

Abstract
Alcohol exposure during development can cause brain malformations and neurobehavioral abnormalities. In view of the teratogenicity of ethanol, identification of molecules that could counteract the neurotoxic effects of alcohol deserves high priority. Here, we report that pituitary adenylate cyclase-activating polypeptide (PACAP) can prevent the deleterious effect of ethanol on neuronal precursors. Exposure of cultured cerebellar granule cells to ethanol inhibited neurite outgrowth and provoked apoptotic cell death. Incubation of granule cells with PACAP prevented ethanol-induced apoptosis, and this effect was not mimicked by vasoactive intestinal polypeptide, suggesting that PAC1 receptors are involved in the neurotrophic activity of PACAP. Ethanol exposure induced a strong increase of caspase-2, -3, -6, -8, and -9 activities, DNA fragmentation, and mitochondrial permeability. Cotreatment of granule cells with PACAP provoked a significant inhibition of all of the apoptotic markers investigated although the neurotrophic activity of PACAP could only be ascribed to inhibition of caspase-3 and -6 activities. These data demonstrate that PACAP is a potent protective agent against ethanol-induced neuronal cell death. The fact that PACAP prevented ethanol toxicity even when added 2 h after alcohol exposure, suggests that selective PACAP agonists could have potential therapeutic value for the treatment of fetal alcohol syndrome.
Keywords: cerebellum‖fetal alcohol syndrome‖development‖caspases
Excessive alcohol consumption during pregnancy can provoke severe growth retardation associated with behavioral and cognitive deficits that are referred to as fetal alcohol syndrome (1). Epidemiological studies indicate that fetal alcohol syndrome is a major public health problem that concerns all industrialized countries (2). Neuronal vulnerability to ethanol coincides with a period of intense synaptogenesis that, in humans, starts during the third trimester of pregnancy (1). However, the mechanisms underlying the deleterious effects of ethanol on the developing brain remain largely unknown and no efficient treatment is currently available.
The cerebellar cortex of the newborn rat, that undergoes differentiation during the postnatal period, represents a unique model in which to investigate the impact of toxic agents on the developing brain (3). Indeed, it has long been known that exposure of rats to ethanol during gestation and lactation causes excessive death of cerebellar granule cells (4). In vitro studies have recently shown that exposure of cultured granule cells to ethanol inhibits neurite outgrowth and induces apoptosis through activation of caspase-3 (5–7). Therefore, molecules able to stimulate neurite outgrowth and/or to delay apoptosis of cerebellar granule cells represent potential candidates for the treatment of fetal alcohol syndrome.
We have recently shown that pituitary adenylate cyclase-activating polypeptide (PACAP), a member of the vasoactive intestinal polypeptide (VIP)-secretin-glucagon family (8), acts as a neurotrophic factor in the cerebellum during development (9). PACAP and its receptors are actively expressed in the rat cerebellar cortex from postnatal day 4 (P4) to P20 (9–12). In particular, high concentrations of PACAP receptors are found in the external granule cell layer during the period of intense neurogenesis (12, 13). In vivo studies have shown that injection of PACAP in the subarachnoid space increases the thickness of the internal granule cell layer and the number of granule neurons (14). Concurrently, in vitro experiments performed on cultured granule neurons have shown that PACAP acts as an antiapoptotic agent that inhibits caspase-3 activation (15–17). These observations prompted us to investigate the ability of PACAP to counteract the neurotoxic effects of ethanol on cerebellar granule neurons.
Materials and Methods
Chemicals.
The 38-amino acid form of PACAP was synthesized by solid-phase methodology as described (18). VIP was obtained from American Peptide (Sunnyvale, CA). Caspase inhibitors were supplied by R&D Systems. Phorbol 12-myristate 13-acetate, chelerythrine chloride, DTT, Ham's F-12 medium, and the N-1 supplement were purchased from Sigma. DMEM and the antibiotic-antimycotic solution were from Life Technologies (Cergy Pontoise, France).
Cell Culture.
Granule cell suspensions were prepared from cerebellums of 8-day-old Wistar rats, as described (19). For cell survival experiments, dispersed cells were seeded in multiwell plates (Costar) at a density of 3.5 × 105 cells per well. For measurement of caspase activity, cells were plated in Falcon 3003 dishes (Becton Dickinson) at a density of 1 × 107 cells per dish. For all experiments, cells were cultured in a chemically defined medium consisting of 75% DMEM and 25% Ham's F-12 supplemented with 2 mM glutamine/1 mM sodium pyruvate/25 mM KCl/1% N-1 supplement (×100) and 1% antibiotic-antimycotic solution. Cells were grown at 37°C in a humidified incubator with an atmosphere of 5% CO2/95% air.
Cell Survival.
Cells were preincubated for 3 h before treatment without or with ethanol, and then cultured for 24 h in the absence or presence of PACAP or VIP (10−11 to 10−6 M). Cells were incubated for 5 min with 15 μg/ml calcein and 15 μg/ml ethidium bromide and examined under a confocal laser scanning microscope (CLSM Noran OZ, Middleton, WI). For quantification of surviving neurons, cells were incubated for 8 min with 15 μg/ml fluorescein diacetate, rinsed once with PBS, and lysed with a Tris⋅HCl solution. Fluorescence intensity was measured with a FL600 microplate reader (Bio-Tek, Winooski, VT). Pilot experiments have shown that the fluorescence intensity is proportional to the cell number (in the range 5 × 104 to 1 × 106 cells per ml).
Cell Differentiation.
Immature granule cells were fixed with 4% paraformaldehyde in PBS at room temperature for 30 min. Fixed cells were incubated at 4°C overnight with a neurofilament 200 antiserum (Sigma) diluted 1:100 in PBS containing 3% Triton X-100 and 0.1% BSA. After several rinses in PBS, the cells were incubated at room temperature for 2 h with the peroxidase-antiperoxidase complex (Sigma) diluted 1:200 in PBS. The cells were washed twice with PBS and rinsed with 0.1 M Tris⋅HCl buffer. The immunoreactivity was revealed by addition of 0.1 M Tris⋅HCl buffer containing (0.5 mg/ml) diaminobenzidine (Sigma) and 0.015% H2O2. The reaction was stopped by removing the diaminobenzidine solution and by rinsing in a large volume of water.
The length of the neurites borne by granule cells was measured by means of a confocal laser scanning microscope (NORAN equipped with the INTERVISION SOFTWARE). The course of each neuronal process was traced manually and the length of the neurites was measured. The percentage of cells devoid of neurites and the number of neurites per cells were also calculated. A minimum of 300 neurites were measured in each well.
DNA Fragmentation.
Internucleosomal DNA cleavage was assessed by conventional gel electrophoresis after extraction of nuclear DNA, by using the Wizard Plus Minipreps DNA purification system (Promega). Control or treated granule cells were incubated for 10 min at room temperature in a lysis buffer consisting of 50 mM Tris⋅HCl, 10 mM EDTA, 1% Triton X-100, and 50 μg/ml RNase A. After a 15-min centrifugation at 14,000 × g, the cleaved DNA was separated from the intact genomic DNA. DNA fragments were mixed with the Wizard resin and transferred into a Vacuum manifold column. After three washes with the column-wash solution, DNA fragments were eluted with 50 μl of water. DNA ladders were visualized on a 1.5% agarose gel.
Caspase Activity.
Cultured cells were washed twice with PBS at 37°C and resuspended in PBS at 4°C. Cells were harvested by centrifugation (350 × g; 4°C; 10 min) and treated with the fluorometric caspase assay systems (Promega and R&D Systems). In brief, the cell pellet was resuspended in 10 μl of hypotonic cell lysis buffer and then incubated with 10 μl of the appropriate caspase substrate at 30°C for 1 h. The substrates used for the different caspases were, respectively: Ac-YVAD-AMC for caspase-1, Ac-VDVAD-AFC for caspase-2, Ac-DEVD-AMC for caspase-3, Ac-VEID-AFC for caspase-6, Ac-IETD-pNA for caspase-8, and Ac-LEHD-AFC for caspase-9. Fluorescence intensity was measured with a Bio-Tek FL600 microplate reader. The specificity of the assay was verified by adding 2.5 mM caspase-1 inhibitor Ac-YVAD-CHO or the caspase-3 inhibitor Ac-DEVD-CHO to the incubation mixture.
Visualization of Caspase Activity.
Granule cells treated during 12 h with ethanol (200 mM) in the absence or presence of PACAP were incubated for 30 min with the fluorescent caspase inhibitor FITC-VAD-FMK (Promega) that only binds the active form of caspases. Cells were then washed twice with PBS to remove the excess of probe and fixed for 30 min in a 4% paraformaldehyde solution. Granule cells were visualized by phase contrast, and active caspase-3 (green fluorescence) was revealed by UV excitation. The two images were superimposed by using the video imaging software METAMORPH (Princeton Instruments, Evry, France).
Mitochondrial Activity.
Cultured cells were incubated in the presence of the JC-1 probe (7.5 μg/ml; Molecular Probes) at 37°C for 15 min and then washed with PBS. In healthy granule cells, the intact membrane potential allows the lipophilic dye JC-1 to enter into the mitochondria where it accumulates and aggregates, producing an intense orange signal. In apoptotic cells, the mitochondrial membrane potential collapses so that the monomeric JC-1 probe remains cytosolic and stains the cell in green. Fluorescence intensity was measured with a Bio-Tek FL600 microplate reader and expressed as a ratio of the emission at 590 nm (orange) over 530 nm (green). Images were acquired on a computer-assisted microscope (Biocom 2000, Les Ulis, France).
Results
Effect of PACAP on Ethanol-Induced Toxicity.
A 24-h incubation of granule cells with graded concentrations of ethanol provoked a dose-dependent decrease of the proportion of surviving cells (Fig. 1A). A 25% reduction in the number of living cells was observed after incubation with 200 mM ethanol, a concentration that was used in all subsequent experiments. Coincubation of granule cells with ethanol (200 mM) and graded concentrations of PACAP for 24 h resulted in a dose-related increase in the number of surviving cells (Fig. 1B). Complete reversal of the toxic effect of ethanol was observed with 10−7 M PACAP. Incubation of granule cells with VIP, at concentrations up to 10−7 M, had no effect on ethanol-evoked cell death (data not shown). At a concentration of 10−6 M, VIP only induced a modest neuroprotective effect.
Figure 1.

Effect of PACAP on ethanol-induced cerebellar granule cell death. (A) Effect of graded concentrations of ethanol on survival of cultured granule cells. (B) Effect of graded concentrations of PACAP on ethanol-induced cell death. Each value represents the mean (±SEM) of three independent experiments performed in triplicate. **, P < 0.01; ***, P < 0.001 vs. control. #, P < 0.05; ##, P < 0.01 vs. ethanol-treated cells.
The neurotoxic effect of ethanol was associated with modifications of granule cell morphology that were suggestive of apoptotic cell death, such as cell shrinkage and nuclear condensation (Fig. 2 A and B). Coincubation of granule cells with ethanol and PACAP (10−7 M) restored the typical shape of differentiated neurons with bipolar fusiform cell bodies and long neurites (Fig. 2 A and C). In basal culture conditions, almost no DNA laddering was observed (Fig. 2D). Exposure of cells to ethanol for 12 h provoked a marked increase in DNA cleavage (Fig. 2D). Addition of PACAP (10−7 M) totally suppressed the effect of ethanol on DNA fragmentation (Fig. 2D).
Figure 2.
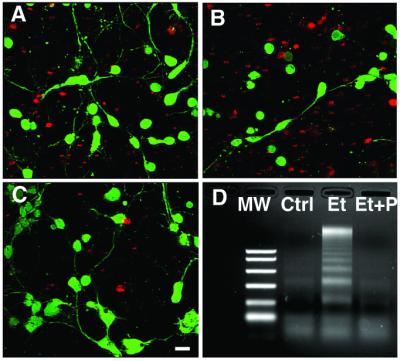
Visualization of the effect of PACAP on ethanol-induced cerebellar granule cell death. (A–C) Typical microphotographs of granule cells cultured in control conditions (A), in the presence of 200 mM ethanol (B), or in the presence of 200 mM ethanol plus 10−7 M PACAP (C). Living cells were labeled with calcein (green fluorescence), and dead cells were labeled with propidium iodide (red fluorescence). (Bar = 10 μm.) (D) DNA laddering in cerebellar granule cells cultured in control conditions (Ctrl) and in cells treated with 200 mM ethanol (Et) or with 200 mM ethanol plus 10−7 M PACAP (Et + P). MW, molecular weight markers.
Effect of PACAP on Ethanol-Induced Inhibition of Neurite Outgrowth.
After 24 h of culture in control conditions, only 17% of granule cells were devoid of neurites (Fig. 3A). Ethanol treatment caused a 2.5-fold increase in the number of neurite-lacking cells. The inhibitory effect of ethanol on neurite outgrowth was reversed by PACAP in a concentration-dependent manner (Fig. 3A). Likewise, ethanol induced a 2-fold decrease of the total length of neurites and the number of neurites per cell (Fig. 3 B and C). These deleterious effects of ethanol were totally prevented by nanomolar concentrations of PACAP (Fig. 3 B and C).
Figure 3.

Effects of PACAP on ethanol-induced inhibition of neurite outgrowth in cerebellar granule cells. The histograms represent the effect of ethanol (200 mM) in the absence or presence of graded concentrations of PACAP on the percentage of cells devoid of neurites (A), the cumulated length of neurites per cell (B), and the total number of neurites per cell (C). Each value represents the mean (±SEM) of three independent experiments performed in triplicate. **, P < 0, 01; ***, P < 0.001 vs. ethanol alone.
Effect of PACAP on Ethanol-Induced Caspase Activation.
Incubation of cultured granule cells with ethanol for various durations (1–12 h) had no effect on caspase-1 activity (Fig. 4). In contrast, ethanol provoked a significant increase in caspase-3 and -6 activity within the first 3 h of incubation. Ethanol induced a sustained activation of caspase-3 that was still observed after 12 h, whereas activation of caspase-6 was transient and vanished after 6 h of treatment. Activation of caspase-2 and -8 occurred after 6 h, whereas activation of caspase-9 was observed only after 9 h of ethanol treatment.
Figure 4.

Time course of the effect of ethanol on caspase-1, -2, -3, -6, -8, and -9 activities in cerebellar granule cells. Cultured granule cells were exposed to 200 mM ethanol for durations ranging from 1 to 12 h. Each value represents the mean (±SEM) of three independent experiments performed in triplicate. **, P < 0.01; ***, P < 0.001 vs. respective controls.
The effects of PACAP on ethanol-induced caspase activation was visualized by video microscopy with the fluorescent caspase inhibitor FITC-VAD-FMK (Fig. 5). Fluorescent neurons were characterized by round cell bodies devoid of neurites. Treatment of granule cells with ethanol for 12 h resulted in intense labeling of most granule cell bodies (Fig. 5B). Coincubation of granule cells with ethanol and PACAP (10−7 M) markedly reduced the proportion of fluorescent cells (Fig. 5C). Fluorescent neurons were characterized by round cell bodies devoid of neurites.
Figure 5.

Visualization of the effects of PACAP on ethanol-induced caspase activation in cerebellar granule cells. Cultured cells were incubated with the fluorescent probe FITC-ZVAD-FMK in control conditions (A), in the presence of 200 mM ethanol (B), or in the presence of 200 mM ethanol plus 10−7 M PACAP (C). (Bar = 10 μm.)
A 3-h incubation of granule cells with ethanol in the absence or presence of graded concentrations of PACAP (10−11 to 10−6 M) did not affect caspase-1 activity (Fig. 6A). Conversely, PACAP induced a concentration-dependent decrease of ethanol-evoked activation of caspase-2, -3, -6, -8, and -9 (Fig. 6 B–F). Blockage of caspase-3 activity with the specific inhibitor Z-DEVD-FMK, totally abolished the activation of caspase-9 induced by ethanol (Fig. 7A). Exposure of cultured granule neurons for 24 h to the caspase-3 inhibitor Z-DEVD-FMK suppressed the neurotoxic effect of ethanol (Fig. 7B). Inhibition of caspase-6 with Z-VEID-FMK attenuated ethanol-induced cell death, whereas the caspase-2 inhibitor Z-VDVAD-FMK, the caspase-8 inhibitor Z-IETD-FMK, and the caspase-9 inhibitor Z-LEHD-FMK had no effect (Fig. 7B).
Figure 6.

Effect of graded concentrations of PACAP on ethanol-induced caspase activation in cerebellar granule cells. Cultured cells were exposed to 200 mM ethanol for 3 h (A, C, and D) or 12 h (B, E, and F) in the absence or presence of graded concentrations of PACAP. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. ethanol alone.
Figure 7.

Effect of caspase inhibitors on ethanol-induced caspase activation and cerebellar granule cell death. (A) Effect of the caspase-3 inhibitor Z-DEVD-FMK (10 μM) on ethanol-induced caspase-9 activation. (B) Effect of 10 μM caspase-2 (Z-VDVAD-FMK), caspase-3 (Z-DEVD-FMK), caspase-6 (Z-VEID-FMK), caspase-8 (Z-IETD-FMK), and caspase-9 (Z-LEHD-FMK) inhibitors on ethanol-induced cell death. Each value represents the mean (±SEM) of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. ethanol alone.
Effect of PACAP on Ethanol-Induced Alteration of Mitochondrial Activity.
To determine whether apoptosis of granule cells induced by ethanol can be ascribed to impairment of mitochondrial activity, the membrane potential of mitochondria was measured by using the ratiometric probe JC-1. In control conditions, granule cells exhibited many active mitochondria in the cell body and along the neurites (orange fluorescence; Fig. 8A). Treatment of granule cells with ethanol for 12 h resulted in a marked decrease of mitochondrial activity (Fig. 8B). Coincubation with PACAP restored mitochondrial activity (Fig. 8C). Quantitative analysis revealed that ethanol induced a significant reduction of the 590 nm/530 nm ratio and that PACAP (10−7 M) totally suppressed the deleterious effect of ethanol on mitochondrial membrane potential (Fig. 7D).
Figure 8.

Effect of PACAP on ethanol-induced alteration of mitochondrial membrane potential in cerebellar granule cells. (A–C) Cultured granule cells were incubated for 12 h in control conditions (A), in the presence of 200 mM ethanol (B), or in the presence of 200 mM ethanol plus 10−7 M PACAP (C). Aggregated (red signal) and monomeric (green signal) fluorescent JC-1 dye revealed active and inactive mitochondria, respectively. (Bar = 25 μm.) (D) Effect of graded concentrations of PACAP on ethanol-induced alteration of the 590/530 nm fluorescent signal. Each value represents the mean (±SEM) of three independent experiments performed in quadruplicate. **, P < 0.01; ***, P < 0.001 vs. ethanol alone.
Effect of Delayed Administration of PACAP on Ethanol-Induced Toxicity.
Granule cells were treated with ethanol, and PACAP (10−7 M) was added 0, 1, 2, 3, or 6 h later. Measurements of cell survival 24 h after ethanol exposure indicated that PACAP (10−7 M) could still prevent ethanol-induced apoptosis when added 2 h after the onset of ethanol exposure (Fig. 9).
Figure 9.

Effect of delayed treatment with PACAP on ethanol-induced cerebellar granule cell death. Cultured cells were incubated with 200 mM ethanol, and PACAP was added 0, 1, 2, 3, or 6 h after the onset of ethanol exposure. Cell survival was measured after a 24-h incubation with ethanol. Each value represents the mean (±SEM) of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01 vs. ethanol alone.
Discussion
Ethanol exposure during the perinatal period has caused several neuronal dysfunctions including reduced proliferation, errors in migration, abnormal cellular differentiation, inappropriate connectivity, alteration in the neuronal versus glial cell ratio, and excessive neuronal cell death (20–22). It has recently been reported that PACAP promotes survival and activates differentiation of granule cells in the cerebellum of newborn rat (14–17, 23, 24). In the present study we demonstrate that PACAP can protect immature granule neurons against ethanol-induced apoptosis, and we describe some of the transduction mechanisms involved.
Effects of PACAP on Ethanol-Induced Granule Cell Apoptosis.
In agreement with previous reports performed on granule cells from 8-day-old rat, we observed that ethanol provokes apoptosis of granule neurons in primary culture (25). The ethanol-induced neuronal death exhibited the characteristic features of apoptosis including cell shrinkage, nuclear condensation, and DNA fragmentation (26). Coincubation of ethanol-treated granule cells with the neuropeptide PACAP resulted in a concentration-dependent increase in the number of surviving neurons, indicating that PACAP can protect neurons against the deleterious effects of alcohol.
Two PACAP receptors are expressed by cerebellar granule neurons: the PACAP-specific receptor PAC1-R (12, 13, 16) and the VIP/PACAP mutual receptor VPAC1-R (13). The present data showed that VIP, at concentrations up to 10−6 M, did not mimic the effect of PACAP on cell survival. This observation indicates that the neuroprotective action of PACAP against ethanol-induced granule cells apoptosis is mediated through activation of PAC1-R.
It has been recently reported that ethanol affects, in vitro, development of dendrites and synapses in various neuronal cell models (5, 27). Our data confirmed that ethanol provoked inhibition of neurite outgrowth on cerebellar granule cells, and showed that PACAP prevented the deleterious effects of ethanol, i.e., PACAP increased the proportion of cells bearing neurites, the total length of neurites, and the number of processes per cell, in a concentration-dependent manner. Previous studies have shown that ethanol exposure alters the expression of certain neurotrophic factors and/or their receptors (28, 29). In particular, prenatal alcohol exposure causes a decline of the density of trkA receptors in the cerebellum of newborn rat (30). This loss of neurotrophic receptors may in turn affect the differentiation of neural cells, impairing the establishment of proper connections, and thereby causing apoptosis of granule neurons (27, 30). Hence, administration of PACAP may compensate a shortage of trophic support, enhance neurite outgrowth, and thus inhibit apoptosis.
Mechanisms Involved in the Antiapoptotic Effect of PACAP.
The second objective of the present study was to determine the mechanisms implicated in the neuroprotective effect of PACAP on ethanol-induced toxicity.
Caspases are critical effectors of apoptotic cell death in the developing brain (31, 32). Because caspase-3 plays a prominent role in apoptosis of cerebellar granule neurons (33) and because ethanol activates caspase-3 in granule neurons (34), we have investigated the possible effects of PACAP on ethanol-induced caspase-3 activation in cerebellar granule cells. A 3-h treatment with ethanol induced a massive increase in caspase-3 activity and this effect was inhibited by PACAP in a concentration-dependent manner, in coincidence with the increase of cell survival and neurite outgrowth. The fact that the caspase-3-inhibitor Z-DEVD-FMK almost completely prevented ethanol-induced apoptosis indicates that caspase-3 activation is a major pathway through which ethanol induces neuronal toxicity.
Although the detailed signaling mechanisms that can trigger apoptosis are not completely elucidated, it is widely accepted that diverse initiator pathways can lead to activation of caspase-3. One of these pathways, recently described in ethanol intoxication of cultured hepatocytes, involves permeabilization of the mitochondrial outer membrane, yielding to leakage of cytochrome C in the cytosol (35, 36). Cytochrome C then causes activation of caspase-9 that subsequently activates caspase-3. Measurements of mitochondrial activity with a membrane potential-sensitive probe revealed that treatment of cerebellar granule neurons with ethanol resulted in a marked decrease of active mitochondria and this effect was prevented with PACAP. However, because caspase-9 activity was only detected 6 h after caspase-3 activation, and because caspase-9 activation could be prevented by the caspase-3 inhibitor Z-DEVD-FMK, whereas the caspase-9 inhibitor Z-LEHD-FMK did not protect granule cells from ethanol-induced apoptosis, it appears that activation of the cytochrome C/caspase 9 cascade is the consequence rather than the cause of caspase-3 activation.
We next tested the possible involvement of other caspases, including caspase-1, -2, -6, and -8, that represented other potential initiators of the effect of ethanol on caspase-3 activation. The implication of caspase-1 was excluded inasmuch as neither ethanol nor PACAP affected its activity. Conversely, caspase-2 and caspase-8 were activated by ethanol, and PACAP dose-dependently inhibited their activity. However, two observations indicate that caspase-2 and caspase-8 are not likely to be involved in the neuroprotective effect of PACAP: (i) activation of these two caspases only occurred after 6 h of treatment with ethanol (versus 3 h for caspase-3), and (ii) the caspase-2 inhibitor Z-WEHD-FMK and the caspase-8 inhibitor Z-IETD-FMK did not prevent ethanol-evoked toxicity. In contrast, a 3-h incubation with ethanol transiently activated caspase-6 and the caspase-6 inhibitor Z-VEID-FMK partially protected granule cells from ethanol-induced apoptosis, suggesting that this latter caspase may contribute to the neurotoxic effect of ethanol. In support of this notion, it has been shown recently that transient activation of caspase-6 is sufficient to initiate apoptosis of human primary neurons (37) and that caspase-6 is capable of processing pro-caspase-3 in cerebellar granule neurons (38). Collectively, these data indicate that caspase-6 may mediate ethanol-induced activation of caspase-3. The neuroprotective effects of PACAP on granule neurons are primarily mediated through the cAMP/protein kinase A signaling cascade (16, 23, 24, 39). Because the inhibitory effect of PACAP on caspase-3 activity is mediated through the protein kinase A pathway (17), it is possible that inactivation of caspase-6 by PACAP can be ascribed to a protein kinase A-dependent phosphorylation (40). Alternatively, PACAP could activate endogenous blockers of caspase-6-like CIF (caspase-inhibitory factor) (40) or the p35 protein (41).
It has been established that ethanol induces cell death by inhibition of N-methyl-D-aspartate receptor function (6, 25, 42). In particular, during a critical period of brain development, ethanol impairs N-methyl-D-aspartate-mediated brain-derived neurotrophic factor expression, leading to excessive granule cell loss (6, 43, 44). The transduction pathway that mediates the antiapoptotic effect of brain-derived neurotrophic factor requires activation of the phosphatidylinositol 3-kinase and phosphorylation of Akt, but does not involve the protein kinase A pathway (6). Because caspase-6 activity cannot be inhibited by Akt (45), the neuroprotective effect of brain-derived neurotrophic factor on cerebellar granule cell survival seems to be mediated through a caspase-6-independent pathway. This latter observation points out the necessity to plan multiple drug treatments to efficiently protect neurons from apoptosis.
Physiopathological Considerations.
High concentrations of PACAP and its receptors are present in the brain during development (11–13). In the rat cerebellar cortex, PACAP is synthesized by Purkinje cells and PAC1-R are intensively expressed by granule cells (11, 13). Experiments using transgenic mice lacking Purkinje cells have shown that Purkinje neurons actually promote the survival of cerebellar granule cells (46). It has also been shown that administration of PACAP during development increases the number of granule neurons in the internal granule cell layer (14). These observations indicate that PACAP likely acts as a neurotrophic factor in vivo, suggesting that nonpeptidic PACAP agonists may prove useful for the treatment of excessive neuronal cell death. The fact that PACAP can still efficiently prevent apoptosis when administered only 2 h after ethanol exposure generates hopes for the development of therapeutic agents that could be delivered after alcohol consumption to block the harmful effects of ethanol.
In conclusion, the present study has demonstrated that PACAP, acting through PAC1-R, exerts a potent neuroprotective effect against ethanol-induced programmed cell death. The antiapoptotic action of PACAP can be ascribed to inhibition of ethanol-induced activation of caspase-3 and likely caspase-6. These data suggest that PAC1-R agonists, which would selectively inhibit neuronal cell death provoked by alcohol consumption, could have a therapeutic value for the treatment of fetal alcohol syndrome.
Acknowledgments
This research was supported by the Institut National de la Santé et de la Recherche Médicale U-413, the Institut de Recherches Scientifiques sur les Boissons (2001/22), an exchange program from the Institut National de la Santé et de la Recherche Médicale-Fonds de la Recherche en Santé du Québéc (to A.F. and H.V.) and the Conseil Régional de Haute-Normandie. H.V. is Affiliated Professor at the Institut National de la Santé et de la Recherche Medicale-Institut Armand Frappier.
Abbreviations
- PACAP
pituitary adenylate cyclase-activating polypeptide
- VIP
vasoactive intestinal polypeptide
References
- 1.Ikonomidou C, Bittigau P, Ishimaru M J, Wozniak D F, Koch C, Genz K, Price M T, Stefovska V, Hörster F, Tenkova T, et al. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 2.Cornelius M D, Goldshmidt L, Taylor P M, Day N L. Alcoholism Clin Exp Res. 1999;23:1238–1244. doi: 10.1111/j.1530-0277.1999.tb04284.x. [DOI] [PubMed] [Google Scholar]
- 3.Komuro H, Rakic P. J Neurosci. 1998;18:1478–1490. doi: 10.1523/JNEUROSCI.18-04-01478.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borges S, Lewis P D. Brain Res. 1983;271:388–391. doi: 10.1016/0006-8993(83)90308-6. [DOI] [PubMed] [Google Scholar]
- 5.Bearer C F, Swick A R, O'Riordan M A, Cheng G. J Biol Chem. 1999;274:13264–13270. doi: 10.1074/jbc.274.19.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhave S V, Ghoda L, Hoffman P L. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saito M, Saito M, Berg M J, Marks N. Neurochem Res. 1999;24:1107–1115. doi: 10.1023/a:1020704218574. [DOI] [PubMed] [Google Scholar]
- 8.Arimura A. Jpn J Physiol. 1998;48:301–331. doi: 10.2170/jjphysiol.48.301. [DOI] [PubMed] [Google Scholar]
- 9.Vaudry D, Gonzalez B J, Basille M, Yon L, Fournier A, Vaudry H. Pharmacol Rev. 2000;52:269–52324. [PubMed] [Google Scholar]
- 10.Basille M, Gonzalez B J, Leroux P, Jeandel L, Fournier A, Vaudry H. Neuroscience. 1993;57:329–338. doi: 10.1016/0306-4522(93)90066-o. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen H S, Hannibal J, Fahrenkrug J. NeuroReport. 1998;9:2639–2642. doi: 10.1097/00001756-199808030-00039. [DOI] [PubMed] [Google Scholar]
- 12.Zhou C J, Shioda S, Shibanuma M, Nakajo S, Funahashi H, Nakai Y, Arimura A, Kikuyama S. Neuroscience. 1999;93:375–391. doi: 10.1016/s0306-4522(99)00108-6. [DOI] [PubMed] [Google Scholar]
- 13.Basille M, Vaudry D, Coulouarn Y, Jégou S, Lihrman I, Fournier A, Vaudry H, Gonzalez B J. J Comp Neurol. 2000;425:495–509. [PubMed] [Google Scholar]
- 14.Vaudry D, Gonzalez B J, Basille M, Fournier A, Vaudry H. Proc Natl Acad Sci USA. 1999;96:9415–9420. doi: 10.1073/pnas.96.16.9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez B J, Basille M, Vaudry D, Fournier A, Vaudry H. Neuroscience. 1997;78:419–430. doi: 10.1016/s0306-4522(96)00617-3. [DOI] [PubMed] [Google Scholar]
- 16.Villalba M, Bockaert J, Journot L. J Neurosci. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaudry D, Gonzalez B J, Basille M, Pamantung TF, Fontaine M, Fournier A, Vaudry H. Proc Natl Acad Sci USA. 2000;97:13390–13395. doi: 10.1073/pnas.97.24.13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chartrel N, Tonon M C, Vaudry H, Conlon J M. Endocrinology. 1991;129:3367–3371. doi: 10.1210/endo-129-6-3367. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez B, Leroux P, Lamacz M, Bodenant C, Balazs R, Vaudry H. Proc Natl Acad Sci USA. 1992;89:9627–9631. doi: 10.1073/pnas.89.20.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller M W. Alcohol Clin Exp Res. 1995;19:1359–1363. doi: 10.1111/j.1530-0277.1995.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 21.Guerri C. Alcohol Clin Exp Res. 1998;22:304–312. doi: 10.1111/j.1530-0277.1998.tb03653.x. [DOI] [PubMed] [Google Scholar]
- 22.Levitt P. Drug Alcohol Depend. 1998;51:109–125. doi: 10.1016/s0376-8716(98)00070-2. [DOI] [PubMed] [Google Scholar]
- 23.Cavallaro S, Copani A, D'Agata V, Musco S, Petralia S, Ventra C, Stivala F, Travali S, Canonico P L. Mol Pharmacol. 1996;50:60–66. [PubMed] [Google Scholar]
- 24.Campard P K, Crochemore C, René F, Monnier D, Koch B, Loeffler J P. DNA Cell Biol. 1997;16:323–333. doi: 10.1089/dna.1997.16.323. [DOI] [PubMed] [Google Scholar]
- 25.Bhave S V, Hoffman P L. J Neurochem. 1997;68:578–586. doi: 10.1046/j.1471-4159.1997.68020578.x. [DOI] [PubMed] [Google Scholar]
- 26.Joza N, Susin S A, Daugas E, Stanford W L, Cho S K, Li C Y, Sasaki T, Elia A J, Cheng H Y, Ravagnan L, et al. Nature (London) 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 27.Yanni P A, Lindsley T A. Dev Brain Res. 2000;120:233–243. doi: 10.1016/s0165-3806(00)00015-8. [DOI] [PubMed] [Google Scholar]
- 28.Heaton M B, Mitchell J J, Paiva M. Alcohol Clin Exp Res. 1999;23:1637–1642. [PubMed] [Google Scholar]
- 29.Tapia-Arancibia L, Rage F, Givalois L, Dingeon P, Arancibia S, Beauge F. J Neurosci Res. 2001;63:200–208. doi: 10.1002/1097-4547(20010115)63:2<200::AID-JNR1012>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 30.Dohrman D P, West J R, Pantazis N J. Alcohol Clin Exp Res. 1997;21:882–893. [PubMed] [Google Scholar]
- 31.Marks N, Berg M J. Neurochem Int. 1999;35:195–220. doi: 10.1016/s0197-0186(99)00061-3. [DOI] [PubMed] [Google Scholar]
- 32.D'Mello S R, Kuan C Y, Flavell R A, Rakic P. J Neurosci Res. 2000;59:24–31. [PubMed] [Google Scholar]
- 33.Kuida K, Zheng T S, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell R A. Nature (London) 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 34.Oberdoerster J, Rabin R A. Eur J Pharmacol. 1999;385:273–282. doi: 10.1016/s0014-2999(99)00714-1. [DOI] [PubMed] [Google Scholar]
- 35.Higuchi H, Adachi M, Miura S, Gores G J, Ishii H. Hepatology. 2001;34:320–328. doi: 10.1053/jhep.2001.26380. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Z, Sun X, Kang Y J. Am J Pathol. 2001;159:329–338. doi: 10.1016/S0002-9440(10)61699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Goodyer C, LeBlanc A. J Neurosci. 2000;20:8384–8389. doi: 10.1523/JNEUROSCI.20-22-08384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allsopp T E, McLuckie J, Kerr L E, Macleod M, Kelly J S. Cell Death Differ. 2000;7:984–993. doi: 10.1038/sj.cdd.4400733. [DOI] [PubMed] [Google Scholar]
- 39.Vaudry D, Gonzalez B J, Basille M, Anouar Y, Fournier A, Vaudry H. Neuroscience. 1998;84:801–812. doi: 10.1016/s0306-4522(97)00545-9. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Tounekti O, Akerman B, Goodyer C G, LeBlanc A. J Neurosci. 2001;21:RC176. doi: 10.1523/JNEUROSCI.21-20-j0007.2001. , 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ekert P G, Silke J, Vaux D L. Cell Death Differ. 1999;6:1081–1086. doi: 10.1038/sj.cdd.4400594. [DOI] [PubMed] [Google Scholar]
- 42.Snell L D, Bhave S V, Tabakoff B, Hoffman P L. J Neurochem. 2001;78:396–405. doi: 10.1046/j.1471-4159.2001.00424.x. [DOI] [PubMed] [Google Scholar]
- 43.Pierce D R, Goodlett C R, West J R. Teratology. 1989;40:113–126. doi: 10.1002/tera.1420400205. [DOI] [PubMed] [Google Scholar]
- 44.Bhave S V, Snell L D, Tabakoff B, Hoffman P L. J Neurochem. 2000;75:1035–1044. doi: 10.1046/j.1471-4159.2000.0751035.x. [DOI] [PubMed] [Google Scholar]
- 45.Cardone M H, Roy N, Stennicke H R, Salvesen G S, Franke T F, Stanbridge E, Frisch S, Reed J C. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 46.Smeyne R J, Chu T, Lewin A, Bian F, Crisman S S, Kunsch C, Lira S A, Oberdick J. Mol Cell Neurosci. 1995;6:230–251. doi: 10.1006/mcne.1995.1019. [DOI] [PubMed] [Google Scholar]