indistinguishable from normal (Fig. 4). These findings taken together with the normal sensitivity of rod photoresponses in a T4R/T4R dog support the notion that the mutation at T4 is not critical for targeting of the mature RHO protein to the photoreceptor outer segment and the mutant molecule functions normally during phototransduction activation.
Figure 5.

A secondary structure model of rhodopsin indicating the transmembrane helices (I–VII), interhelical loops of the extracellular domain (E1–E3), the intracellular and extracellular domains, and the locations of residues (circles) altered in canine (black) and human (gray) RHO mutations leading to a similar phenotype.
Of the seven human pathogenic RHO point mutations that we identified as having very prolonged recovery from light exposure (5, 27, 28), five are in the N-terminal and interhelical loops of the extracellular domain (Fig. 5). Two others are in transmembrane (TM) helices I and II. Evidence from in vitro studies (37) and the crystal structure of bovine RHO (3, 8) suggests that the extracellular surface and these TM domains closely interact with the bound chromophore. Misfolding of the mutant RHO molecules and abnormal chromophore binding have been among the mechanisms suggested to contribute to the pathophysiology in patients, but the exact molecular sequence leading to dysfunction and rod cell death remains unknown (8).
The extremely prolonged dark adaptation of photoreceptors harboring certain mutant alleles is likely to have a complex basis. After decades of debate about the sites in the visual system involved in normal recovery of sensitivity in the dark after exposure to light (38–42), there is recent consensus that latent transduction activity originating from bleach products within rods (32, 43) leads to bleaching adaptation in vivo. Several different bleach products probably contribute to varying extents at different times during adaptation (42, 44). Inactivation of these active bleach products and regeneration of RHO through the visual cycle pathway (45) define the observed kinetics of sensitivity recovery during bleaching adaptation. Among the active bleach products are photoisomerized rhodopsin, as well as phosphorylated and arrestin-bound forms. In addition, transduction activity originating from opsin and noncovalently bound forms of opsin and retinoid has been described. The relative activity and effective lifetimes of these moieties are currently not well established. Abnormally prolonged bleaching adaptation, such as in the RHO T4R/+ dog or in human RHO mutations, could be the result of slowed or deficient deactivation of one of these intermediate bleach products. Because the abnormality occurs relatively late during adaptation and after intense adapting lights, a late intermediate with relatively low activity is likely responsible. Alternatively, long-lived products resulting from photoreversal of mutant rhodopsin could cause the abnormal adaptation (46). Whether photoreceptor degeneration in the RHO mutant dog leads from the incomplete recovery after light exposure (7, 47–49) awaits further study.
Why should there be regions of severe structural damage surrounded by areas of structurally normal retina when the mutant rhodopsin is presumably distributed uniformly? The regional nature of observed structural damage may arise from topographic expression of genes (50–52), nonuniform distribution of light absorption across the retina, or both. The initial focus of disease in the canine T4R RHO mutant retina is mainly in the temporal tapetal retina. This region is the area of central vision in the dog and the area of highest photoreceptor population density. It may well be that this area receives the greatest light exposure naturally. Light damage has been suggested to cause regional disease in RHO mutations in humans (6, 7). Thus, the history of exposure to light may be a critical factor controlling the observed regional and temporal variation in the onset and progression of retinal degeneration in Mastiff autosomal dominant PRA.
Identification of this nonhuman RHO mutant large animal offers opportunities for evaluation of mechanism and therapy in humans affected with RP as a result of RHO mutations. The specific class of mutations includes the P23H RHO allele that accounts for a major percentage of adRP in the U.S. (4). The effect of the mutant (missense) allele dosage on disease severity, a controversial topic in the human literature (e.g., refs. 53–55), may be resolvable in the canine model. Adverse effects of environmental light and benefits of supplemental nutrients found in transgenic rodent models of human RHO mutations (e.g., refs. 47 and 56) can now be extended to a large animal. The temporal, functional, and structural progression of disease in the canine model offers an ideal time window of opportunity for gene therapy (16, 57). Affected dogs are clinically normal for at least several months before retinal degeneration develops, but at this stage retinal dysfunction can be clearly monitored by dark adaptometry. The therapeutic approach in the heterozygote, assuming a deleterious effect of the mutant allele, would require a vector designed to deliver a “knockdown” construct capable of destroying the mutant allele rather than simply replacing it with the wild-type allele. The RHO mutant dog thus provides an invaluable tool to evaluate such therapies before commencement of human clinical trials.
Acknowledgments
The support of the Mastiff Club of America is gratefully acknowledged. We thank Ms. J. Alling, Mr. K. Watamura, Ms. L. Gardner, and Dr. J. Huang for technical, graphical, and editorial assistance; and Drs. E. Banin and Y. Huang for involvement in patient studies. Antibodies were kindly provided by Drs. G. Adamus (Oregon Health and Science University, Portland, OR) and P. Hargrave (University of Florida, Gainesville, FL). This work was supported by grants from the National Institutes of Health (EY06855, EY13132, EY05627, EY13203, and EY13369), the Foundation Fighting Blindness, the Morris Animal Foundation/The Seeing Eye, the Mackall Foundation, and by the F. M. Kirby Foundation. A.V.C. is a Research to Prevent Blindness William and Mary Greve Scholar. S.G.J. is a Research to Prevent Blindness Senior Scientific Investigator. J.W.K. is supported by a grant from the Cornell University Center for Biotechnology and by the New York State Office of Science, Technology, and Academic Research.
Abbreviations
- RP
retinitis pigmentosa
- PRA
progressive retinal atrophy
- ERG
electroretinogram
- OCT
optical coherence tomography
- LRP
longitudinal reflectivity profile
- ONH
optic nerve head
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AY092841).
References
- 1.Spiegel A M. Annu Rev Physiol. 1995;58:143–170. doi: 10.1146/annurev.ph.58.030196.001043. [DOI] [PubMed] [Google Scholar]
- 2.Rattner A, Sun H, Nathans J. Annu Rev Genet. 1999;33:89–131. doi: 10.1146/annurev.genet.33.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K, Kumasaka T, Hori T, Behnke C A, Motoshima H, Fox B A, Le Trong I, Teller D C, Okada T, Stenkamp R E, et al. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 4.Gal A, Apfelstedt-Sylla E, Janecke A R, Zrenner E. Prog Ret Eye Res. 1997;16:51–79. [Google Scholar]
- 5.Cideciyan A V, Hood D C, Huang Y, Banin E, Li Z-Y, Stone E M, Milam A H, Jacobson S G. Proc Natl Acad Sci USA. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heckenlively J R, Rodriguez J A, Daiger S P. Arch Ophthalmol. 1991;109:84–91. doi: 10.1001/archopht.1991.01080010086038. [DOI] [PubMed] [Google Scholar]
- 7.Li Z-Y, Jacobson S G, Milam A H. Exp Eye Res. 1994;58:397–408. doi: 10.1006/exer.1994.1032. [DOI] [PubMed] [Google Scholar]
- 8.Menon S T, Han M, Sakmar T P. Physiol Rev. 2001;81:1659–1688. doi: 10.1152/physrev.2001.81.4.1659. [DOI] [PubMed] [Google Scholar]
- 9.Acland G M, Ray K, Mellersh C S, Gu W, Langston A A, Rine J, Ostrander E A, Aguirre G D. Proc Natl Acad Sci USA. 1998;95:3048–3053. doi: 10.1073/pnas.95.6.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acland G, Ray K, Mellersh C, Langston A, Rine J, Ostrander E, Aguirre G. Genomics. 1999;59:134–142. doi: 10.1006/geno.1999.5842. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre G, Baldwin V, Pearce-Kelling S, Narfstrom S, Ray K, Acland G. Mol Vis. 1998;4:23–29. [PubMed] [Google Scholar]
- 12.Aguirre G, Baldwin V, Weeks K, Acland G, Ray K. J Hered. 1999;90:143–147. doi: 10.1093/jhered/90.1.143. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Q, Acland G M, Zangerl B, Johnson J L, Mao Z, Zeiss C J, Ostrander E A, Aguirre G D. Invest Ophthalmol Visual Sci. 2001;42:2466–2471. [PubMed] [Google Scholar]
- 14.Gould D J, Peterson-Jones S M, Sohal A, Barnett K C, Sargan D R. Anim Genet. 1995;26:261–267. doi: 10.1111/j.1365-2052.1995.tb03253.x. [DOI] [PubMed] [Google Scholar]
- 15.Acland G M, Aguirre G D. Exp Eye Res. 1987;44:491–521. doi: 10.1016/s0014-4835(87)80160-4. [DOI] [PubMed] [Google Scholar]
- 16.Acland G M, Aguirre G D, Ray J, Zhang Q, Aleman T S, Cideciyan A V, Pearce-Kelling S E, Anand V, Zeng Y, Maguire A M, et al. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 17.Cideciyan A V. Methods Enzymol. 2000;316:611–626. doi: 10.1016/s0076-6879(00)16753-9. [DOI] [PubMed] [Google Scholar]
- 18.Cideciyan A V, Jacobson S G. Vision Res. 1996;36:2609–2621. doi: 10.1016/0042-6989(95)00327-4. [DOI] [PubMed] [Google Scholar]
- 19.Pugh E N, Jr, Lamb T D. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- 20.Murphy C J, Howland H C. Vision Res. 1987;27:599–607. doi: 10.1016/0042-6989(87)90045-9. [DOI] [PubMed] [Google Scholar]
- 21.Huang D, Swanson E A, Lin C P, Schuman J S, Stinson W G, Chang W, Hee M R, Flotte T, Gregory K, Puliafito C A, et al. Science. 1991;254:1178–1181. doi: 10.1126/science.1957169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Cideciyan A V, Papastergiou G I, Banin E, Semple-Rowland S L, Milam A H, Jacobson S G. Invest Ophthalmol Visual Sci. 1998;39:2405–2416. [PubMed] [Google Scholar]
- 23.Huang Y, Cideciyan A V, Aleman T S, Banin E, Huang J, Syed N A, Petters R M, Wong F, Milam A H, Jacobson S G. Exp Eye Res. 2000;70:247–251. doi: 10.1006/exer.1999.0793. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Mieziewska K, Philp N, van Veen T, Aguirre G. J Neurosci Methods. 1993;47:227–234. doi: 10.1016/0165-0270(93)90085-6. [DOI] [PubMed] [Google Scholar]
- 25.Aguirre G D, Acland G M. Exp Eye Res. 1988;46:663–687. doi: 10.1016/s0014-4835(88)80055-1. [DOI] [PubMed] [Google Scholar]
- 26.Adamus G, Zam Z S, Arendt A, Palczewski K, McDowell J H, Hargrave P A. Vision Res. 1991;31:17–31. doi: 10.1016/0042-6989(91)90069-h. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson S G, Kemp C M, Sung C-H, Nathans J. Am J Ophthalmol. 1991;112:256–271. doi: 10.1016/s0002-9394(14)76726-1. [DOI] [PubMed] [Google Scholar]
- 28.Kemp C M, Jacobson S G, Roman A J, Sung C-H, Nathans J. Am J Ophthalmol. 1992;113:165–174. doi: 10.1016/s0002-9394(14)71529-6. [DOI] [PubMed] [Google Scholar]
- 29.Cideciyan A V, Haeseleer F, Fariss R N, Aleman T S, Jang G F, Verlinde C L, Marmor M F, Jacobson S G, Palczewski K. Visual Neurosci. 2000;17:667–678. doi: 10.1017/s0952523800175029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobson S G, Cideciyan A V, Iannaccone A, Weleber R G, Fishman G A, Maguire A M, Affatigato L M, Bennett J, Pierce E A, Danciger M, et al. Invest Ophthalmol Visual Sci. 2000;41:1898–1908. [PubMed] [Google Scholar]
- 31.Bunge S, Wedemann H, David D, Terwilliger D J, van den Born L I, Aulehla-Scholz C, Samanns C, Horn M, Ott J, Schwinger E, et al. Genomics. 1993;17:230–233. doi: 10.1006/geno.1993.1309. [DOI] [PubMed] [Google Scholar]
- 32.Thomas M M, Lamb T D. J Physiol. 1999;518:479–496. doi: 10.1111/j.1469-7793.1999.0479p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stone E M, Kimura A E, Nichols B E, Khadivi P, Fishman G A, Sheffield V C. Ophthalmology. 1991;98:1806–1813. doi: 10.1016/s0161-6420(91)32046-3. [DOI] [PubMed] [Google Scholar]
- 34.Cha K, Reeves P J, Khorana H G. Proc Natl Acad Sci USA. 2000;97:3016–3021. doi: 10.1073/pnas.97.7.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferretti L, Karnik S S, Khorana H G, Nassal M, Oprian D D. Proc Natl Acad Sci USA. 1986;83:599–603. doi: 10.1073/pnas.83.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung C-H, Schneider B G, Agarwal N, Papermaster D S, Nathans J. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garriga P, Liu X, Khorana H G. Proc Natl Acad Sci USA. 1996;93:4560–4564. doi: 10.1073/pnas.93.10.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stiles W S, Crawford B H Phys. Soc. London, editors. Report of a Joint Discussion on Vision. Cambridge, U.K.: Cambridge Univ. Press; 1932. pp. 194–211. [Google Scholar]
- 39.Dowling J. Nature (London) 1960;188:114–118. doi: 10.1038/188114a0. [DOI] [PubMed] [Google Scholar]
- 40.Barlow H B. Vision Res. 1964;4:47–58. doi: 10.1016/0042-6989(64)90031-8. [DOI] [PubMed] [Google Scholar]
- 41.Lamb T D. Vision Res. 1981;21:1773–1782. doi: 10.1016/0042-6989(81)90211-x. [DOI] [PubMed] [Google Scholar]
- 42.Leibrock C S, Reuter T, Lamb T D. Eye. 1998;12:511–520. doi: 10.1038/eye.1998.139. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy M J, Lee K A, Niemi G A, Craven K B, Garwin G G, Saari J C, Hurley J B. Neuron. 2001;31:87–101. doi: 10.1016/s0896-6273(01)00340-3. [DOI] [PubMed] [Google Scholar]
- 44.Fain G L, Matthews H R, Cornwall M C, Koutalos Y. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- 45.Rando R R. Chem Rev. 2001;101:1881–1896. doi: 10.1021/cr960141c. [DOI] [PubMed] [Google Scholar]
- 46.Grimm C, Reme C E, Rol P O, Williams T P. Invest Ophthalmol Visual Sci. 2000;41:3984–3990. [PubMed] [Google Scholar]
- 47.Naash M I, Peachey N S, Li Z-Y, Gryczan C C, Goto Y, Blanks J, Milam A H, Ripps H. Invest Ophthalmol Visual Sci. 1996;37:775–782. [PubMed] [Google Scholar]
- 48.Chen C-K, Burns M E, Spencer M, Niemi G A, Chen J, Hurley J B, Baylor D A, Simon M I. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi S, Hao W, Chen C-K, Simon M I. Proc Natl Acad Sci USA. 2001;98:13096–13101. doi: 10.1073/pnas.201417498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Timmers A M, Wintjes E T, Hauswirth W W. Invest Ophthalmol Visual Sci. 1995;36:2008–2019. [PubMed] [Google Scholar]
- 51.van Ginkel P R, Timmers A M, Szel A, Hauswirth W W. Dev Brain Res. 1995;89:146–149. doi: 10.1016/0165-3806(95)00122-t. [DOI] [PubMed] [Google Scholar]
- 52.Sakuta H, Suzuki R, Takahashi H, Kato A, Shintani T, Iemura S, Yamamoto T S, Ueno N, Noda M. Science. 2001;293:111–115. doi: 10.1126/science.1058379. [DOI] [PubMed] [Google Scholar]
- 53.Kumaramanickavel G, Maw M, Denton M J, John S, Srikumari C R S, Orth U, Oehlmann R, Gal A. Nat Genet. 1994;8:10–11. doi: 10.1038/ng0994-10. [DOI] [PubMed] [Google Scholar]
- 54.Ayuso C, Trujillo M J, Robledo M, Ramos C, Benitez J, Martin-Oses F, del Rio T, Garcia-Sandoval B. Hum Genet. 1996;98:51–54. doi: 10.1007/s004390050158. [DOI] [PubMed] [Google Scholar]
- 55.Reig C, Trujillo M J, Martinez-Gimeno M, Garcia-Sandoval B, Calvo M T, Ayuso C, Carballo M. Ophthalmic Genet. 2000;21:79–87. [PubMed] [Google Scholar]
- 56.Li T, Sandberg M A, Pawlyk B S, Rosner B, Hayes K C, Dryja T P, Berson E L. Proc Natl Acad Sci USA. 1998;95:11933–11938. doi: 10.1073/pnas.95.20.11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewin A S, Drenser K A, Hauswirth W W, Nishikawa S, Yasumura D, Flannery J G, LaVail M M. Nat Med. 1998;8:967–971. doi: 10.1038/nm0898-967. [DOI] [PubMed] [Google Scholar]