

ABSTRACT
Autophagy is a crucial cellular process responsible for the degradation and recycling of damaged or unnecessary components, maintaining cellular homeostasis and protecting against stress. Dysregulation of autophagy has been implicated in a variety of neurodegenerative diseases, including multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease. Various types of autophagy exist, each with distinct mechanisms, such as macroautophagy, mitophagy, lipophagy, and chaperone-mediated autophagy. These processes are essential for the removal of toxic substrates like protein aggregates and dysfunctional mitochondria, which are vital for neuronal health. In neurodegenerative diseases, the impairment of these clearance mechanisms leads to the accumulation of harmful substances, which accelerate disease progression. Modulating autophagy has emerged as a promising therapeutic strategy, with ongoing studies investigating molecules that can either stimulate or regulate this process. However, despite its potential, significant challenges remain in translating preclinical findings into clinically effective treatments. In this review, we will explore the different types of autophagy, their roles in neurodegenerative diseases, and the therapeutic potential associated with modulating these processes.
KEYWORDS: Multiple sclerosis, autophagy, mitophagy, ferritinophagy, mielinophagy, lipophagy, therapy
Introduction to autophagy and multiple sclerosis
Autophagy is an evolutionarily conserved intracellular process responsible for the degradation and recycling of cellular components. It plays a crucial role in human diseases, as its mechanisms are essential for maintaining cellular homeostasis and responding to various stressors [1]. In mammals, three distinct types of autophagy have been identified, each defined by the mechanism through which cellular cargo is transported to lysosomes for degradation: chaperone-mediated autophagy, microautophagy, and macroautophagy [2]. Among these, macroautophagy, often referred to as bulk autophagy (or simply autophagy, as referred to herein), is the most extensively studied and well characterized. Autophagy is generally considered a nonselective process. This form of autophagy involves the indiscriminate sequestration of cytosolic components, including proteins, lipids, and organelles, within autophagosomes. This process is particularly prominent during nutrient starvation, when the cell relies on autophagy to degrade and recycle intracellular materials to meet its metabolic and energy demands [3]. However, autophagy also encompasses several distinct and selective forms, each tailored to specific cellular tasks, but all sharing the common goal of maintaining cellular integrity through the degradation and recycling of damaged components [4]. Selective autophagy, unlike its nonselective counterpart, is a highly specialized process that targets specific cellular components for degradation. This includes damaged organelles such as mitochondria (mitophagy), lysosomes (lysophagy), the endoplasmic reticulum (ER-phagy), and ribosomes (ribophagy), as well as aggregated proteins (aggrephagy), invading pathogens like bacteria (xenophagy), and other specialized substrates [2]. Other important forms of selective autophagy, include ferritinophagy, which regulate iron homeostasis by targeting ferritin [5]; myelinophagy, involved in the degradation of myelin sheaths and playing a crucial role in neuronal remodeling and repair [6]; and lipophagy, which refers to the autophagic degradation of lipid droplets [7].
Multiple sclerosis (MS) is a chronic demyelinating autoimmune disorder of the central nervous system (CNS), arising from a complex interplay of environmental, genetic, and immunological factors. The disease is characterized by the convergence of neuroinflammatory and neurodegenerative processes, leading to progressive neurological disability in affected individuals [4]. Traditionally, MS was considered a T-cell-mediated autoimmune disease; however, recent research has revealed its multifaceted nature, involving various immune and CNS cell types. In addition to T-cells, immune cells such as dendritic cells and B-cells, as well as CNS-resident cells like neurons, astrocytes, oligodendrocytes, and microglia, contribute to MS pathogenesis [8]. Most MS patients experience a relapsing-remitting course (RRMS), characterized by episodes of acute inflammation and neurodegeneration followed by partial recovery. Over time, RRMS often transitions into secondary progressive MS (SPMS), marked by a steady accumulation of neurological deficits with reduced inflammatory activity [9]. Despite advances in understanding, the precise molecular mechanisms underlying MS, as well as the factors driving the transition from relapsing-remitting to progressive forms, remain incompletely understood. Currently, there is no cure for MS. Existing treatments primarily focus on managing the relapsing-remitting phase by modulating the immune response to reduce inflammation. Therapeutic options for progressive MS remain limited, highlighting the need for further research into disease mechanisms and novel treatment strategies [10]. In MS, autophagy exerts cellular quality control by targeting and eliminating potentially harmful or dysfunctional components, such as damaged organelles, protein aggregates, and myelin debris. This process is vital for preserving cellular homeostasis and ensuring proper neuronal and immune system function. However, dysregulated autophagy has been increasingly implicated in the pathogenesis of MS [11,12]. Aberrant autophagic processes contribute to the accumulation of dysfunctional organelles and misfolded proteins, exacerbating neuroinflammation and demyelination. For example, impaired lipophagy disrupts the clearance of damaged myelin [13], a hallmark of MS pathology. Similarly, disruptions in ferritinophagy can lead to iron dysregulation, promoting oxidative stress and further neuronal damage [14]. These findings emphasize the significance of selective autophagy in MS and suggest that modulating autophagic pathways could contribute to controlling disease progression and enhancing repair mechanisms.
In this review, we will examine the role of autophagy and selective autophagy in the context of multiple sclerosis, focusing on their contributions to disease pathology, including protein aggregation, organelle dysfunction, and neuronal damage. Dysregulated autophagy has been implicated as a key factor in these processes, highlighting its potential as a therapeutic target. We will also discuss conventional strategies and emerging therapeutic approaches aimed at modulating autophagy to prevent or mitigate disease progression. This includes an evaluation of autophagy-inducing agents and their efficacy in preclinical models, both in vitro and in vivo. By providing a comprehensive overview of the interplay between autophagy and multiple sclerosis, we aim to shed light on novel opportunities for therapeutic intervention.
Multiple sclerosis: pathophysiological insights
MS is a chronic autoimmune and inflammatory disease of the CNS, affecting approximately 2.3 million people worldwide [15]. This disease is characterized by demyelination and axonal damage, resulting in a wide range of neurological deficits. The clinical course of the disease is highly unpredictable, with symptoms ranging from sensory disturbances and motor impairments to cognitive dysfunction and vision problems [16].
MS is most commonly diagnosed between the ages of 20 and 50 years and shows a strong female predominance (approximately 2–3 times more common in women than in men). Geographically, MS is more prevalent in populations of Northern European descent and its incidence tends to decrease closer to the equator. In recent years, the number of cases has been increasing in various regions, particularly in Western countries [17].
The exact etiology of MS remains unclear. However, it is generally believed that a complex interplay of genetic, environmental, and possibly infectious factors contributes to its development [8]. Although MS is not a Mendelian disease, genome-wide association studies have identified multiple genetic variants that increase its susceptibility. Key environmental risk factors include low vitamin D levels, smoking, childhood obesity, and a history of Epstein-Barr virus infection. Ethnicity and geographic distribution also play roles: MS is more common among individuals of European descent living at higher latitudes, where sunlight exposure is reduced, while it is less frequent in individuals of Asian, Black, or Hispanic descent, although these groups may experience more severe disease forms once diagnosed [17,18]. MS manifests in various clinical subtypes, each with distinct courses. These include Relapsing-Remitting MS (RRMS), Secondary Progressive MS (SPMS), Primary Progressive MS (PPMS), and Progressive-Relapsing MS (PRMS)[9].
RRMS is the most common form, affecting approximately 85% of MS patients. It is characterized by episodes of neurological symptoms (relapses), followed by periods of partial or complete recovery (remission). Over time, RRMS may evolve into SPMS, marked by a gradual, steady accumulation of disability, with or without superimposed relapse. SPMS is now considered a later phase of the same disease [19].
In about 10% of cases, patients present PRMS, a form distinguished by gradual worsening of symptoms from the outset, without distinct relapses or remissions, although temporary plateaus may occur. PPMS tends to be more resistant to conventional therapies used for other MS subtypes. The rarest form of MS, PRMS, affects fewer than 5% of patients and is defined by progressive disease from onset with intermittent relapses and no remission periods[20]. Another important clinical presentation is the clinically isolated syndrome (CIS), which refers to a first episode of neurological symptoms suggestive of MS, lasting at least 24 hours. While not all CIS cases evolve into MS, it is considered a potential precursor, especially in individuals with magnetic resonance imaging (MRI) evidence of demyelination.
MS diagnosis relies on a combination of clinical evaluation and supportive diagnostic tests. The most widely used tools include magnetic resonance imaging, to detect characteristic lesions or plaques, cerebrospinal fluid (CSF) analysis to identify oligoclonal bands and elevated immunoglobulin G levels, and the exclusion of other neurological disorders that may mimic MS [20].
Inflammation is a central feature of MS, and there is a broad consensus that the disease is immune-mediated. However, it remains debated whether inflammation is the primary pathologic or a secondary reaction to an infectious agent or intrinsic CNS degeneration[21].
Although MS does not meet all criteria of a classic autoimmune disease, most evidence supports the notion that self-reactive immune cells attack the myelin sheath of CNS neurons. This breakdown in immune tolerance leads to activation, proliferation, and CNS infiltration of autoactivate lymphocytes. T-cells, upon activation by antigen-presenting cells (APCs) such as dendritic cells and macrophages proliferate in lymphoid tissues and enter the bloodstream, guided by sphingosine-1-phosphate. They then bind to upregulated adhesion molecules and secrete matrix metalloproteinases, which disrupt the blood-brain barrier (BBB) and permit immune cells entry into the CNS.
Within the CNS, T-cells interact with resident APCs and continue to expand. The attack on myelin leads to T helper cells differentiation into pro-inflammatory Th1 cells and anti-inflammatory Th2 subset. An imbalance between Th1 and Th2 level response is observed in MS. Th2 cells release cytokines that activate macrophages and microglial cells. In parallel, autoreactive T cells stimulate B cells to produce myelin-specific antibodies that cross the damaged BBB. These antibodies activate the complement system, amplifying myelin damage. These immune processes are thought to drive the relapsing nature of the disease, making inflammation a primary therapeutic target. Microglial cells, the innate immune cells of the CNS, also contribute to the inflammatory cascade by responding to pro-inflammatory signals and participating in demyelination and neuronal injury. MS is histopathologically characterized by the formation of plaques in the CNS. These are commonly found in the white matter surrounding the ventricles, optic nerves and tracts, corpus callosum, cerebellar peduncles, and long tracts of the spinal cord and brainstem, although gray matter involvement is also recognized. Plaques are observed in all disease forms but vary in quantity distribution, and immunopathological characteristics over time. This reflects the heterogeneity of demyelination and oligodendrocyte pathology, particularly when comparing relapsing and progressive MS subtypes [19,22].
Types of autophagy and molecular mechanisms
The most extensively studied form is macroautophagy (autophagy). This cellular process involves the formation of a double-membrane vesicle, known as the autophagosome, which engulfs damaged proteins and organelles. These vesicles fuse with lysosomes, where their contents are degraded and recycled. Autophagy is traditionally considered a bulk, nonselective process, responsible for the indiscriminate recycling of cytoplasmic components to maintain cellular energy levels and provide biosynthetic precursors. However, it is now well established that autophagy can also operate in a selective manner, targeting specific substrates through dedicated receptors and adaptor proteins. Depending on the cargo, selective autophagy includes processes such as mitophagy (mitochondria), nucleophagy (nuclei), pexophagy (peroxisomes), reticulophagy or ER-phagy (endoplasmic reticulum), lipophagy (lipids), aggrephagy (protein aggregates), and ferritinophagy (ferritin). This section will examine the molecular mechanisms that govern these different forms of autophagy.
Autophagy
Autophagy begins with the formation of a phagophore, a cup-shaped membrane structure that expands and engulfs cytoplasmic material to form an autophagosome. The initiation of autophagy is regulated by the ULK1 complex, which includes ULK1 (UNC-51-like kinase 1), autophagy-related (ATG) 13/200-kDa focal adhesion kinase family-interacting protein (FIP200) and ATG101. Under normal energy and amino acid levels, the mechanistic target of rapamycin (MTOR), a central nutrient sensor, inhibits autophagy by phosphorylating ULK1 at Ser 757, preventing its activation. Conversely, during nutrient deprivation, hypoxia, or mitochondrial dysfunction, AMPK promotes autophagy by phosphorylating ULK1 at Ser 317 and Ser 777. AMPK also inhibits MTOR by phosphorylating its upstream regulators, further enhancing autophagic activity. The interaction between MTOR and AMPK tightly regulates autophagy in response to the cellular energy status. Additionally, ULK1 can undergo autophosphorylation at Thr 180, as well as phosphorylate other key autophagy-related proteins, including FIP200, ATG13, and ATG101. These phosphorylation events are tightly regulated by protein phosphatases, such as protein phosphatase 2A and PP2C phosphatases (Ptc2 and Ptc3). The ULK1 complex also regulates another critical regulator, which is Beclin 1, (BECN1), a component of the class III phosphatidylinositol 3-kinase (PI3K) complex. Once activated, BECN1 forms a complex with PI3K and its regulatory subunits, including vacuolar protein sorting 34, p150, and ATG14L, which mediates the nucleation and elongation of autophagosomes. The ATG12–ATG5–ATG16L1 complex and the LC3–phosphatidylethanolamine (PE) conjugate are essential for the elongation and closure of autophagosomes, although the precise mechanisms remain unclear. A key step in autophagosome maturation is the lipidation of LC3, which binds to PE on the expanding membrane. Once incorporated, lipidated LC3 (LC3-II) facilitates the recruitment of adaptor proteins, enabling cargo recognition and promoting vesicle elongation and closure. The subsequent fusion of the autophagosome with the lysosome results in acidification of the lysosomal compartment, degradation of macromolecules by hydrolases and lipases, and the recycling of basic cellular components (Figure 1). Autophagy is known to be dysregulated in several neurodegenerative diseases. For instance, autophagy can degrade pathologic forms of amyloid β (Aβ) and tau protein in Alzheimer’s disease (AD) [23]. Consistently, genetic deletion of ATG7 or loss of autophagy function leads to plaque formation [24]. Moreover, mutations in Presenilin-1, an enzyme involved in the Aβ production and associated with familial AD, impair autophagosome clearance [25]. Interestingly, autophagy has also been implicated in AD-related dementias, as reductions in autophagy-related markers have been detected in the biofluids of patients affected by various types of dementia. These findings suggest that monitoring of autophagy components could serve as a novel approach for the diagnosis and prognosis of AD and related disorders [26–28]. Impaired autophagosome clearance has also been observed in Amyotrophic Lateral Sclerosis (ALS) where accumulations of autophagosomes and pathological proteins are detected in patients. Consistently, ALS models expressing mutant SOD1 (superoxide disumutase 1), a familial ALS gene, show impaired autophagy flux due to the interaction between mutant SOD1 to BECN1 [29]. Supporting the importance of autophagy in ALS, mutations in sequestosome 1 (p62) may contribute to disease onset and act as genetic susceptibility factors [30]. Similarly, the genetic ablation of Atg5 or Atg7 in motor neurons leads to protein aggregation and motor dysfunction [31]. Huntington’s disease (HD) is fatal autosomal-dominant neurodegenerative disorder caused by a mutation in the Huntingtin (HTT) gene. Wild-type HTT interacts with ULK1 acts as a scaffold for the MTORC1-ULK1 complex without impairing autophagy [32]. Opposite, mutant HTT shows reduced affinity for ULK1, leading to impaired initiation of autophagy [33]. Regarding Parkinson’s disease (PD), mitophagy is the main autophagy mechanisms involved in the onset and progression of this neurodegenerative condition. Nevertheless, several studies demonstrate that also bulk autophagy concurs to PD. Accordingly, aberrant α-synuclein (α-syn) leads to an increase in autophagy and BECN1 expression to enhance lysosomal breakdown and ameliorate the neurodegenerative condition [34]. Moreover, evidence of impaired autophagy has been found in the substantia nigra of PD brains [35,36].
Figure 1.
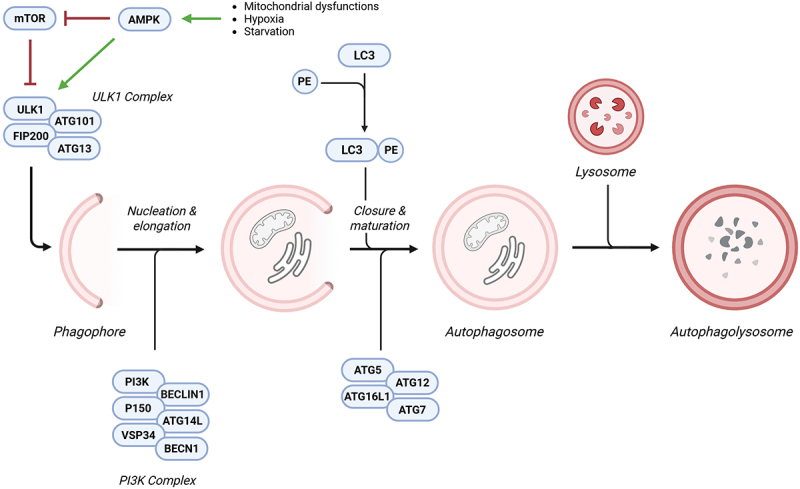
Molecular mechanism of autophagy. Various stress conditions, such as mitochondrial dysfunction, hypoxia, and starvation, can activate AMPK. In turn, AMPK inhibits MTOR and stimulates the ULK1 complex, promoting phagophore formation. Once the phagophore is formed, the PI3K complex mediates its nucleation and elongation into an autophagosome. LCE-PE, along with the ATG12–ATG5–ATG16L1 complex, plays a crucial role in autophagosome maturation and closure. Finally, the fusion of the autophagosome with the lysosome results in the formation of the autophagolysosome, where engulfed macromolecules undergo degradation. Abbreviations: AMP-activated protein kinase (AMPK), mechanistic target of rapamycin (MTOR), UNC-51-like kinase 1 (ULK1), phosphatidylinositol 3-kinase (PI3K), LC3–phosphatidylethanolamine (LC3-PE), autophagy related 12 (ATG12), autophagy related 5 (ATG5), autophagy related 16 like 1 (ATG16L1). Created with Biorender.
Mitophagy
Mitophagy is a specialized form of selective autophagy dedicated to the elimination of dysfunctional, aged, or surplus mitochondria. This process ensures that impaired mitochondria are sequestered within autophagosomes and subsequently degraded through lysosomal activity. By removing compromised mitochondria, mitophagy prevents the accumulation of harmful reactive oxygen species (ROS), preserves cellular energy efficiency, and maintains mitochondrial quality control. Moreover, mitophagy plays a pivotal role in adjusting mitochondrial content to meet cellular demands under various stress conditions, including nutrient deprivation, hypoxia, or oxidative stress. This dynamic regulation is essential for maintaining cellular homeostasis and supporting cell survival and function in response to environmental and metabolic challenges.
Over the past decade, substantial progress has been made in elucidating the molecular mechanisms underlying mitophagy, revealing the involvement of multiple, often redundant, pathways. Among these, the PTEN-induced kinase 1 (PINK1)/Parkinson protein 2, E3 ubiquitin protein ligase (Parkin) axis is the most extensively characterized mitophagy pathway in metazoan cell types. This pathway coordinates the selective ubiquitination of damaged mitochondria, promoting their recognition and clearance by the autophagic machinery. Under basal conditions, PINK1 levels are tightly controlled through mitochondrial import and subsequent proteolytic degradation. Specifically, PINK1 is imported into the mitochondria and processed by the matrix protein mitochondrial processing peptidase and the inner mitochondrial membrane protease presenilin-associated rhomboid-like (PARL). However, in damaged mitochondria, membrane potential dissipation impairs PINK1 import and cleavage, leading to its accumulation and stabilization on the outer mitochondrial membrane (OMM). Once stabilized, PINK1 phosphorylates both the E3 ubiquitin ligase Parkin and ubiquitin, facilitating the recruitment of Parkin to the mitochondria. Activated Parkin ubiquitinates several OMM proteins, labeling them with “eat-me” signals for selective autophagic degradation. These ubiquitin tags are recognized by adaptor proteins containing both LC3-interacting region (LIR) motifs and ubiquitin-binding domains. Key adaptors include p62, optineurin (OPTN), NDP52, NBR1, and TAX1BP1. Their activity is further modulated by phosphorylation via TANK-binding kinase 1, which enhances their binding affinities for ubiquitin chains and ATG8 family proteins. For instance, OPTN is phosphorylated at Ser177 to increase its interaction with LC3, while phosphorylation at Ser473 and Ser513 increases its affinity for polyubiquitin chains, thereby optimizing its function in cargo recognition and autophagosome formation. The specific autophagic elimination of mitochondria can also occur independently of Parkin. Consistently, in cells deficient for either PINK1 or Parkin, the clearance of dysfunctional mitochondria still takes place. In response to different stimuli, specific mitophagy receptors located in the OMM, such as FUN14 domain-containing protein 1, NIX, BCL2 interacting protein 3, and FKBP prolyl isomerase 8, interact directly with LC3 via a conserved LIR motif located in their N-terminal regions. Moreover, recent studies have shown that IMM proteins, such as cardiolipin and prohibitin 2, can externalize to the OMM and directly bind to LC3 for mitophagy induction. Alterations in mitophagy activities are frequently observed during neurodegeneration. Genetic mutations or loss of mitophagy-related factors, including PINK1 and Parkin, are associated with familial forms of PD. In PD models, α-syn disrupts Parkin function and delay mitophagy. At the same time, specific mutation in α-syn (such as A53T) can provoke neuronal death through an excessive mitophagy activation. Mouse models of AD, as well as postmortem human brain samples, provide clear evidence of mitophagy impairment. In line with this, both in vitro and in vivo studies have shown that Tau and Aβ species induce a blockade of mitophagy flux, resulting in the consequent accumulation of wasted mitochondria and neuronal injury. Dysregulated mitochondrial turnover has been also observed in the pathogenesis of HD. Consistently, mutant HTT impairs mitophagy at the cargo recognition step [37]. Restoring of the mitophagy pathway (particularly the PINK-Parkin axis) has been shown to alleviate mitochondrial defects and neuronal damage in HD models [38] (Figure 2).
Figure 2.
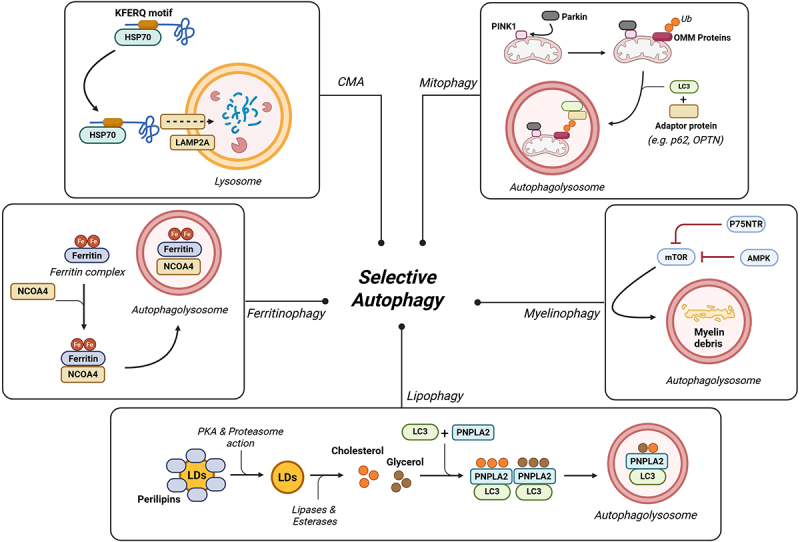
Selective autophagy pathways in neurodegeneration. Autophagy can function in a highly selective manner, defining distinct pathways depending on the macromolecules or organelles targeted for degradation. Mitophagy is tightly regulated by the PINK1/Parkin axis, where PINK1 facilitates the recruitment of Parkin, an E3 ubiquitin ligase that ubiquitinates OMM proteins. Ubiquitinated OMM proteins are recognized by adaptor proteins, which, in turn, recruit LC3, promoting mitochondrial engulfment into autophagosomes. Myelinophagy is influenced by the p75NTR/AMPK/MTOR axis, where p75NTR and AMPK inhibit MTOR, thereby stimulating myelin-specific autophagy in Schwann cells. Lipophagy regulates intracellular lipid storage. Normally, perilipins are removed by protein-kinase a and the proteasome, allowing LDs to be hydrolyzed into glycerol and cholesterol. These components are recognized by PNPLA2 in conjunction with LC3, facilitating their sequestration into autophagosomes for degradation. Ferritinophagy specifically targets intracellular ferritin, a protein responsible for iron storage. Under conditions of iron imbalance, NCOA4 selectively binds ferritin complex, enabling its transport to autophagosomes for degradation. CMA is responsible for the selective degradation of proteins containing a KFERQ-like motif. This motif is recognized by the constitutive chaperone HSC70, which facilitates the translocation of the protein into the lysosome through LAMP2A. Abbreviations: PTEN-induced kinase 1 (PINK1), outer mitochondrial membrane (OMM), p75 neurotrophin receptor (p75NTR), AMP-activated protein kinase (AMPK), mechanistic target of rapamycin (MTOR), protein-kinase a (PKA), lipid droplets (LDs), patatin-like phospholipase domain-containing 2 (PNPLA2), nuclear receptor coactivator 4 (NCOA4), chaperone-mediated autophagy (CMA), heat shock-cognate protein of 70 kDa (Hsp70), lysosome-associated membrane protein type 2A (LAMP2A). Created with Biorender.
Lipophagy
In 2009, it was discovered that macroautophagy can selectively degrade lipids in hepatocytes, leading to the coining of the term “lipophagy” to describe this process. Lipophagy plays a key role in regulating intracellular lipid storage, controlling the levels of free lipid levels (such as fatty acids), and maintaining energy homeostasis. It can be triggered by nutrient deprivation and is transcriptionally regulated by factors that promote lysosomal and mitochondrial biogenesis. At the molecular perspective, lipophagy is coordinated with other catabolic pathways, including chaperone-mediated autophagy (CMA) and lipolysis. Its execution involves a variety of autophagy receptors containing ATG8-family interaction motifs or LC3-interaction regions (such as ATG32, Nix, and BNIP) as well as LIR-containing adaptor proteins, including OPTN, NBR1, TAX1B1, NDP52, and p62, which are also implicated in the selective autophagy of other components. Perilipins are a family of proteins that coat intracellular lipid droplets (LDs), protecting them from unnecessary lipolysis and preserving lipid storage. During lipophagy, perilipins are initially phosphorylated by protein-kinase A and subsequently degraded via the proteasome. Following this, they are recognized by the heat shock 70 kDa protein 8 (HSC70) and delivered to the lysosome-associated membrane protein type 2A (LAMP-2A), targeting them for degradation via CMA. This process initiates a cascade involving lipases and esterases that hydrolyze into fatty acids, glycerol, and cholesterol. These lipid catabolites interact with Patatin-like phospholipase domain-containing 2 (PNPLA2), also known as adipose triglyceride lipase, to promote further lipolysis. PNPLA2 can bind LC3B, facilitating the tethering of lipid droplets (LDs) to the autophagosome membrane or alternatively interact with the cargo receptor p62 to mediate LDs recruitment. Concurrently, the recruitment of LC3-positive autophagic membranes to LDs is also supported by Rab10, in cooperation with adaptor proteins such as EH domain binding protein 1 and the membrane remodeling ATPase EH domain-containing 2. Finally, LC3 positive droplets are recognized by specific receptors that serve as adaptors, linking lipids to the autophagy machinery and enabling their sequestration of lipid within autophagosomes for degradation (Figure 2).
Myelinophagy
Myelin disruption due to trauma or disease leads to the accumulation of myelin debris, which must be efficiently removed to prevent pathological consequences. Recent studies have demonstrated that autophagy selectively contributes to myelin clearance, mainly within Schwann cells. Despite this, the precise molecular mechanisms uniquely governing this process remain under investigation. Current evidence suggests that this form of selective autophagy, referred to as myelinophagy, may be regulated by the JNK1/c-Jun signaling pathway and the p75NTR/AMPK/MTOR axis [6,39]. Nevertheless, the effective role of MTOR should be further investigated, as some findings indicate that myelin clearance can also occur in the presence of MTOR activation. In such cases, MTOR activation might reflect the activation of major biosynthetic pathways aimed at promoting cellular growth and regeneration after injury, rather than an inhibition of autophagy itself [6]. Similarly, the autophagy receptors are specifically responsible for targeting myelin remain to be fully characterized. Among the candidates, p62 is likely to play a key role in the autophagic degradation of myelin. Interestingly, TAX1BP1, a well-established autophagy receptor involved in the selective form of autophagy, has been shown to prevent proteotoxic stress in the brain by mediating the clearance of aggregated proteins [40]. Although a direct role for TAX1BP1 in myelinophagy has not yet been demonstrated, its known function in selective autophagy, especially in the nervous system, suggests that it could also be involved in the recognition and removal of damaged myelin. Further investigations are required to confirm the involvement of p62 and to identify additional receptors, such as TAX1BP1 that may mediate the selective clearance of myelin [41] (Figure 2).
Ferritinophagy
Ferritinophagy is a selective form of autophagy that specifically targets intracellular ferritin for degradation, playing a pivotal role in maintaining iron homeostasis. Ferritin, a protein complex composed of ferritin heavy chain (FTH1) and ferritin light chain subunits, binds and stores excess iron within cells, thereby mitigating oxidative damage caused by iron overload [1]. This process ensures the degradation and recycling of ferritin, allowing the release of bioavailable iron for essential cellular functions while preventing iron-induced toxicity.
At the molecular level, extracellular Fe3+ is imported into cells through transferrin receptor 1 (TFR1) and subsequently reduced to Fe2+ within endosomes by the iron reductase six-transmembrane epithelial antigen of the prostate 3. The reduced Fe2+ is then transported into the cytoplasm via the divalent metal transporter 1, contributing to the labile iron pool (LIP). Cytoplasmic Fe2+ can be oxidized and stored as Fe3+ within ferritin, or alternatively, it may be exported from the cell via ferroportin. These tightly regulated processes of iron intake, storage, and export ensure the maintenance of redox-active iron levels within the LIP. When intracellular iron levels are altered, nuclear receptor coactivator 4 (NCOA4) selectively binds to the FTH1 subunit of the ferritin complex, enabling its delivery to the autophagosome. It is important to note that NCOA4 does not contain a canonical LIR motif. Nevertheless, studies suggest that its recruitment mechanism is related to ATG8-family proteins and may involve alternative motifs for ATG proteins [42]. At the same time, alternative biological pathways distinct from the classical autophagy machinery have been implicated in ferritin degradation. These include the ubiquitin–proteasome system, ATG9A, FIP200, Tax1, and the endosomal sorting complex required for transport pathway. Finally, the autophagosome fuses with the lysosome to form the autolysosome, where the ferritin complex is degraded, releasing ferrous iron (Fe2+) for cellular use (Figure 2).
Chaperone-Mediated Autophagy (CMA)
One of the defining characteristics of CMA is its selective nature, whereby proteins targeted for degradation are individually recognized based on the KFERQ-like motif within their amino acid sequences [43]. Through this selective mechanism, CMA not only contributes to protein quality control but also participates in maintaining cellular homeostasis. Its ability to fine-tune the degradation of specific proteins ensures a dynamic balance between synthesis and degradation, supporting the adaptation of cells to changing physiological demands and environmental stresses. This regulatory role underscores the importance of CMA in preserving cellular function and preventing the accumulation of toxic protein aggregates. CMA occurs in the cytosol where target proteins are recognized and bound by the constitutive chaperone HSC70. The resulting complex is directed to the lysosome membrane, where it binds to the cytosolic tail of LAMP-2A. This interaction promotes the multimerization of LAMP-2A and its association with luminal chaperones, leading to the unfolding of the substrate protein. Once unfolded, the protein is translocated into the lysosomal lumen, where it undergoes complete degradation. CMA fulfills several essential physiological functions, primarily by regulating the turnover of protein complexes through the selective removal of damaged or oxidized proteins. Moreover, CMA is upregulated in response to various stress conditions, including oxidative damage, hypoxia, and exposure to toxic agents, and has also been implicated in antigen presentation [44]. Importantly, CMA dysfunction has been associated with the pathogenesis of several human disorders, particularly neurodegenerative disorders. For instance, many PD-related proteins, including α-syn, possess CMA-targeting motifs. Mutant forms of these proteins not only resist degradation via CMA substrates but also interfere with the processing of other CMA substrates, thereby disrupting CMA function and contributing to the accumulation of undegraded proteins. In a similar fashion, tau protein is a known CMA substrate, and its pathogenic variants exhibit impaired translocation into the lysosomal matrix, resulting in defective degradation and the formation of toxic tau oligomers (Figure 2).
Autophagy and MS
The last few decades have been marked by significant advancements in understanding how autophagy plays a crucial role in neurodegeneration. Moreover, recent years have seen particular progress in uncovering the autophagy-dependent pathways involved in the onset and progression of multiple sclerosis.
Autophagy
The involvement of autophagy in MS was first suggested in 2009, when a strong correlation was identified between the expression of ATG5 and the clinical disability observed in the experimental autoimmune encephalomyelitis (EAE) animal model of MS [45]. Interestingly, ATG5 was also overexpressed in T cells from RRMS patients and in postmortem brain tissue with a progressive MS form. Subsequently, the increase of ATG5 in T cells was confirmed in another investigation where the expression of autophagy marker was linked to the inflammatory status. In line with this, differentiated expression of mRNA and protein levels (such as ATG5, ATG7, ULK1, ATG4C, BCL2 and ATG9A) of autophagy-related elements were also found in human biofluids, mainly in serum and CSF [11,14], as well as an increased presence of autophagolysosomes in autoptic brain of MS patients [12]. However, these studies did not establish whether increases in autophagy were correlated with the clinical activity of the disease. This gap was addressed in a recent study, which demonstrated that elevated levels of autophagy markers were predominantly detected in patients experiencing the active phase of the disease. These findings were further supported by evidence of high inflammatory activity, as indicated by MRI results and the release of pro-inflammatory cytokines [46]. Despite these findings, research reports also demonstrate that autophagy-related elements could be downregulated in human fluids. Consistently, reduced amounts of mRNA of the autophagy genes BECN1, AMBRA, ATG16L, LAMP2, and ATG9A were detected in blood of MS patients [11], suggesting the possibility that autophagy may be a protective event against the progression of the diseases. Consistent with the possibility that autophagy could be deregulated in MS, excessive soluble oligomers, normally associated with protein-misfolding diseases (PMDs) was detected in MS and normal control brain samples obtained at autopsy. In line with this, pathological protein aggregation in MS was also detected in the spinal cord of mice with EAE [47]. It is important to highlight that this great protein aggregation was associated with a diminished proteasomal activity, as already observed in two independent studies conducted in humans [48] and in rodent mouse model [49]. Briefly, in these investigations, reduced functions of the activities of the three peptidases of the 20S proteasome (i.e. chymotrypsin-like, caspase-like and trypsin-like) was observed. MS rodent mouse also displayed a progressive reduction of the expression of autophagy-related proteins LC3-II, BECN1 and a parallel increase of p62 levels in the spinal cord during the progression of the EAE model, which could reflect a reduction of autophagy activity. Nevertheless, when these elements were measured in the same experimental model but in different regions of the CNS (hippocampus), the results obtained were in contrast. Indeed, BECN1 and LC3 II reductions were accompanied by a significant decrease in p62 levels, suggesting a sustained autophagy flux [50]. These findings suggest that autophagy may be differentially activated and play distinct roles in damaged neurons depending on the CNS region involved. Also the expression level of the pathway Akt/MTOR resulted in contrast among the CNS region investigated during EAE. While the levels of (p)-Akt and p-MTOR were lower in the hippocampus of EAE mice than controls, in spinal cords were augmented. The fact that the role of MTOR has conflicting results in the pathogenesis of the MS is not a novelty. Corruption of the MTOR pathway can reduce the disease severity in EAE mice. In this context, a recent study highlighted a new protective mechanism by which MTOR signaling is modulated in MS. Specifically, activation of the AXL receptor tyrosine kinase (AXL) induces autophagy and mitigates EAE by suppressing microglial inflammation via the PI3K/AKT/MTOR signaling pathway [51]. AXL activation was associated with reduced microglial activation and pro-inflammatory cytokine production, indicating that enhancing autophagy through AXL may have therapeutic potential in neuroinflammatory conditions such as MS. Moreover, complementary evidence from a recent study showed that a ketogenic diet attenuates microglia-mediated neuroinflammation in EAE by inhibiting NLR family pyrin domain containing 3 (NLRP3) inflammasome activation through histone deacetylase 3 inhibition, which in turn activates autophagy through the AMPK/MTOR pathway [52]. Conversely, in the cuprizone model of demyelination, following negative modulation of MTOR pathway, which should reflect autophagy activation, the disease status of treated mice worsened [53–55]. Interestingly, this effect was observed following both pharmacologic and genetic MTOR inactivation. Consistently, autophagy activation following cuprizone addition was confirmed in other investigations [14]. It is, however, important to note that these recent reports did not assess the possible involvement of MTOR signaling. Such differences may be attributable to several reasons. On one hand, although both models are widely used to study MS, they capture distinct aspects of the disease. The cuprizone model primarily simulates demyelination and remyelination processes through oligodendrocyte toxicity, without directly involving the immune response. In contrast, the EAE model closely mirrors the autoimmune and inflammatory aspects of multiple sclerosis, driven by T-cell-mediated pathology. On the other hand, the differences observed – particularly regarding the role of MTOR – can be explained by the fact that MTOR regulates more than just autophagy. MTOR serves as a key regulator of cellular development, survival, and growth. Consistent with this, evidence suggests that MTOR signaling modulates demyelination events by controlling the metabolic state of oligodendrocytes, the activation and recruitment of infiltrating cells, and electrical signal transmission [53,54]. However, only a few studies have attempted to correlate these modulations with changes in autophagy [56].
Selective autophagy: mitophagy
Disarrangements in mitochondria functioning and dynamics are common aspects of MS. Therefore, it is not surprising that mitophagy could be involved in this disease. As reported above, AMPK is the primary energy sensor of cells and a modulator of diverse selective autophagy responses, including mitophagy. In the context of MS, numerous studies have shown that AMPK signaling is associated with myelin degradation and impaired mitochondrial function. Furthermore, it has been established that under MS-like conditions, mitochondrial function is disrupted, leading to an increase in ROS production and autophagic processes, mediated by AMPK signaling [57]. However, the effective involvement of mitophagy process during the pathogenesis of MS has remained unclear. Recent studies addressed this gap, revealing that both in vitro and in vivo MS models are characterized by severely impaired mitochondrial activity. This dysfunction shifts cellular metabolism toward lactic acid production and drives an increased mitophagic flux through the PINK1-Parkin axis [14,57]. A subsequent report confirmed the association between the PINK1-Parkin axis and demyelination in MS, providing an additional piece of evidence. Indeed, it was demonstrated that the chaperone FK506 binding protein 5, in association with PPAR-γ regulates both mitochondrial activity and structure and promotes mitophagy by acting on PINK1/Parkin axis in demyelinating pathological settings [58]. Furthermore, reactive nitrogen species such as nitric oxide and peroxynitrite can post-translationally modify key mitophagy-related proteins, exacerbating mitochondrial dysfunction [59]. In particular, a recent study demonstrated that nitrosative stress induces the S-nitrosylation of dynamin-related protein 1 and Parkin, two essential regulators of mitochondrial fission and mitophagy, respectively [60]. Supporting the role of mitophagy in MS pathogenesis, a recent study demonstrated that C-type lectin domain containing 16A (CLEC16A), a gene previously associated with autoimmune diseases, exerts a protective function in astrocytes by promoting mitophagy. Conditional deletion of CLEC16A in astrocytes leads to impaired mitophagic clearance, accumulation of damaged mitochondria, increased oxidative stress, and enhanced inflammatory signaling, ultimately exacerbating pathology in the EAE model. Notably, human brain tissue from MS patient samples also showed defective mitophagy and CLEC16A downregulation, reinforcing its potential clinical relevance [61]. In line with these findings, polymorphisms in the CLEC16A gene have been identified in MS patients and are believed to contribute to disease susceptibility [62]. Furthermore, a comprehensive genetic and histopathological analysis of 179 genotyped MS brain donors revealed a significant association between CLEC16A variants and the presence of chronic active lesions in CNS tissue, suggesting a functional role for this gene in lesion progression and mitochondrial homeostasis in MS [63]. Moreover, another study reported that astrocytes in the EAE model undergo profound metabolic reprogramming, with increased glycolysis and altered mitochondrial dynamics. Although mitophagy was not directly examined, the upregulation of mitochondrial and lysosomal genes suggests a possible, though speculative, activation of astrocytic mitophagy in response to inflammation and mitochondrial stress [64]. Interestingly, increased lactate and mitophagy-related elements were also found in both CSF and sera of MS patients (mainly identified in patients with the relapsing-remitting form (RRMS), further supporting of the critical role of autophagy processes in MS. This finding is consistent with other studies that have reported elevated levels of mitophagy-related components, such as PINK1 and Parkin, as well as biomarkers of axonal damage, like neurofilament light chain, and mitochondrial dysfunction, which were differentially expressed in MS patients [65–67]. These alterations were associated with patients’ inflammatory status and disease activity, suggesting that excessive mitophagy may contribute to MS pathogenesis. However, a recent study specifically investigating PPMS revealed a distinct pattern characterized by reduced levels and activity of autophagy and mitophagy machinery exclusively in PPMS patients [68]. This investigation highlights two possible interpretations of this reduction: it may either reflect a true loss of function of these catabolic pathways or, conversely, an uncontrolled overactivation leading to excessive degradation of autophagic vesicles and their markers. This distinction is critical for therapeutic strategies, as it implies that PPMS treatment might require either enhancement or inhibition of autophagy/mitophagy depending on the underlying mechanism. Despite these observations, some evidence indicates that mitophagy could exert beneficial effects in MS. For example, PINK1-deficient mice exhibit an earlier onset and more severe acute symptoms compared to controls, with the disease persisting throughout the recovery phase [69]. Interestingly, this phenotype was observed only in adult mice. Younger PINK1−/− mice displayed a milder disease course, suggesting a PINK1-dependent age-related mechanism in EAE. Parkin−/− mice also showed earlier onset, a sustained inflammatory profile and greater severity of EAE than wild-type, although this study did not explore potential age-related effects [70]. Finally, by quantitatively and qualitatively analyzing the components of PINK1-Parkin axis in a cohort of MS patients, this double face of mitophagy in MS was further supported. On one hand, patients have increased levels of PINK1 and Parkin along with mitochondrial dysfunctions in peripheral blood mononuclear cells. On the other hand, in the same cohort, a potentially protective variant of NDP52 was identified. Indeed, NDP52 G140E substitution showed increased binding to LC3C compared to wild-type NDP52WT. In vitro investigations demonstrated that this variant favors efficient mitophagy and reduces the production of inflammatory mediators [71] (Figure 3).
Figure 3.
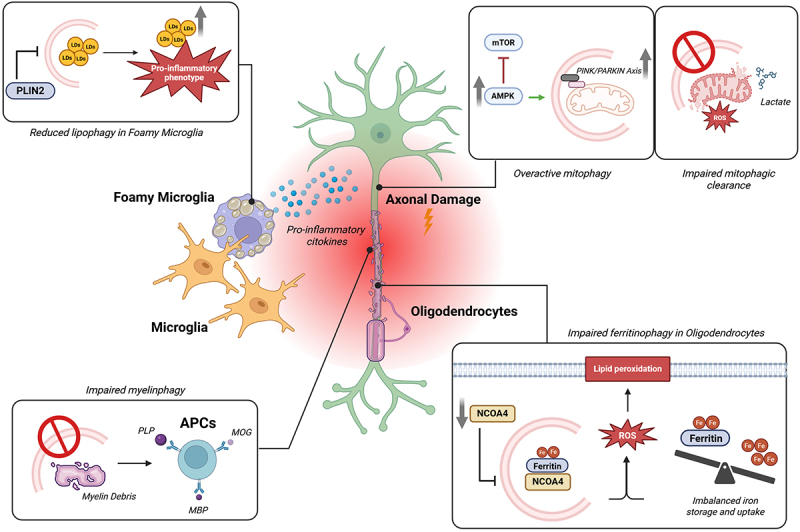
Dysregulated autophagy in multiple sclerosis: pathological mechanisms. Several autophagy-dependent pathways are involved in MS pathogenesis. Direct axonal damage is caused by the accumulation of dysfunctional mitochondria, leading to excessive production of lactate and ROS. Conversely, increased AMPK activity and enhanced activation of the PINK1/Parkin axis promote overactive mitophagy, which further contributes to axonal injury. Additionally, reduced levels of NCOA4 impair ferritinophagy, resulting in disrupted iron storage and uptake, which further increases ROS production. In oligodendrocytes, elevated ROS levels trigger lipid peroxidation and deterioration of the myelin sheath. In parallel, myelin debris, including myelin-related proteins, are not efficiently cleared due to defective myelinophagy. This persistence of debris activates antigen-presenting cells (APCs), thereby initiating an adaptive autoimmune response. Finally, the neurodegenerative environment is exacerbated by the presence of foamy microglia, cells with impaired lipophagy which accumulate lipid droplets and shift toward a proinflammatory phenotype. Abbreviations: reactive oxygen species (ROS), AMP-activated protein kinase (AMPK), nuclear receptor coactivator 4 (NCOA4). Created with Biorender.
Selective autophagy: ferritinophagy
Also ferritinophagy plays a crucial role in regulating the inflammatory response, the oxidative stress and demyelination in MS. Consistently, it has been demonstrated that when oligodendrocytes are exposed to proinflammatory cytokines, the loss of myelin basic protein (MBP) is accompanied by reduced activation of NCOA4 and TFR1, potentially reflecting the involvement of the ferritinophagy pathway. Supporting this, altered expression of ferritin-related markers, increase of oxidative stress (with consequent lipid peroxidation) and compensatory antioxidant mechanisms were observed in the corpus callosum of mice fed a cuprizone diet and in active state of demyelination [72]. Conversely, mice treated with molecule capable to prevent lipid peroxidation and ferroptosis decreased the progressive demyelination induced by cuprizone [72]. Recent findings have further strengthened the link between ferroptosis and MS progression. For instance, it has been shown that inhibition of ferroptosis with UAMC-3203 in a chronic EAE model significantly reduced demyelination and ferroptosis-associated damage, suggesting a causal role of ferroptosis in oligodendrocyte loss in progressive MS [73]. In addition, multi-omics analyses have identified ferroptosis-associated gene signatures involved in neurodegenerative and inflammatory pathways in MS brains [74]. Differentiated expressions of ferritinophagy-related markers have also been reported in humans. MS patients displayed increased levels of lipid peroxidation and iron metabolism indicators with respect to healthy control [75]. Interestingly, this trend was particularly present in patients in the active state [75]. Moreover, when comparing RRMS and PPMS, significant differences were mainly observed in levels of the marker of lipid peroxidation 4-hydroxynonenal [76]. Interestingly, a study further confirmed this finding, reporting that ferroptosis-related lipid peroxidation biomarkers distinguish between MS phenotypes and may reflect disease progression [76]. Finally, dysregulated iron homeostasis and altered iron metabolism have consistently been observed in the brains of MS patients [77–79]. Additionally, MRI-based studies have demonstrated that iron accumulation in deep gray matter regions (such as the basal ganglia and thalamus) may serve as a key driver of ferroptosis and subsequent neurodegeneration in MS [80–82]. However, recent evidence using macromolecular proton fraction and T2* mapping revealed that subcortical gray matter demyelination progresses independently of iron deposition, suggesting that myelin loss is more strongly associated with clinical disability than iron overload in these regions [83] (Figure 3).
Selective autophagy: lipophagy
Foamy microglia (FM) are microglial cells, the immune cells of the central nervous system, characterized by a foamy or spongy appearance. This distinctive morphology results from the accumulation of LDs within their cytoplasm. It has been demonstrated that MS lesions (active and chronic) are characterized by the presence of FM containing myelin debris [84]. Early studies suggested that FM predominantly drives MS disease progression as they could improve the presenting brain-derived autoantigens and the inflammation. Consistent with this, myelin engulfment by adult human-derived microglia provokes a great increase in oxidative stress, release of pro-inflammatory cytokines (such as IL-1β, TNFα, and IL-6) and switch from M2 to M1 polarization of macrophages [85,86]. However, more recent evidence suggests that myelin internalization can also induce a protective phenotype in these cells. Indeed, FM which reside in MS lesions as well as in vitro cultures and in vivo experimental model reduce the autoreactive T cell proliferation and the inflammatory phenotype [87–89]. Overall, the effective role of FM during MS and the specific molecular mechanisms involved remain unclear. As reported in the previous section, autophagy within microglia controls lipid homeostasis, particularly through the process of lipophagy, which mediates the selective degradation of LD. Recent studies have demonstrated that active MS lesions are characterized by impaired levels of lipophagy, leading to the accumulation of LDs and promoting a disease-associated inflammatory phenotype. Restoration of lipophagy through trehalose treatment, known to reduce lipid accumulation in atherosclerotic macrophages [90], results in decreased inflammation and a progressive reduction in the lipid load of foamy macrophages. Notably, trehalose was found to improve clinical score and promote remyelination in experimental mouse models of MS [13]. Conversely, a subsequent study observed increased lipophagy levels in the demyelinated areas, alongside LDs accumulation. The authors suggested that the lipophagy activation serves as a protective mechanism aimed at removing excess LDs. Consistently, they demonstrated that a specific miRNA (miR-233) enhances lipophagy, resulting in reduced LDs accumulation and inflammatory environments [91]. Furthermore, recent research has identified the LDs-associated protein perilipin-2 (PLIN2) as a regulator of LDs metabolism in myelin-containing phagocytes. PLIN2 protects LDs from lipolysis-mediated degradation, impairing the processing of myelin-derived lipids. Loss of PLIN2 stimulates LDs turnover in foamy phagocytes, driving them toward a less inflammatory phenotype and markedly improving remyelination in both ex vivo and in vivo models [92]. Together, these findings highlight the pivotal function of lipophagy in regulating lipid metabolism and the inflammatory response of microglia within the context of MS. Modulating lipophagy-related pathways may represent a promising therapeutic approach to attenuate neuroinflammation and enhance remyelination in this disease (Figure 3).
Selective autophagy: myelinophagy and CMA
Finally, to date, no specific studies have directly demonstrated the involvement of myelinophagy or CMA in MS or their contribution to disability progression. However, several findings suggest their involvement in MS. In the context of myelinophagy, during the onset and progression of MS, the immune system attacks myelin, resulting in demyelination and the generation of myelin fragments that must be cleared to prevent further damage and facilitate neuronal recovery. Accordingly, MBP, proteolipid protein (PLP), and other myelin-related proteins, such as myelin oligodendrocyte glycoprotein (MOG), act as novel neuronal antigens that stimulate specific antigen-presenting cells (mainly astrocytes and macrophages [93,94]), as well as nonprofessional APCs (such as endothelial cells [95,96]), triggering an adaptive autoimmune response in MS. Myelinophagy could play a crucial role in removing these myelin fragments. If this process is disrupted, the accumulation of myelin debris could contribute to chronic inflammation and accelerate MS progression [97] (Figure 3). Furthermore, axonal injury in MS is characterized by a process called Wallerian degeneration (WD), which involves the breakdown of the distal axonal segment and the subsequent distortion and fragmentation of the myelin sheath. Reports suggest that WD occurs in the early phases of MS and contributes to the development of disability. The fact that, during WD, autophagy and myelinophagy are differentially expressed [6] could further suggest that this specific type of autophagy is involved in MS. Similarly, the precise role of CMA in MS remains an ongoing area of research; however, several indications suggest an interconnection. As CMA regulates the activity of inflammatory mediators, its dysregulation may contribute to the chronic inflammation seen in MS by allowing the accumulation of pro-inflammatory proteins. Recent research has identified that deficient CMA promotes microglial activation through the p300/NF-κB/NLRP3 pathway, leading to increased CNS inflammation [98]; although these findings have been observed in models other than MS, and no direct studies in MS exist yet, it can be speculated that a similar mechanism could contribute to MS pathogenesis, especially considering that NLRP3 inflammasome activation has been demonstrated to play a significant role in MS [99]. In line with this, CMA is reported to control immune tolerance and activation. Dysfunctions in these processes can enhance the autoimmune response in MS. Additionally, CMA plays a role in maintaining the homeostasis of myelin-forming cells. Therefore, efficient CMA could help prevent demyelination and reduce the impact of the demyelinating attacks in MS. Further research into the molecular mechanisms of CMA and myelinophagy will be essential to bridge these gaps and explore their potential as therapeutic targets.
Therapeutic strategies targeting autophagy
Targeting autophagy as a therapeutic strategy has been extensively explored in oncology. In recent years, however, this approach has garnered growing interest in the context of neurodegenerative diseases, including MS, where autophagy dysregulation is increasingly recognized as a contributor to disease pathogenesis.
Currently, numerous studies are focused on identifying novel compounds capable of modulating autophagy with the aim of restoring cellular homeostasis and slowing disease progression. These efforts offer promising avenues for the development of innovative therapeutic strategies. In MS, as in other neurodegenerative disorders, the activation of autophagy has shown beneficial effects, including the clearance of toxic protein aggregates, attenuation of oxidative stress and improved neuronal survival under adverse conditions [11]. Conversely, in specific pathological settings, autophagy inhibition may also offer therapeutic advantages. For instance, regulating autophagy can influence the survival of stress-tolerant or damaged cells, potentially promoting their removal and preventing further tissue injury. This dual and context-dependent role of autophagy underscores the need for a nuanced understanding of its mechanisms to effectively harness its therapeutic potential.
In the following section, we provide a comprehensive overview of the key autophagy-modulating compounds. We will summarize studies assessing their efficacy in vitro and in vivo, and briefly discuss the few available clinical trials, which offer insights into their pharmacological properties, safety, and potential for clinical translation (Table 1).
Table 1.
Summary Table of Key Molecules Capable of Modulating Autophagy, Their Mechanism of action, Potential Adverse Effects, In Vivo and In Vitro Evidence of Efficacy on MS, and Corresponding Clinical Trials.
| Autophagy modulators | Mechanism of action | Potential Adverse Effects | In vitro/In vivo evidence | Clinical Trials |
|---|---|---|---|---|
| Rapamycin and Rapalogs (Autophagy inducer) |
MTOR inhibition | Mucocutaneous toxicities (mouth ulcerations, stomatitis, skin rashes). Systemic effects (menstrual dysfunction, delayed wound healing), Immunosuppression increasing infection risk. Pharmacokinetic challenges (long half-life, variable bioavailability) | ↓ Myelin-autoreactive T cells (Th1 and Th17), Neuroinflammation, Neuronal cell death ↑ Tregs |
Non-randomized prospective study (n = 6 RRMS patients) Non-randomized prospective study (n = 8 RRMS patients) Double blind phase II trial (n = 296 patients with active and relapsing MS) |
| Metformin (Autophagy inducer) |
AMPK activation | Vitamin B12 deficiency risk with long-term use. Gastrointestinal side effects (diarrhea, nausea, abdominal discomfort) affecting adherence (20–30%) | ↓ Th17 cells, Neuroinflammation ↑ Tregs, Remyelination, Protection from oxidative stress |
Randomized prospective phase II trial (n = 80 patients with RRMS) Non-randomized prospective trial (n = 50 MS patients with metabolic syndrome) Interventional study c/o Antwerp Hospital (Currently underway) |
| Resveratrol (Autophagy inducer) |
SIRT1 activation | Generally well tolerated; no major adverse effects clearly reported. Challenges with systemic exposure due to pharmacokinetics | ↑ Neurobehavioral outcomes,Myelin repair ↓ Neuronal damage, Neuroinflammation |
Double-blind randomized trial (n = 55 RRMS patients) |
| Antipsychotic Drugs (Autophagy and mitophagy inhibitors) |
Interfering with AMPK/MTOR/ULK1 pathway | Mechanisms not fully understood. Conflicting data on effects on myelin-specific T cells. Lack of clinical data and trials in MS patients | ↓ Mitochondrial stress, Pro-inflammatory cytokines ↑ Axonal myelination |
Not reported |
| Spermidine (Autophagy inducer) |
Epigenetic regulator of key autophagy-related genes | Generally well tolerated at experimental doses Limited clinical studies in humans, so the safety profile is not fully established. Theoretical risk of promoting cell proliferation in some contexts, but not confirmed in neurological settings | ↓ Demyelination ↑ Axonal integrity |
Not reported |
| Trehalose (Lipophagy inducer) |
TFEB translocation and activation | - Considered safe as a food additive and in preclinical studies, Low toxicity reported in experimental models, Potential issues related to bioavailability and metabolism, but no serious systemic side effects documented, Lack of specific clinical data in MS to fully assess safety profile | ↓ Intracellular lipid load in myelin-laden macrophages | Not reported |
Rapamycin
Rapamycin, also known as Sirolimus, is a macrocyclic lactone originally isolated from Streptomyces hygroscopius found in soil samples from Easter Island. Initially utilized as an antifungal agent and for the prevention of organ transplant rejection, rapamycin has gained recognition as an autophagy inducer [100]. Its primary mechanism of action involves the inhibition of MTORC1 by disrupting its association with Raptor. Notably, MTORC2 is generally resistant to short-term rapamycin treatment, though prolonged exposure may also inhibit this complex [56].
Rapamycin is considered a promising candidate for the treatment of MS due to its lipophilic nature, which facilitates its ability to cross the BBB. Studies have consistently demonstrated that rapamycin can inhibit the induction and progression of EAE. Although the results are more variable, similar effects have been observed also in the cuprizone model, further supporting the efficacy of rapamycin in both primary animal models of MS.
Rapamycin plays a crucial role in modulating the infiltration of peripheral immune cells into CNS and the activation of innate immune systems. In EAE, rapamycin has been shown to ameliorate MS pathological conditions by reducing the recruitment of myelin-autoreactive Th1 and Th17 cells while increasing the presence of regulatory T cells (Tregs), which are known for their role in restraining autoimmunity [101,102]. Notably, the reduction of Th1 and Th17 cells is caused by a rapamycin-induced blockade of the MTOR-STAT3 pathway [103]. This results in decreased CNS demyelination and axonal loss.
Other studies have shown that rapamycin treatment promotes autophagy, thereby reducing neuroinflammation and neuronal cell death. Regarding inflammation, recent investigations have focused on the intricated relationship between autophagy and neuroinflammation in MS pathogenesis. Specifically, impaired autophagy due to MTOR activation can enhance NLRP3 inflammasome activity, a process reversed by rapamycin treatment. Supporting this, rapamycin improves the efficacy of MCC950, a well-characterized and highly selective NLRP3 inhibitor [104,105]. Controlling neuroinflammatory processes is critical for delaying MS progression and promoting myelin repair.
Despite the extensive preclinical data from EAE models, studies investigating the effects of rapamycin in patients with MS remain limited. These include two small clinical trials and a phase II study conducted in 2005 [106–108]. Both clinical trials were non-randomized, prospective studies that evaluated the effects of 2 mg/day of rapamycin over six months in cohorts of six and eight RRMS patients, respectively. In the first study, reduced IFNγ levels and increased TGFβ levels were observed compared to controls. The second study reported enhanced Treg proliferation and a reduction of MRI-detected lesional area. However, the small sample sizes in these trials precluded definitive conclusions. In contrast, the 2005 phase II clinical trial included 296 patients and assessed three different doses of temsirolimus, a structural analog of rapamycin, in individuals with active and relapsing MS. The highest dose group (8 mg) demonstrated approximately a 50% reduction in active lesions and relapse rates. Nevertheless, the risk-benefit profile prevented progression to phase III trials.
Regarding the risk/benefit profile, clinical trials have reported a range of adverse effects associated with rapamycin and its analogs. These include mucocutaneous toxicities such as mouth ulcerations, stomatitis, and skin rashes, as well as systemic effects like menstrual dysfunctions and delayed wound healing [109]. In addition, rapamycin’s inhibition of MTORC1 contributes to a dose-dependent immunosuppressive profile [56]. This effect significantly increases the risk of opportunistic infections, especially in immunocompromised or elderly individuals [56]. Furthermore, rapamycin exhibits challenging pharmacokinetic properties, including a long half-life and variable oral bioavailability, which complicate dose optimization and therapeutic monitoring.
Metformin
Metformin, a synthetic derivative of guanidine, is widely used as an oral antidiabetic agent [7]. Beyond its conventional role in glucose regulation, metformin has demonstrated considerable potential as a modulator of autophagy, particularly in the context of MS. Its primary mechanism of action involves the activation of AMP-activated protein kinase (AMPK). Specifically, metformin inhibits complex I of the mitochondrial electron chain, thereby reducing oxidative phosphorylation and ATP production. Given the tight link between cellular energy status and autophagy, this inhibition leads to AMPK activation, which in turn suppresses MTOR signaling and promotes autophagy [110].
Similarly to rapamycin, metformin has been shown to exert neuroprotective agent in murine models of MS by ameliorating diseases symptoms, reducing neuroinflammation and promoting remyelination. In EAE, evidence indicates that modulation of AMPK/MTOR pathway plays a critical role in regulating immune responses, including the suppression of pro-inflammatory Th17 cells and the expansion of Tregs [111,112]. Furthermore, AMPK-mediated MTOR inhibition has been shown to protect neurons and glial cells from oxidative stress, a major contributor to neurodegeneration in MS. Notably, the AMPK/MTOR signaling axis has also been implicated in supporting endogenous oligodendrogenesis, thereby facilitating remyelination processes [113]. Additionally, metformin-induced AMPK activation reveals similar effects in the cuprizone model, facilitating the recruitment of oligodendrocyte precursor cells and accelerating myelin recovery [114]. As with rapamycin, MTOR inhibition by metformin reduces NLRP3 inflammasome activitation, mitigating neuroinflammatory responses [56].
Regarding clinical applications, the role of metformin in MS remains poorly explored. Recently, a prospective phase II randomized controlled trial investigated the impact of metformin on RRMS patients [115]. This study evaluated its efficacy as an adjunctive therapy to IFNβ-1a, one of the most commonly used disease modifying therapy for MS. The trial, conducted in a cohort of 80 patients, demonstrated a potential effect of metformin in reducing oxidative stress, as evidenced by levels of malondialdehyde. However, no significant effects on immunological or clinical outcomes were observed. Another prospective study explored the therapeutic potential of metformin in MS, focusing on a smaller cohort of 20 obese RRMS patients with metabolic syndrome [116]. Even so, the presence of this comorbidity could confound the results, making it challenging to isolate the specific effects of metformin on MS. In addition, an interventional study is currently underway at Antwerp University Hospital (NCT05893225), aiming to assess whether metformin can prevent clinical disability progression in MS patients by enhancing endogenous remyelination. In this study, metformin is administrated as an add-on therapy alongside the patients’ current disease modifying therapy.
Finally, regarding its pharmacological profile, metformin is a widely used and generally well-tolerated oral antidiabetic agent, primarily prescribed for the management of type 2 diabetes mellitus. However, long-term metformin use has been associated with vitamin B12 deficiency, a well-documented side effect that is of particular concern in MS patients due to the critical role of B12 in myelin synthesis and neuronal function. Deficiency in B12 has been linked to exacerbation of neurological symptoms, including fatigue, cognitive impairment, and peripheral neuropathy, potentially complicating disease progression and management in this population [117]. Moreover, while generally safe, metformin is not without risks: common gastrointestinal side effects, such as diarrhea, nausea, and abdominal discomfort, occur in 20–30% of patients and can impact adherence [118].
Resveratrol
Resveratrol, a polyphenolic compound recognized for its antioxidant and anti-inflammatory properties since the French Paradox in 1992, has recently garnered considerable attention also for its neuroprotective effects [119]. Although the precise mechanisms underlying these effects remain incompletely understood, resveratrol is known to modulate multiple biological processes, including autophagy.
To date, only a limited number of studies directly linked the effects of resveratrol on autophagy to the pathophysiology of MS. Similar to rapamycin and metformin, resveratrol acts upstream as an autophagy inducer. Its primary molecular target is Sirtuin 1 (SIRT1), a NAD±dependent deacetylase that regulates numerous cellular processes, including autophagy. SIRT1 promotes autophagy by deacetylating key transcription factors, such as the forkhead box class O (FoxO) family, which in turn enhance the expression of various autophagy-related genes [120].
Samy et al. demonstrated that resveratrol modulates autophagy in the cuprizone mouse model of demyelination. Specifically, resveratrol has been shown to reverse the downregulation of SIRT1 and FoxO observed in cuprizone-treated mice, leading to improved neurobehavioral outcomes and enhanced histological recovery [121]. Additional studies have reported that resveratrol alleviates neuronal damage in the EAE model [122,123]. However, its efficacy in EAE remains controversial and appears less consistent than in the cuprizone model.
While the therapeutic potential of resveratrol in MS management is not yet fully established, accumulating evidence supports its neuroprotective role in other neurodegenerative diseases [124]. SIRT1 activation by resveratrol has demonstrated beneficial effects on neuronal function and survival, including the alleviation of protein aggregation, oxidative stress, neuroinflammation and mitochondrial disfunction [125,126]. Thus, resveratrol, through SIRT1 activation, may represent a promising strategy for the treatment of neurodegenerative conditions such as MS. Nonetheless, the complex interplay between resveratrol, autophagy and MS warrants further investigation.
Concerning clinical studies, the therapeutic application of resveratrol in MS therapy is still marginally explored. Recently, a double-blind randomized placebo-controlled trial investigated the effects of resveratrol supplementation on inflammatory markers in RRMS patients [127]. Notably, this study, which included a cohort of 55 participants, reported a significant reduction in malondialdehyde levels, mirroring results obtained with metformin.
One of the principal challenges of resveratrol is its poor pharmacokinetic profile, characterized by low oral bioavailability, rapid metabolism, and short plasma half-life, all of which severely limit its systemic exposure [128]. As noted by Shaito et al., ongoing clinical trials aim to better define its pharmacological properties and explore its repositioning from a dietary supplement to therapeutic agent. These studies are investigating optimized formulations, delivery systems (e.g. liposomes and nanoparticles), and combination therapies to overcome bioavailability constraints and enhance tissue penetration. At now, the majority of completed human trials have yielded modest or inconclusive results, especially in terms of clinically meaningful outcomes [129]. Thus, until the results of current trials are published and validated, the clinical utility of resveratrol remains largely speculative, particularly in complex, multifactorial diseases like MS.
Antipsychotic drugs
Second-generation antipsychotics, also known as atypical antipsychotics (AAPs), have shown notable efficacy in alleviating neuroinflammation associated with psychiatric disorders [130]. Recently, these immunomodulatory effects have been explored in the context of MS, where neuroinflammatory processes are central to driving disease progression.
Studies have demonstrated the therapeutic effects of APPs in both EAE and cuprizone models. These studies confirm their ability to delay disease onset, reduce severity, and suppress the production of pro-inflammatory cytokines [131–133]. However, the precise mechanism underlying these effects remains incompletely understood, as not all studies agree on their impact on the expansion and differentiation of myelin-specific T cells [134].
Regarding the mechanism of action, one particularly insightful study provides an explanation of how antipsychotic drugs may counteract autophagy and mitophagy in MS [14]. Elevated levels of autophagic markers, such as LC3-II and BECN1, were observed, as previously reported in another study by the same research group [135]. Moreover, the study reported that proinflammatory cytokines drive autophagic activation, myelin loss in vitro and mitochondrial stress associated with mitophagy. These pathological conditions were reversed by the ability of clozapine and haloperidol to modulate autophagy. According to the authors, antipsychotic drugs act as inhibitors of autophagy, potentially interfering with the AMPK-MTORC-ULK1 pathways. Clozapine and Haloperidol have been shown to remodulate autophagic processes, restoring myelin production and axonal myelination. Notably, these compounds were able to reverse cuprizone-induced MBP loss in vivo and motor performance.
These findings represent a promising avenue for MS therapy. However, further research is essential to address these gaps and to overcome challenges related to their clinical application, for which no significant studies are yet available.
Other autophagy modulators: spermidine, trehalose, lithium, valproic acid, and vitamin D
After discussing the most well-known compounds, it is possible to mention other autophagy – modulating molecules that are less investigated in the context of multiple sclerosis, such as spermidine and trehalose. Additionally, there are molecules that have been shown to separately influence both autophagic processes and MS progression, although studies directly examining their role in autophagy as a potential therapeutic target in multiple sclerosis are lacking.
Spermidine, a natural polyamine, is a well-documented inducer of autophagy. It functions as an epigenetic regulator by modulating the expression of key autophagy-related genes, such as BECN1, LC3-II and p62 [136]. These molecular effects enhance cellular resilience against oxidative stress and inflammation. Moreover, spermidine has demonstrated neuroprotective effects in EAE, where it significantly alleviates disease severity, improves axonal integrity, and reduces demyelination [137,138]. These benefits are achieved through immunomodulatory effects on CD4+ T cells and macrophages. Taken together, these collective findings suggest its potential as a therapeutic candidate in MS.
Similarly, trehalose, a natural disaccharide, has gained attention for its ability to induce autophagy and mitigate neurodegeneration. Notably, trehalose induces lipophagy, which when is dysregulated, drives the inflammatory phenotype into macrophages. It has been shown to reduce intracellular lipid load in myelin-laden macrophages, thereby shifting their phenotype toward a reparative one that promotes CNS remyelination [13]. This leads to improved remyelination in the cuprizone model. Beyond this role, trehalose has demonstrated neuroprotective effects in other neurodegenerative diseases by facilitating autophagic clearance of protein aggregates and protecting against lysosomal dysfunction [139]. Here as well, these findings highlight trehalose as valuable molecules for consideration in MS therapy.
In addition to spermidine and trehalose, other molecules such as lithium, valproic acid and vitamin D deserve mention in this section. While studies exploring a direct correlation between autophagy and MS are limited for spermidine and trehalose, for these additional molecules it is challenging to find studies directly linking the two topics. However, lithium, valproic acid and Vitamin D are recognized for their role in regulating autophagy and have also demonstrated efficacy in MS through other cellular mechanisms [140–144]. Therefore, investigating their potential role in MS from an autophagic prospective could yield important insights into their mechanisms of action and reveal novel therapeutic opportunities for the future.
Challenges and future prospectives
Limitations of animal models
Most in vivo studies investigating molecular mechanisms and therapeutic efficacy in MS have relied on two main experimental mouse models: EAE and the cuprizone model. Despite their extensive use, both models present significant limitations that hinder direct translation to the human disease.
The cuprizone model induces demyelination by selectively targeting mature oligodendrocytes, without engaging the peripheral immune system – an essential component of MS pathology. Moreover, outcomes in this model are highly influenced by factors such as species, strain, age, and sex, leading to variability and reduced reproducibility [145,146].
By contrast, the EAE model recapitulates immune-mediated demyelination but differs from human MS in key aspects. Notably, EAE lesions are predominantly infiltrated by CD4+ T cells, whereas MS lesions are characterized by a greater presence of CD8+ T cells and macrophages [147]. Furthermore, EAE pathology primarily affects the spinal cord, while MS typically involves demyelinating lesions in the cerebrum and cerebellum [148].
These discrepancies raise concerns about the translational relevance of findings derived from animal models, especially considering that several therapeutic strategies effective in EAE or cuprizone models have failed in clinical trials. Therefore, it is essential to interpret preclinical data with caution and to validate critical findings using human tissues and clinical studies [149]. To address these challenges, complementary approaches such as patient-derived iPSC-based models, 3D brain organoids, and advanced in vitro co-culture systems are being increasingly explored.
Nanoparticles delivery systems
The use of nanoparticles is emerging as a promising approach to overcome the limitations of both novel and conventional therapies. Nanoparticles offer a drug delivery system capable of improving bioavailability, pharmacological stability, and tissue selectivity, while minimizing side effects.
One innovative study evaluated intranasal delivery of rapamycin encapsulated in PEGylated nanoparticles. Intriguingly, this method facilitated direct nose-to-brain transport, bypassing the BBB and enhancing rapamycin accumulation in the brain [150]. Furthermore, in aging-related studies, rapamycin-loaded solid lipid nanoparticles have demonstrated efficacy in mitigating oxidative stress and neurodegeneration [151].
Similarly, resveratrol-loaded nanoparticles have shown enhanced neuroprotective effects, reducing oxidative stress and inflammation [152]. In addition, resveratrol nanoparticles have been reported to reduce neuronal loss in EAE, highlighting their potential in demyelinating diseases, such as MS [153].
Notably, many similar nanoparticle-based approaches have already been explored in other fields, such as cancer, to improve the delivery of autophagy-modulating compounds. Thus, the broader application of nanomedicine suggests that nanoparticle-based strategies may represent an important tool for advancing the clinical use of these compounds.
Gene editing and omics technologies
In a medical landscape highly focused on personalized medicine, the potential use of gene editing and omics technologies to modulate autophagy in the context MS remains largely theoretical. The first studies have emerged exploring the possibility of using gene therapy to reprogram T lymphocytes in MS, suggesting new avenues for intervention. However, no studies have addressed the modulation of autophagy through these approaches in the context of MS [154].
Although the translational applicability of these strategies as therapeutic intervention is still very limited, it is important to highlight the value of these technologies. The integration of multi-omics approaches including transcriptomics, proteomics, and metabolomics could provide valuable insights into the complex network linking autophagy and MS, facilitate the identification of autophagy-related signatures specific to MS, and ultimately accelerate the discovery or repurposing of existing drugs with therapeutic potential [155,156]. Once autophagy-related signatures in MS are identified, gene editing can be used to restore proper autophagic flux, reduce neuroinflammation, and promote remyelination. For example, manipulating MTOR pathway in immune cells may shift their phenotype toward an anti-inflammatory profile. However, the application of gene editing to modulate autophagy in MS is still in an exploratory phase and without concrete results.
Conclusion
Bulk autophagy and its selective forms play a crucial role in maintaining cellular homeostasis and ensuring proper function of both neuronal and immune system in MS. Although considerable progress has been made in elucidating the role of autophagy in MS, its involvement remains complex and incompletely understood.
Early studies linked autophagy markers with disease progression and inflammatory activity. Subsequent investigations, however, revealed that autophagy may be either upregulated or impaired depending on the region of CNS and the stage of the disease phase. These conflicting findings underscore the heterogeneous and context-dependent nature of autophagy in MS pathophysiology. Such discrepancies are likely due to differences in experimental models, methodologies, and the intricate interplay of cellular pathways regulating autophagy and related processes (Table 2).
Table 2.
Overview of Selected Studies on Multiple Sclerosis (MS) Analyzing Different Clinical Forms, Types of Human Samples, Biomarkers Investigated, and Main Findings.
| Study (reference) | MS Clinical Form | Human Sample | Markers Studied | Main Findings |
|---|---|---|---|---|
| 45 | RRMS, SPMS | T cells; postmortem brain | ATG5 | ↑ ATG5 expression in T cells and brain tissue in both MS forms |
| 47 | RRMS, Progressive MS | mRNA from blood | Autophagy, mitophagy and lysosomal markers | Differential expression of autophagy markers |
| 14 | RRMS | Serum and CSF | Autophagy, mitophagy ferritinophagy markers | ↑ Autophagy, mitophagy ferritinophagy |
| 12 | RRMS and Progressive MS | Postmortem brain | Autophagolysosomes | ↑ Autophagolysosomes in MS brains |
| 134 | RRMS | Serum and CSF | Autophagy and mitophagy markers | ↑ Autophagy correlated with inflammation (cytokines) |
| 46 | RRMS | Serum and CSF | Autophagy and mitophagy markers | ↑ Autophagy correlated with inflammation (cytokines and MRI) and with the active state of MS |
| 64 | PRSM and SPMS | CSF, serum | Autophagy, mitophagy markers | ↑ Autophagy and mitophagy markers (correlation with axonal damage markers) |
| 65 | RRMS | Serum | Antioxidant status and oxidative levels; autophagy and mitophagy markers | ↑ Autophagy and mitophagy markers; oxidative stress ↓ Antioxidant status |
| 66 | RRMS and Clinically isolated syndrome (CIS) | CSF and serum | Mitophagy markers | ↑ Mitophagy (no distinction between RRMS and CIS) |
| 67 | RRMS, PPMS | Brain tissue | Autophagy and mitophagy markers | ↓ Autophagy and mitophagy in PPMS (correlation with axonal damage markers); ↑ Autophagy, mitophagy in RRMS |
| 62 | Not specified | Brains | autophagy markers | ↓ Autophagy |
| 70 | RRMS | PBMCs | Mitophagy | Differential expression of mitophagy markers |
| 74 | RRMS and progressive MS* in active and non-active state | Serum | Lipid peroxidation, iron, ferritinophagy-related markers | ↑ Lipid peroxidation and ferritinophagy in active state |
| 75 | RR-MS vs progressive MS* | Serum | lipid peroxidation ferritinophagy-related markers | ↑ Lipid peroxidation and ferritinophagy in RRMS compared to PPMS |
| 73 | RRMS | Brain, CSF. serum | Ferroptosis/Ferritinophagy gene signatures | ↑ Ferroptosis/Ferritinophagy pathways in MS brains |
| 78 | RRMS (adolescent patients) | Brain (Susceptibility-Weighted Imaging) | Iron | ↑ Iron content |
| 76 | RRMS | Brain (Susceptibility-Weighted Imaging) | Iron | ↑ Iron content |
| 79 | RRMS | Brain (Magnetic Field Correlation Imaging) | Iron | ↑ Iron content |
| 82 | RRMS, and SPMS | Brain (Macromolecular Proton Fraction Mapping) | Iron | No variation in iron content in demyelinated area |
| 80 | RRMS | Brain (T2-weighted gradient echo imaging) and serum | Iron | ↑ Iron in the putamen, globus pallidus and cortex ↓ iron in the thalamus and serum. |
| 83 | Active and chronic MS | Brain | Foamy microglia | ↑ FM present in lesions |
| 91 | Not specified | Brain | Lipophagy marker (PLIN2) | PLIN2 stabilizes LDs; its deletion improves remyelination |
| 13 | Active (unspecified the type) | Brain | Lipophagy | ↑ Lipophagy |
| 6–96 | MS (hypothesized) | Indirect evidence | MBP, PLP, MOG fragments; APC activation | Myelin debris triggers immune response; defective clearance (myelinophagy) may fuel chronic inflammation |
| 97–98 | MS (hypothesized) | Indirect evidence | CMA markers, NLRP3, p300/NF-κB | Deficient CMA may ↑ microglial inflammation; CMA linked to immune tolerance, myelin cell homeostasis |
The Table Highlights the Methodological and Clinical Diversity of Approaches Used to Identify Potential Diagnostic, Prognostic, or Pathogenic Markers of the Disease. Abbreviations: ↑ = Increased; ↓ = Decreased; FM = Foamy Microglia; LD = Lipid Droplet; CMA = Chaperone-Mediated Autophagy; MBP = Myelin Basic Protein; MOG = Myelin Oligodendrocyte Glycoprotein; PLP = Proteolipid Protein; APC = Antigen Presenting Cell; NA = Not Applicable (Preclinical); PRMS: Progressive-Relapsing Multiple Sclerosis; SPMS: Secondary Progressive Multiple sclerosis*no Distintion Between PRMS and SPMS Have Been Described.
Nevertheless, there is compelling evidence that autophagy contributes to both onset and progression of MS, supporting its potential as a promising therapeutic target. Numerous in vitro and in vivo studies have demonstrated the neuroprotective and immunomodulatory potential of autophagy regulation. However, the multifactorial and heterogeneous nature of MS, with its various clinical subtypes and progression patterns, presents major challenges for the development of effective autophagy-based therapies and their successful clinical translation.
One critical barrier is the limited pharmacological specificity and safety of available autophagy modulators. Several compounds tested in preclinical models, such as rapamycin, metformin, and resveratrol, exhibit off-target effects and unfavorable pharmacokinetic profiles, thereby limiting their clinical applicability.
In conclusion, despite these challenges, advances in understanding the molecular mechanisms governing autophagy in MS, coupled with the development of more targeted and safer therapeutic agents, offer promising prospects for improving clinical outcomes in affected individuals.
Acknowlegment
GR, SP, LS, KK and VZ wrote the main manuscript text. GR prepared the figures. SP, PP and CG edited and reviewed the manuscript.
Biographies
Dr. Giulio Righes graduated in Pharmacy and is currently pursuing his PhD in Medical, Omics and Oncological Sciences at the University of Ferrara. His research focuses on inflammatory processes in neurodegenerative diseases, particularly in multiple sclerosis.
Dr. Luana Semenzato is a neurologist at the San Camillo IRCCS Hospital, where she works in the Divisional Neurology Outpatient Clinic. She is part of the Neurorehabilitation Department and contributes to the multidisciplinary management of patients with cerebrovascular diseases in a neurorehabilitation setting. Dr. Semenzato is also involved in continuing medical education, serving as a lecturer in accredited training courses on neurological diseases and related comorbidities organized by San Camillo IRCCS.
Dr. Konstantinos Koutsikos specializes in Physical and Rehabilitation Medicine with extensive experience in Neurorehabilitation. He obtained his Medical Degree in 2003 from the University of Padua, where he later completed his residency in the Department of Neurosciences in 2014. Actively involved in research since 2007, he has participated in various rehabilitation studies as well as clinical trials on neurodegenerative diseases, particularly ALS and movement disorders within IRCCS San Camillo and the University of Padua.
Dr. Veronica Zanato graduated in Medicine from the University of Padua in 2009 and completed her residency in Internal Medicine with a focus on ultrasound diagnostics, with honors, in 2015. During her training, she specialized in endocrine-metabolic disorders and worked at the Bariatric Unit of the Padua University Hospital, managing patients with severe and complicated obesity and assessing their cardio- and cerebrovascular risk. Since September 2021, she has been part of the medical staff at San Camillo IRCCS, where she is responsible for managing internal medicine conditions in hospitalized patients.
Prof. Paolo Pinton has been Full Professor of General Pathology at the University of Ferrara since 2015, within the Department of Medical Sciences. He has extensive research experience in signal transduction, and he is recognized as one of the world’s leading experts in calcium homeostasis and the role of mitochondria in pathophysiological contexts. His works have been published in leading scientific journals, receiving over 50,000 citations.
Prof. Carlotta Giorgi is Full Professor of General Pathology at the University of Ferrara. Her research is funded by national and international agencies, including the Italian Association for Cancer Research (AIRC) and the European Research Council (ERC). Her works have been published in leading scientific journals, receiving over 19,000 citations.
Dr. Simone Patergnani is a senior researcher at the University of Ferrara’s Department of Medical Sciences. His research focuses on autophagy, particularly in its role in mitochondrial function, neurodegenerative diseases such as Multiple Sclerosis and cancer. He authored several works in high-impact scientific journals, and he was supported by fellowship from the Italian Multiple Sclerosis Foundation (FISM) and the Italian Association for Cancer Research (AIRC).
Funding Statement
The study was funded by the European Research Council (853057-InflaPML to CG), the Fondazione Italiana Sclerosi Multipla (FISM, cod.2022/R-Multi/050 and co-financed with the ‘5 per mille’ public funding to SP), the Associazione Ricerca Oncologica Sperimentale Estense (A-ROSE), the Italian Association for Cancer Research (IG-23670 to PP, IG-19803 to CG and MFAG 2023 - ID. 29087 to SP), Progetti di Rilevante Interesse Nazionale (2022-PRIN-PP_001, 2023-PRIN-PP_001, 2023-PRIN-PNRR-PP_001, 2024_PNRR_TINY_RIC to PP and 2023-PRIN-GC_001, 2023-PRIN-PNRR-GC_001, 2024-PNRR-RIC-GC_001 to CG), and local funds from the University of Ferrara to CG, PP and SP.
Abbreviations
- (AD)
Alzheimer’s disease
- (AMPK)
AMP-activated protein kinase
- (Aβ)
amyloid β
- ALS)
Amyotrophic Lateral Sclerosis (
- (APCs)
antigen-presenting cells
- (AAPs)
atypical antipsychotics
- (ATG)
autophagy-related
- (FIP200)
ATG 13/200-kDa focal adhesion kinase family-interacting protein
- (AXL)
AXL receptor tyrosine kinase
- (BECN1)
Beclin 1
- (BBB)
blood-brain barrier
- (CLEC16A)
C-type lectin domain containing 16A
- (CNS)
central nervous system
- (CMA)
chaperone-mediated autophagy
- (CIS),
clinically isolated syndrome
- (EAE)
experimental autoimmune encephalomyelitis
- (FTH1)
ferritin heavy chain
- (FoxO)
forkhead box class O
- (HSC70)
heat shock 70 kDa protein 8
- (HTT)
Huntingtin
- (LAMP-2A)
lysosome-associated membrane protein type 2A
- (LIR)
LC3-interacting region
- (MTOR)
mechanistic target of rapamycin
- (LC3)
microtubule-associated proteins 1A/1B light chain 3B
- (MS)
Multiple sclerosis
- (MBP)
myelin basic protein
- (NLRP3)
NLR family pyrin domain containing 3
- (NCOA4)
nuclear receptor coactivator 4
- (OMM)
outer mitochondrial membrane
- (OPTN)
optineurin
- (PD)
Parkinson’s disease
- (Parkin)
Parkinson protein 2, E3 ubiquitin protein ligase
- (PLIN2)
perilipin-2
- (PE)
phosphatidylethanolamine
- (PPMS)
Primary Progressive MS
- (PRMS)
Progressive-Relapsing MS
- (PINK1)
PTEN-induced kinase 1
- (ROS)
reactive oxygen species
- (RRMS)
relapsing-remitting course
- (SPMS)
secondary progressive MS
- (p62)
sequestosome 1
- (SIRT1)
Sirtuin 1
- (ULK1)
UNC-51-like kinase 1
- (WD)
Wallerian degeneration
Data availability
No datasets were generated or analyzed during the current study.
Competing interests
The authors declare no competing interests.
Author approval statement
All authors confirm that they have read, reviewed, and approved the final version of this manuscript for submission.
References
- 1.Klionsky DJ, Petroni G, Amaravadi RK, et al. Autophagy in major human diseases. EMBO J. 2021;40(19):e108863. doi: 10.15252/embj.2021108863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy. 2021;17(1):1–29. doi: 10.1080/15548627.2020.1797280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deretic V, Kroemer G.. Autophagy in metabolism and quality control: opposing, complementary or interlinked functions?. Autophagy. 2022;18(2):283–292. doi: 10.1080/15548627.2021.1933742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vargas JNS, Hamasaki M, Kawabata T, et al. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24(3):167–185. doi: 10.1038/s41580-022-00542-2 [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Tsvetkov AS, Shen HM, et al. International consensus guidelines for the definition, detection, and interpretation of autophagy-dependent ferroptosis. Autophagy. 2024;20(6):1213–1246. doi: 10.1080/15548627.2024.2319901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol. 2015;210(1):153–168. doi: 10.1083/jcb.201503019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakimovski D, Bittner S, Zivadinov R, et al. Multiple sclerosis. The Lancet. 2024;403(10422):183–202. doi: 10.1016/S0140-6736(23)01473-3 [DOI] [PubMed] [Google Scholar]
- 9.Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol. 2014;27(3):271–278. doi: 10.1097/WCO.0000000000000094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olejnik P, Roszkowska Z, Adamus S, et al. Multiple sclerosis: a narrative overview of current pharmacotherapies and emerging treatment prospects. Pharmacol Rep. 2024;76(5):926–943. doi: 10.1007/s43440-024-00642-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Igci M, Baysan M, Yigiter R, et al. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene. 2016;588(1):38–46. doi: 10.1016/j.gene.2016.04.042 [DOI] [PubMed] [Google Scholar]
- 12.Albert M, Barrantes-Freer A, Lohrberg M, et al. Synaptic pathology in the cerebellar dentate nucleus in chronic multiple sclerosis. Brain Pathol. 2017;27(6):737–747. doi: 10.1111/bpa.12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haidar M, Loix M, Vanherle S, et al. Targeting lipophagy in macrophages improves repair in multiple sclerosis. Autophagy. 2022;18(11):2697–2710. doi: 10.1080/15548627.2022.2047343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patergnani S, Bonora M, Ingusci S, et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc Natl Acad Sci U S A. 2021;118(24). doi: 10.1073/pnas.2020078118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haki M, Al-Biati HA, Al-Tameemi ZS, et al. Al-hussaniy HA: review of multiple sclerosis: epidemiology, etiology, pathophysiology, and treatment. Medicine (baltimore). 2024;103(8):e37297. doi: 10.1097/MD.0000000000037297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portaccio E, Magyari M, Havrdova EK, et al. Multiple sclerosis: emerging epidemiological trends and redefining the clinical course. Lancet Reg Health Eur. 2024;44:100977. doi: 10.1016/j.lanepe.2024.100977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haki M, Al-Biati HA, Al-Tameemi ZS, et al. Review of multiple sclerosis: epidemiology, etiology, pathophysiology, and treatment. Medicine (Baltimore). 2024;103(8):e37297. doi: 10.1097/MD.0000000000037297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward M, Goldman MD. Epidemiology and pathophysiology of multiple sclerosis. Continuum (minneap Minn). 2022;28(4):988–1005. doi: 10.1212/CON.0000000000001136 [DOI] [PubMed] [Google Scholar]
- 19.Cree BAC, Arnold DL, Chataway J, et al. Secondary progressive multiple sclerosis: new insights. Neurology. 2021;97(8):378–388. doi: 10.1212/WNL.0000000000012323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldenberg MM. Multiple sclerosis review. P T. 2012;37(3):175–184. [PMC free article] [PubMed] [Google Scholar]
- 21.Nourbakhsh B, Mowry EM. Multiple sclerosis risk factors and pathogenesis. Continuum (minneap Minn). 2019;25(3):596–610. doi: 10.1212/CON.0000000000000725 [DOI] [PubMed] [Google Scholar]
- 22.Huang WJ, Chen WW, Zhang X. Multiple sclerosis: pathology, diagnosis and treatments. Exp Ther Med. Exp & Ther Med. 2017;13(6):3163–3166. doi: 10.3892/etm.2017.4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boland B, Kumar A, Lee S. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilsson P, Loganathan K, Sekiguchi M, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5(1):61–69. doi: 10.1016/j.celrep.2013.08.042 [DOI] [PubMed] [Google Scholar]
- 25.Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castellazzi M, Patergnani S, Donadio M, et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci Rep. 2019;9(1):20009. doi: 10.1038/s41598-019-56614-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longobardi A, Catania M, Geviti A, et al. Autophagy markers are altered in Alzheimer’s disease, dementia with Lewy Bodies and frontotemporal dementia. Int J Mol Sci. 2024;25(2):1125. doi: 10.3390/ijms25021125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veverova K, Laczo J, Katonova A, et al. Alterations of human CSF and serum-based mitophagy biomarkers in the continuum of Alzheimer disease. Autophagy. 2024;20(8):1868–1878. doi: 10.1080/15548627.2024.2340408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nassif M, Valenzuela V, Rojas-Rivera D, et al. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy. 2014;10(7):1256–1271. doi: 10.4161/auto.28784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teyssou E, Takeda T, Lebon V, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–522. doi: 10.1007/s00401-013-1090-0 [DOI] [PubMed] [Google Scholar]
- 31.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- 32.Ochaba J, Lukacsovich T, Csikos G, et al. Potential function for the huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. 2014;111(47):16889–16894. doi: 10.1073/pnas.1420103111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franco-Iborra S, Plaza-Zabala A, Montpeyo M, et al. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 2021;17(3):672–689. doi: 10.1080/15548627.2020.1728096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spencer B, Potkar R, Trejo M, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-Synuclein models of Parkinson’s and Lewy Body Diseases. J Neurosci. 2009;29(43):13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanji K, Mori F, Kakita A, et al. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol Dis. 2011;43(3):690–697. doi: 10.1016/j.nbd.2011.05.022 [DOI] [PubMed] [Google Scholar]
- 36.Shahmoradian SH, Lewis AJ, Genoud C, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22(7):1099–1109. doi: 10.1038/s41593-019-0423-2 [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13(5):567–576. doi: 10.1038/nn.2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khalil B, El Fissi N, Aouane A, et al. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015;6(1):e1617. doi: 10.1038/cddis.2014.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li R, Li D, Wu C, et al. Nerve growth factor activates autophagy in schwann cells to enhance myelin debris clearance and to expedite nerve regeneration. Theranostics. 2020;10(4):1649–1677. doi: 10.7150/thno.40919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarraf SA, Shah HV, Kanfer G, et al. Loss Of TAX1BP1-Dir Autophagy results in protein aggreg accumulation in the brain Mol Cell. 2020;80(5):779–795 e710. doi: 10.1016/j.molcel.2020.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jo YR, Oh Y, Kim YH, et al. Adaptive autophagy reprogramming in schwann cells during peripheral demyelination. Cell Mol Life Sci. 2023;80(1):34. doi: 10.1007/s00018-022-04683-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.von Muhlinen N, Akutsu M, Ravenhill BJ, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell. 2012;48(3):329–342. doi: 10.1016/j.molcel.2012.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15(8):305–309. doi: 10.1016/0968-0004(90)90019-8 [DOI] [PubMed] [Google Scholar]
- 44.Zhou D, Li P, Lin Y, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22(5):571–581. doi: 10.1016/j.immuni.2005.03.009 [DOI] [PubMed] [Google Scholar]
- 45.Alirezaei M, Fox HS, Flynn CT, et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy. 2009;5(2):152–158. doi: 10.4161/auto.5.2.7348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castellazzi M, Patergnani S, Donadio M, et al. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J Neuroinflammation. 2019;16(1):131. doi: 10.1186/s12974-019-1526-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dasgupta A, Zheng J, Perrone-Bizzozero NI, et al. Increased carbonylation, protein aggregation and apoptosis in the spinal cord of mice with experimental autoimmune encephalomyelitis. ASN Neuro. 2013;5(2):e00111. doi: 10.1042/AN20120088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng J, Bizzozero OA. Decreased activity of the 20S proteasome in the brain white matter and gray matter of patients with multiple sclerosis. J Neurochem. 2011;117(1):143–153. doi: 10.1111/j.1471-4159.2011.07182.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng J, Dasgupta A, Bizzozero OA. Changes in 20S subunit composition are largely responsible for altered proteasomal activities in experimental autoimmune encephalomyelitis. J Neurochem. 2012;121(3):486–494. doi: 10.1111/j.1471-4159.2012.07699.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ceccariglia S, Sibilia D, Parolini O, et al. Altered expression of autophagy biomarkers in hippocampal neurons in a multiple sclerosis animal model. Int J Mol Sci. 2023;24(17):13225. doi: 10.3390/ijms241713225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun M, Gong P, Yuan B, et al. AXL-Induced autophagy mitigates experimental autoimmune encephalomyelitis by suppressing microglial inflammation via the PI3K/AKT/MTOR signaling pathway. Mol Immunol. 2023;159:15–27. doi: 10.1016/j.molimm.2023.05.005 [DOI] [PubMed] [Google Scholar]
- 52.Zhang Q, Zheng M, Sun W, et al. Ketogenic diet attenuates microglia-mediated neuroinflammation by inhibiting NLRP3 inflammasome activation via HDAC3 inhibition to activate mitophagy in experimental autoimmune encephalomyelitis. Food Funct. 2025;16(12):4731–4753. doi: 10.1039/D5FO00422E [DOI] [PubMed] [Google Scholar]
- 53.Jeffries MA, McLane LE, Khandker L, et al. MTOR signaling regulates metabolic function in oligodendrocyte precursor cells and promotes efficient brain remyelination in the cuprizone model. J Neurosci. 2021;41(40):8321–8337. doi: 10.1523/JNEUROSCI.1377-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamate-Morgan H, Lauderdale K, Horeczko J, et al. Functional effects of cuprizone-induced demyelination in the presence of the MTOR-Inhibitor rapamycin. Neuroscience. 2019;406:667–683. doi: 10.1016/j.neuroscience.2019.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sachs HH, Bercury KK, Popescu DC, et al. A new model of cuprizone-mediated demyelination/remyelination. ASN Neuro. 2014;6(5). doi: 10.1177/1759091414551955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vakrakou AG, Alexaki A, Brinia ME, et al. The MTOR signaling pathway in multiple sclerosis; from animal models to human data. Int J Mol Sci. 2022;23(15):8077. doi: 10.3390/ijms23158077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bonora M, De Marchi E, Patergnani S, et al. Tumor necrosis factor-α impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014;21(8):1198–1208. doi: 10.1038/cdd.2014.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun X, Qian M, Li H, et al. FKBP5 activates mitophagy by ablating PPAR-γ to shape a benign remyelination environment. Cell Death Dis. 2023;14(11):736. doi: 10.1038/s41419-023-06260-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jomova K, Raptova R, Alomar SY, et al. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch Toxicol. 2023;97(10):2499–2574. doi: 10.1007/s00204-023-03562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li W, Feng J, Gao C, et al. Nitration of Drp1 provokes mitophagy activation mediating neuronal injury in experimental autoimmune encephalomyelitis. Free Radic Biol Med. 2019;143:70–83. doi: 10.1016/j.freeradbiomed.2019.07.037 [DOI] [PubMed] [Google Scholar]
- 61.Kadowaki A, Wheeler MA, Li Z, et al. CLEC16A in astrocytes promotes mitophagy and limits pathology in a multiple sclerosis mouse model. Nat Neurosci. 2025;28(3):470–486. doi: 10.1038/s41593-025-01875-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berge T, Sørum Leikfoss I, Harbo HF. From Identification to Characterization of the Multiple Sclerosis Susceptibility Gene CLEC16A. Int J Mol Sci. 2013;14(3):4476–97. doi: 10.3390/ijms14034476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fransen NL, Crusius JB, Smolders J, et al. Post-mortem multiple sclerosis lesion pathology is influenced by single nucleotide polymorphisms. Brain Pathol. 2020;30(1):106–119. doi: 10.1111/bpa.12760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das Neves SP, Sousa JC, Magalhaes R, et al. Astrocytes undergo metabolic reprogramming in the multiple sclerosis animal model. Cells. 2023;12(20):2484. doi: 10.3390/cells12202484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hassanpour M, Cheraghi O, Laghusi D, et al. The relationship between ANT1 and NFL with autophagy and mitophagy markers in patients with multiple sclerosis. J Clin Neurosci. 2020;78:307–312. doi: 10.1016/j.jocn.2020.04.122 [DOI] [PubMed] [Google Scholar]
- 66.Joodi Khanghah O, Nourazarian A, Khaki‐Khatibi F, et al. Evaluation of the diagnostic and predictive value of serum levels of ANT1, ATG5, and Parkin in multiple sclerosis. Clin Neurol Neurosurg. 2020;197:106197. doi: 10.1016/j.clineuro.2020.106197 [DOI] [PubMed] [Google Scholar]
- 67.Cossu D, Yokoyama K, Sechi LA, et al. Potential of PINK1 and PARKIN proteins as biomarkers for active multiple sclerosis: a Japanese cohort study. Front Immunol. 2021;12:681386. doi: 10.3389/fimmu.2021.681386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patergnani S, Laudisi M, Bonora M, et al. Pathogenetic involvement of autophagy and mitophagy in primary progressive multiple sclerosis. J Cellular Mol Medi. 2025;29(8):e70455. doi: 10.1111/jcmm.70455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cossu D, Yokoyama K, Sato S, et al. Age related immune modulation of experimental autoimmune encephalomyelitis in PINK1 knockout mice. Front Immunol. 2022;13:1036680. doi: 10.3389/fimmu.2022.1036680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cossu D, Yokoyama K, Sato S, et al. PARKIN modifies peripheral immune response and increases neuroinflammation in active experimental autoimmune encephalomyelitis (EAE). J Neuroimmunol. 2021;359:577694. doi: 10.1016/j.jneuroim.2021.577694 [DOI] [PubMed] [Google Scholar]
- 71.Di Rita A, Angelini DF, Maiorino T, et al. Characterization of a natural variant of human NDP52 and its functional consequences on mitophagy. Cell Death Differ. 2021;28(8):2499–2516. doi: 10.1038/s41418-021-00766-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jhelum P, Santos-Nogueira E, Teo W, et al. Ferroptosis mediates cuprizone-induced loss of oligodendrocytes and demyelination. J Neurosci. 2020;40(48):9327–9341. doi: 10.1523/JNEUROSCI.1749-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jhelum P, Zandee S, Ryan F, et al. Ferroptosis induces detrimental effects in chronic EAE and its implications for progressive MS. Acta Neuropathol Commun. 2023;11(1):121. doi: 10.1186/s40478-023-01617-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu T, Ning S, Zhang H, et al. Role of ferroptosis in neuroimmunity and neurodegeneration in multiple sclerosis revealed by multi-omics data. J Cell Mol Med. 2024;28(10):e18396. doi: 10.1111/jcmm.18396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sfagos C, Makis AC, Chaidos A, et al. Serum ferritin, transferrin and soluble transferrin receptor levels in multiple sclerosis patients. Mult Scler. 2005;11(3):272–275. doi: 10.1191/1352458505ms1171oa [DOI] [PubMed] [Google Scholar]
- 76.Stojkovic L, Djordjevic A, Stefanovic M, et al. Circulatory indicators of lipid peroxidation, the Driver of ferroptosis, reflect differences between relapsing–remitting and progressive multiple sclerosis. Int J Mol Sci. 2024;25(20):11024. doi: 10.3390/ijms252011024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bergsland N, Agostini S, Lagana MM. Serum iron concentration is associated with subcortical deep gray matter iron levels in multiple sclerosis patients. Neuroreport. 2017;28(11):645–648. doi: 10.1097/WNR.0000000000000804 [DOI] [PubMed] [Google Scholar]
- 78.De Lury AD, Bisulca JA, Lee JS, et al. Magnetic resonance imaging detection of deep gray matter iron deposition in multiple sclerosis: a systematic review. J Neurol Sci. 2023;453:120816. doi: 10.1016/j.jns.2023.120816 [DOI] [PubMed] [Google Scholar]
- 79.Hagemeier J, Yeh EA, Brown MH, et al. Iron content of the pulvinar nucleus of the thalamus is increased in adolescent multiple sclerosis. Mult Scler. 2013;19(5):567–576. doi: 10.1177/1352458512459289 [DOI] [PubMed] [Google Scholar]
- 80.Ge Y, Jensen JH, Lu H, et al. Quantitative assessment of iron accumulation in the deep gray matter of multiple sclerosis by magnetic field correlation imaging. AJNR Am J Neuroradiol. 2007;28(9):1639–1644. doi: 10.3174/ajnr.A0646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Al-Radaideh A, El-Haj N, Hijjawi N. Iron deposition and atrophy in cerebral grey matter and their possible association with serum iron in relapsing-remitting multiple sclerosis. Clin Imaging. 2021;69:238–242. doi: 10.1016/j.clinimag.2020.09.006 [DOI] [PubMed] [Google Scholar]
- 82.Bjorklund G, Wallace DR, Hangan T, et al. Cerebral iron accumulation in multiple sclerosis: pathophysiology and therapeutic implications. Autoimmun Rev. 2025;24(4):103741. doi: 10.1016/j.autrev.2025.103741 [DOI] [PubMed] [Google Scholar]
- 83.Yarnykh VL, Krutenkova EP, Aitmagambetova G, et al. Iron-insensitive quantitative assessment of subcortical gray matter demyelination in multiple sclerosis using the macromolecular proton fraction. AJNR Am J Neuroradiol. 2018;39(4):618–625. doi: 10.3174/ajnr.A5542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grajchen E, Hendriks JJA, Bogie JFJ. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol Commun. 2018;6(1):124. doi: 10.1186/s40478-018-0628-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Williams K, Ulvestad E, Waage A, et al. Activation of adult human derived microglia by myelin phagocytosis in vitro. J Of Neuroscience Research. 1994;38(4):433–443. doi: 10.1002/jnr.490380409 [DOI] [PubMed] [Google Scholar]
- 86.Wang X, Cao K, Sun X, et al. Macrophages in spinal cord injury: phenotypic and functional change from exposure to myelin debris. Glia. 2015;63(4):635–651. doi: 10.1002/glia.22774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Zwam M, Samsom JN, Nieuwenhuis EE, et al. Myelin ingestion alters macrophage antigen-presenting function in vitro and in vivo. J Leukoc Biol. 2011;90(1):123–132. doi: 10.1189/jlb.1209813 [DOI] [PubMed] [Google Scholar]
- 88.Liu Y, Hao W, Letiembre M, et al. Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J Neurosci. 2006;26(50):12904–12913. doi: 10.1523/JNEUROSCI.2531-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. 2018;359(6376):684–688. doi: 10.1126/science.aan4183 [DOI] [PubMed] [Google Scholar]
- 90.Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8(1):15750. doi: 10.1038/ncomms15750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma H, Ou ZL, Alaeiilkhchi N, et al. MiR-223 enhances lipophagy by suppressing CTSB in microglia following lysolecithin-induced demyelination in mice. Lipids Health Dis. 2024;23(1):194. doi: 10.1186/s12944-024-02185-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loix M, Wouters E, Vanherle S, et al. Perilipin-2 limits remyelination by preventing lipid droplet degradation. Cell Mol Life Sci. 2022;79(10):515. doi: 10.1007/s00018-022-04547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bogie JF, Stinissen P, Hellings N, et al. Myelin-phagocytosing macrophages modulate autoreactive T cell proliferation. J Neuroinflammation. 2011;8(1):85. doi: 10.1186/1742-2094-8-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee SC, Moore GR, Golenwsky G, et al. Multiple sclerosis: a role for astroglia in active demyelination suggested by class II MHC expression and ultrastructural study. J Neuropathol Exp Neurol. 1990;49(2):122–136. doi: 10.1097/00005072-199003000-00005 [DOI] [PubMed] [Google Scholar]
- 95.Lopes Pinheiro MA, Kamermans A, Garcia-Vallejo JJ, et al. Internalization and presentation of myelin antigens by the brain endothelium guides antigen-specific T cell migration. Elife. 2016;5:5. doi: 10.7554/eLife.13149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yao F, Luo Y, Chen Y, et al. Myelin debris impairs tight junctions and promotes the migration of microvascular endothelial cells in the injured spinal cord. Cell Mol Neurobiol. 2023;43(2):741–756. doi: 10.1007/s10571-022-01203-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hammel G, Zivkovic S, Ayazi M, et al. Consequences and mechanisms of myelin debris uptake and processing by cells in the central nervous system. Cell Immunol. 2022;380:104591. doi: 10.1016/j.cellimm.2022.104591 [DOI] [PubMed] [Google Scholar]
- 98.Noori L, Saqagandomabadi V, Di Felice V, et al. Putative roles and therapeutic potential of the chaperone system in amyotrophic lateral sclerosis and multiple sclerosis. Cells. 2024;13(3):217. doi: 10.3390/cells13030217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Inoue M, Williams KL, Gunn MD, et al. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109(26):10480–10485. doi: 10.1073/pnas.1201836109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–379. doi: 10.1016/j.cmet.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Esposito M, Ruffini F, Bellone M, et al. Rapamycin inhibits relapsing experimental autoimmune encephalomyelitis by both effector and regulatory T cells modulation. J Neuroimmunol. 2010;220(1–2):52–63. doi: 10.1016/j.jneuroim.2010.01.001 [DOI] [PubMed] [Google Scholar]
- 102.DiToro D, Harbour SN, Bando JK, et al. Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity. Immunity. 2020;52(4):650–667.e10. doi: 10.1016/j.immuni.2020.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hou H, Miao J, Cao R, et al. Rapamycin ameliorates experimental autoimmune encephalomyelitis by suppressing the MTOR-STAT3 pathway. Neurochem Res. 2017;42(10):2831–2840. doi: 10.1007/s11064-017-2296-7 [DOI] [PubMed] [Google Scholar]
- 104.Xu L, Zhang C, Jiang N, et al. Rapamycin combined with MCC950 to treat multiple sclerosis in experimental autoimmune encephalomyelitis. J Cell Biochem. 2019;120(4):5160–5168. doi: 10.1002/jcb.27792 [DOI] [PubMed] [Google Scholar]
- 105.Xu L, Zhang C, He D, et al. Rapamycin and MCC950 modified gut microbiota in experimental autoimmune encephalomyelitis mouse by brain gut axis. Life Sci. 2020;253:117747. doi: 10.1016/j.lfs.2020.117747 [DOI] [PubMed] [Google Scholar]
- 106.Moraal B, van den Elskamp IJ, Knol DL, et al. Long-interval T2-weighted subtraction magnetic resonance imaging: a powerful new outcome measure in multiple sclerosis trials. Ann Neurol. 2010;67(5):667–675. doi: 10.1002/ana.21958 [DOI] [PubMed] [Google Scholar]
- 107.Salehi M, Bagherpour B, Shayghannejad V, et al. Th1, Th2 and Th17 cytokine profile in patients with multiple sclerosis following treatment with rapamycin. Iran J Immunol. 2016;13(2):141–147. [PubMed] [Google Scholar]
- 108.Bagherpour B, Salehi M, Jafari R, et al. Promising effect of rapamycin on multiple sclerosis. Mult Scler Relat Disord. 2018;26:40–45. doi: 10.1016/j.msard.2018.08.009 [DOI] [PubMed] [Google Scholar]
- 109.Lee DJW, Hodzic Kuerec A, Maier AB. Targeting ageing with rapamycin and its derivatives in humans: a systematic review. The Lancet Healthy Longevity. 2024;5(2):e152–e162. doi: 10.1016/S2666-7568(23)00258-1 [DOI] [PubMed] [Google Scholar]
- 110.Dziedzic A, Saluk-Bijak J, Miller E, et al. Metformin as a potential agent in the treatment of multiple sclerosis. Int J Mol Sci. 2020;21(17):5957. doi: 10.3390/ijms21175957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun Y, Tian T, Gao J, et al. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J Neuroimmunol. 2016;292:58–67. doi: 10.1016/j.jneuroim.2016.01.014 [DOI] [PubMed] [Google Scholar]
- 112.Duan W, Ding Y, Yu X, et al. Metformin mitigates autoimmune insulitis by inhibiting Th1 and Th17 responses while promoting treg production. Am J Transl Res. 2019;11(4):2393–2402. [PMC free article] [PubMed] [Google Scholar]
- 113.Sanadgol N, Barati M, Houshmand F. Metformin accelerates myelin recovery and ameliorates behavioral deficits in the animal model of multiple sclerosis via adjustment of AMPK/Nrf2/MTOR signaling and maintenance of endogenous oligodendrogenesis during brain self-repairing period. Pharmacol Rep. 2020;72(3):641–658. doi: 10.1007/s43440-019-00019-8 [DOI] [PubMed] [Google Scholar]
- 114.Houshmand F, Barati M, Golab F. Metformin-induced AMPK activation stimulates remyelination through induction of neurotrophic factors, downregulation of NogoA and recruitment of Olig2+ precursor cells in the cuprizone murine model of multiple sclerosis. Daru. 2019;27(2):583–592. doi: 10.1007/s40199-019-00286-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Abdelgaied MY, Rashad MH, El-Tayebi HM, et al. The impact of metformin use on the outcomes of relapse-remitting multiple sclerosis patients receiving interferon beta 1a: an exploratory prospective phase II open-label randomized controlled trial. J Neurol. 2024;271(3):1124–1132. doi: 10.1007/s00415-023-12113-2 [DOI] [PubMed] [Google Scholar]
- 116.Negrotto L, Farez MF, Correale J. Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol. 2016;73(5):520–528. doi: 10.1001/jamaneurol.2015.4807 [DOI] [PubMed] [Google Scholar]
- 117.Miller A, Korem M, Almog R, et al. Vitamin B12, demyelination, remyelination and repair in multiple sclerosis. J Neurol Sci. 2005;233(1–2):93–97. doi: 10.1016/j.jns.2005.03.009 [DOI] [PubMed] [Google Scholar]
- 118.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi: 10.1007/s00125-017-4342-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun AY, Simonyi A, Sun GY. The “French paradox” and beyond: neuroprotective effects of polyphenols1,2 1Guest editor: Arthur Cederbaum 2This article is part of a series of reviews on “alcohol, oxidative stress and cell injury.” the full list of papers may be found on the homepage of the journal. Free Radic Biol Med. 2002;32(4):314–318. doi: 10.1016/S0891-5849(01)00803-6 [DOI] [PubMed] [Google Scholar]
- 120.Zhao H, Chen S, Gao K, et al. Resveratrol protects against spinal cord injury by activating autophagy and inhibiting apoptosis mediated by the SIRT1/AMPK signaling pathway. Neuroscience. 2017;348:241–251. doi: 10.1016/j.neuroscience.2017.02.027 [DOI] [PubMed] [Google Scholar]
- 121.Samy DM, Zaki EI, Hassaan PS, et al. Neurobehavioral, biochemical and histological assessment of the effects of resveratrol on cuprizone-induced demyelination in mice: role of autophagy modulation. J Physiol Biochem. 2023;79(3):583–596. doi: 10.1007/s13105-023-00959-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fonseca-Kelly Z, Nassrallah M, Uribe J, et al. Resveratrol neuroprotection in a chronic mouse model of multiple sclerosis. Front Neurol. 2012;3:84. doi: 10.3389/fneur.2012.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shindler KS, Ventura E, Dutt M, et al. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J Neuroophthalmol. 2010;30(4):328–339. doi: 10.1097/WNO.0b013e3181f7f833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun AY, Wang Q, Simonyi A, et al. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol Neurobiol. 2010;41(2–3):375–383. doi: 10.1007/s12035-010-8111-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Singh V, Ubaid S. Role of silent information regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation. 2020;43(5):1589–1598. doi: 10.1007/s10753-020-01242-9 [DOI] [PubMed] [Google Scholar]
- 126.Monzio Compagnoni G, Di Fonzo A, Corti S, et al. The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer’s disease and Parkinson’s disease. Mol Neurobiol. 2020;57(7):2959–2980. doi: 10.1007/s12035-020-01926-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keramatzadeh S, Hosseini SA, Majdinasab N, et al. Effects of resveratrol supplementation on inflammatory markers, fatigue scale, fasting blood sugar and lipid profile in relapsing-remitting multiple sclerosis patients: a double-blind, randomized placebo-controlled trial. Nutr Neurosci. 2025;28(7):854–862. doi: 10.1080/1028415X.2024.2425649 [DOI] [PubMed] [Google Scholar]
- 128.Walle T. Bioavailability of resveratrol. Ann N Y Acad Sci. 2011;1215(1):9–15. doi: 10.1111/j.1749-6632.2010.05842.x [DOI] [PubMed] [Google Scholar]
- 129.Smoliga JM, Baur JA, Hausenblas HA. Resveratrol and health–a comprehensive review of human clinical trials. Mol Nutr Food Res. 2011;55(8):1129–1141. doi: 10.1002/mnfr.201100143 [DOI] [PubMed] [Google Scholar]
- 130.Patlola SR, Donohoe G, McKernan DP. Anti-inflammatory effects of 2nd generation antipsychotics in patients with schizophrenia: a systematic review and meta-analysis. Journal Of Psychiatric Research. 2023;160:126–136. doi: 10.1016/j.jpsychires.2023.01.042 [DOI] [PubMed] [Google Scholar]
- 131.Stamoula Ε, Ainatzoglou A, Stamatellos VP, et al. Atypical antipsychotics in multiple sclerosis: a review of their in vivo immunomodulatory effects. Mult Scler Relat Disord. 2022;58:103522. doi: 10.1016/j.msard.2022.103522 [DOI] [PubMed] [Google Scholar]
- 132.Green LK, Zareie P, Templeton N, et al. Enhanced disease reduction using clozapine, an atypical antipsychotic agent, and glatiramer acetate combination therapy in experimental autoimmune encephalomyelitis. Mult Scler J Exp Transl Clin. 2017;3(1):2055217317698724. doi: 10.1177/2055217317698724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.O’Sullivan D, Green L, Stone S, et al. Treatment with the antipsychotic agent, risperidone, reduces disease severity in experimental autoimmune encephalomyelitis. PLOS ONE. 2014;9(8):e104430. doi: 10.1371/journal.pone.0104430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zareie P, Connor B, La Flamme AC. Amelioration of experimental autoimmune encephalomyelitis by clozapine is not associated with defective CD4 T cell responses. J Neuroinflammation. 2017;14(1):68. doi: 10.1186/s12974-017-0842-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Patergnani S, Castellazzi M, Bonora M, et al. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J Neurol Neurosurg Psychiatry. 2018;89(4):439–441. doi: 10.1136/jnnp-2017-316234 [DOI] [PubMed] [Google Scholar]
- 136.Satarker S, Wilson J, Kolathur KK, et al. Spermidine as an epigenetic regulator of autophagy in neurodegenerative disorders. Eur J Pharmacol. 2024;979:176823. doi: 10.1016/j.ejphar.2024.176823 [DOI] [PubMed] [Google Scholar]
- 137.Yang Q, Zheng C, Cao J, et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ. 2016;23(11):1850–1861. doi: 10.1038/cdd.2016.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zheng R, Kong M, Wang S, et al. Spermine alleviates experimental autoimmune encephalomyelitis via regulating T cell activation and differentiation. Int Immunopharmacol. 2022;107:108702. doi: 10.1016/j.intimp.2022.108702 [DOI] [PubMed] [Google Scholar]
- 139.Lee HJ, Yoon YS, Lee SJ. Mechanism of neuroprotection by trehalose: controversy surrounding autophagy induction. Cell Death Dis. 2018;9(7):712. doi: 10.1038/s41419-018-0749-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.De Sarno P, Axtell RC, Raman C, et al. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2008;181(1):338–345. doi: 10.4049/jimmunol.181.1.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Niino M. Vitamin D and its immunoregulatory role in multiple sclerosis. Drugs Today (Barc). 2010;46(4):279–290. doi: 10.1358/dot.2010.46.4.1476498 [DOI] [PubMed] [Google Scholar]
- 142.Azuchi Y, Kimura A, Guo X, et al. Valproic acid and ASK1 deficiency ameliorate optic neuritis and neurodegeneration in an animal model of multiple sclerosis. Neurosci Lett. 2017;639:82–87. doi: 10.1016/j.neulet.2016.12.057 [DOI] [PubMed] [Google Scholar]
- 143.Gaffke L, Firyn N, Rintz E, et al. Therapeutic potential of lithium chloride and valproic acid against neuronopathic types of mucopolysaccharidoses through induction of the autophagy process. Arch Biochem Biophys. 2023;747:109754. doi: 10.1016/j.abb.2023.109754 [DOI] [PubMed] [Google Scholar]
- 144.Bhutia SK. Vitamin D in autophagy signaling for health and diseases: insights on potential mechanisms and future perspectives. J Nutr Biochem. 2022;99:108841. doi: 10.1016/j.jnutbio.2021.108841 [DOI] [PubMed] [Google Scholar]
- 145.Valeiras B, Rosato Siri MV, Codagnone M, et al. Gender influence on schizophrenia-relevant abnormalities in a cuprizone demyelination model. Glia. 2014;62(10):1629–1644. doi: 10.1002/glia.22704 [DOI] [PubMed] [Google Scholar]
- 146.Taylor LC, Gilmore W, Matsushima GK. SJL mice exposed to cuprizone intoxication reveal strain and gender pattern differences in demyelination. Brain Pathol. 2009;19(3):467–479. doi: 10.1111/j.1750-3639.2008.00230.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Denic A, Johnson AJ, Bieber AJ, et al. The relevance of animal models in multiple sclerosis research. Pathophysiology. 2011;18(1):21–29. doi: 10.1016/j.pathophys.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dedoni S, Scherma M, Camoglio C, et al. An overall view of the most common experimental models for multiple sclerosis. Neurobiol Dis. 2023;184:106230. doi: 10.1016/j.nbd.2023.106230 [DOI] [PubMed] [Google Scholar]
- 149.Mix E, Meyer-Rienecker H, Hartung HP, et al. Animal models of multiple sclerosis—potentials and limitations. Prog Neurobiol. 2010;92(3):386–404. doi: 10.1016/j.pneurobio.2010.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang X, Yang W, Xia X, et al. Intranasal delivery of BACE1 siRNA and rapamycin by dual targets modified nanoparticles for Alzheimer’s disease therapy. Small. 2022;18(30):e2203182. doi: 10.1002/smll.202203182 [DOI] [PubMed] [Google Scholar]
- 151.Polchi A, Magini A, Mazuryk J, et al. Rapamycin loaded solid lipid nanoparticles as a new tool to deliver MTOR inhibitors: formulation and in vitro characterization. Nanomaterials (Basel). 2016;6(5):87. doi: 10.3390/nano6050087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yu Y, Li S, Kong L, et al. Development of a brain-targeted nano drug delivery system to enhance the treatment of neurodegenerative effects of resveratrol. J Liposome Res. 2024;34(3):435–451. doi: 10.1080/08982104.2023.2290050 [DOI] [PubMed] [Google Scholar]
- 153.Shamsher E, Khan RS, Davis BM, et al. Nanoparticles enhance solubility and neuroprotective effects of resveratrol in demyelinating disease. Neurotherapeutics. 2023;20(4):1138–1153. doi: 10.1007/s13311-023-01378-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Keeler GD, Kumar S, Palaschak B, et al. Gene therapy-induced antigen-specific Tregs inhibit Neuro-inflammation and reverse disease in a mouse model of multiple sclerosis. Mol Ther. 2018;26(1):173–183. doi: 10.1016/j.ymthe.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.La Cognata V, Morello G, Cavallaro S. Omics data and their integrative analysis to support stratified medicine in neurodegenerative diseases. Int J Mol Sci. 2021;22(9):4820. doi: 10.3390/ijms22094820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Carraro C, Montgomery JV, Klimmt J, et al. Tackling neurodegeneration in vitro with omics: a path towards new targets and drugs. Front Mol Neurosci. 2024;17:1414886. doi: 10.3389/fnmol.2024.1414886 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analyzed during the current study.