

Abstract
Stretch-activated channels (SACs) have been found in smooth muscle and are thought to be involved in myogenic responses. Although SACs have been shown to be Ca2+ permeable when Ca2+ is the only charge carrier, it has not been clearly demonstrated that significant Ca2+ passes through SACs in physiological solutions. By imaging at high temporal and spatial resolution the single-channel Ca2+ fluorescence transient (SCCaFT) arising from Ca2+ entry through a single SAC opening, we provide direct evidence that significant Ca2+ can indeed pass through SACs and increase the local [Ca2+]. Results were obtained under conditions where the only source of Ca2+ was the physiological salt solution in the patch pipette containing 2 mM Ca2+. Single smooth muscle cells were loaded with fluo-3 acetoxymethyl ester, and the fluorescence was recorded by using a wide-field digital imaging microscope while SAC currents were simultaneously recorded from cell-attached patches. Fluorescence increases at the cell-attached patch were clearly visualized before the simultaneous global Ca2+ increase that occurred because of Ca2+ influx through voltage-gated Ca2+ channels when the membrane was depolarized by inward SAC current. From measurements of total fluorescence (“signal mass”) we determined that about 18% of the SAC current is carried by Ca2+ at membrane potentials more negative than the resting level. This would translate into at least a 0.35-pA unitary Ca2+ current at the resting potential. Such Ca2+ currents passing through SACs are sufficient to activate large-conductance Ca2+-activated K+ channels and, as shown previously, to trigger Ca2+ release from intracellular stores.
Stretch-activated channels (SACs) have been found in many cell types and are thought to be involved in various mechanical signal transduction mechanisms ranging from hearing, touching, and cell movement to smooth muscle contraction (for reviews, see refs. 1–3). SACs in smooth muscle have also been shown to be Ca2+ permeable (4–6). This conclusion was obtained from patch-clamp ion substitution experiments where Ca2+ was the only available carrier of the inward current. These studies also pointed out that the SAC conductance for Ca2+ is much smaller than for monovalent cations. Therefore, it has not been clearly demonstrated whether significant amounts of Ca2+ can indeed enter the cell through SACs when physiological salt solutions are used, or whether the fraction of the current carried by Ca2+ is large enough to have a significant effect on the intracellular [Ca2+]. Nevertheless, SACs have been considered essential for stretch-induced contractions and vascular smooth muscle myogenic responses (4, 6–8). Two roles for SACs were proposed: to pass cations (mainly Na+) leading to membrane depolarization, and to pass Ca2+ to increase the cytosolic [Ca2+] either directly or indirectly by triggering Ca2+ release from stores.
Indirect evidence that Ca2+ could enter cells through SACs was obtained by using various Ca2+ imaging techniques while assuming SACs were activated by using mechanical or osmotic stimulation (see, e.g., ref. 9 and references in ref. 3). Because SAC unitary currents were not recorded at the same time, various channel inhibitors or blockers were usually used to try to relate any recorded elevation in [Ca2+] to openings of SACs.
To our knowledge there has been only one study where Ca2+ fluorescence was spatially imaged while unitary SAC currents were recorded at the same time. Kirber et al. (8), using fura-2 as the fluorescent Ca2+ indicator, observed with high spatial resolution local fluorescence increases near the pipette tip when SACs opened in cell-attached patches. Nevertheless, this local increase in fluorescence was no longer observed when the intracellular Ca2+ stores were emptied by adding 0.5 mM caffeine and 100 μM ryanodine to the bathing solution. Based on these and other observations, it was suggested that Ca2+ entry through SAC openings could trigger Ca2+ release from intracellular Ca2+ stores. Therefore, the fluorescence increase observed in the images was due, presumably, to the contribution from Ca2+ entering through SACs and Ca2+ released from intracellular stores (mainly the latter), but Ca2+ entry alone without amplification by Ca2+ release from stores could not be detected.
In the present study, carried out with fluo-3 as the Ca2+ indicator at much higher time resolution and for longer time periods than used previously, we provide direct evidence that significant amounts of Ca2+ can indeed enter the cell through SACs in physiological salt solutions. We also demonstrate that when the cell-attached patch is stretched, the local Ca2+ increase due to openings of SACs precedes the global Ca2+ increase due to openings of voltage-gated Ca2+ channels (VGCCs) (8). In addition, we estimate the Ca2+ current passing through the SACs. A brief preliminary report of some of the findings presented here has been published in abstract form†.
Methods
Single Smooth Muscle Cell Preparation.
Smooth muscle cells were enzymatically dispersed from the stomach of the toad Bufo marinus as previously described (10, 11) and used on the same day. All experiments were carried out at room temperature.
Patch-Clamp Recordings, Data Processing, and Ca2+ Imaging.
SACs were activated by applying negative pressure (suction) to the back end of the patch pipette as described previously (4, 8). The single-channel unitary currents were recorded with an Axopatch-1D amplifier (Axon Instruments, Foster City, CA), using the cell-attached patch configuration of the patch-clamp technique. The cell membrane potential was not held constant; inward SAC currents could cause depolarization of the cell membrane as indicated by a decrease in the unitary current amplitude (see ref. 8 for more details). Currents were initially low-pass filtered at 200 Hz and sampled at 1 kHz. Additional filtering by using a subtraction method was sometimes employed to remove 60-Hz noise.
Methods for two-dimensional Ca2+ imaging and data processing were similar to those used by Zou et al. (12). Fluorescence images were acquired by using a custom-built high-speed, wide-field digital imaging microscope (see ref. 13 for a description of the system) with fluo-3 as the Ca2+ indicator (loaded into the cells by using a 1 μM concentration of the acetoxymethyl ester form at room temperature for about 1 h). At each pixel (333-nm square), the fluorescence in the absence of transients (F0), and during a transient (F), was used to construct the ratio images [ΔF/F0 = (F − F0)/F0]. Multiple recordings were generally obtained from the same cell. Camera readout command pulses and channel currents were simultaneously recorded to facilitate the alignment of the fluorescence trace with the corresponding channel-current trace. A bright-field image of the cell with patch pipette was acquired before each experiment to verify the location of the pipette tip.
Method for Estimating Ca2+ Current from Total Fluorescence Measurements.
Ca2+ entry through SACs was estimated from the change in “signal mass,” or total fluorescence (ΔFLtotal), that occurs when SACs open and Ca2+ binds to fluo-3 (12, 14, 15). ΔFLtotal was obtained by calculating the difference in fluorescence just before and just after the fluorescence increase from an image area that was large enough to cover the entire fluorescence change as determined from the ΔF/F0 images (as outlined in ref. 12 and illustrated in Fig. 1). To do this measurement the original raw images were used instead of the ratio images described in the previous section.
Figure 1.

Illustration of the method for estimating the Ca2+ current from the total fluorescence increase when a SAC opens. (A) To collect the entire fluorescence increase (ΔFLtotal) caused by Ca2+ entry through a channel opening, measurements have to be obtained from a large enough image area (outlined by the yellow box). (B) The unitary current recording (with inward currents indicated by a downward deflection) and the change in fluorescence with time at the location of the channel (ΔF/F0) are plotted together. ΔFLtotal was measured from the baseline total fluorescence before the channel opened, to the near steady state in fluorescence that occurs briefly after the channel closed (to allow some time for equilibration of Ca2+ with fluo-3 and other Ca2+ buffers). The actual fluorescence measurements were obtained by summing over all of the pixels in the raw images within the relevant box. Channel current was integrated to obtain the total charge (ΔQ) passing through the channel. Note (in Inset) that there is a linear relationship between ΔFLtotal and ΔQ (plotted every ms, interpolating between the 5-ms images), as indicated by the black line fitted to the data. ΔFLtotal also increases nearly linearly with time (as predicted in ref. 12) even with the short closures and openings of the channel. For the purpose of illustrating the method, a ratio image (ΔF/F0) instead of the raw fluorescence image is shown (although the latter was used for obtaining ΔFLtotal). Images were acquired continuously with a 5-ms exposure time, and the image shown is that at the first fluorescence peak. The pixel size is 333 nm, and the calibration bar is 5 μm. The standard solution and the Ca2+-free solution with 1 μM thapsigargin were used for pipette and bath solutions, respectively. The patch membrane potential was 100 mV more negative than the cell membrane potential.
Determination of the Ca2+ influx (ΔCa2+) from ΔFLtotal requires the transfer function between the two. This transfer function takes into account both the relationship between the change in fluorescence and the increase in Ca2+-bound fluo-3 and the relationship among Ca2+ influx, free Ca2+, and the binding of Ca2+ to fluo-3 and other buffers.
For SAC openings in a physiological salt solution (standard solution, see below), only a fraction of the total current is carried by Ca2+. Integrating the current will give the total charge entry, ΔQ, during the channel openings. As shown in the Inset to Fig. 1, there is a linear relationship between ΔFLtotal and ΔQ as would be predicted from the simulations carried out in ref. 12, assuming that the current carried by Ca2+ is a constant fraction of the SAC current. Therefore, the transfer function is simply a constant over the range of conditions in these experiments, i.e.,
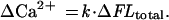 |
The value of this constant, k, or conversion factor, was obtained by using a 90 mM Ca2+ pipette solution (see below), where Ca2+ was the only cation available to carry the inward SAC current. The conversion factor can be calculated as the ratio of total Ca2+ entry [in this case, equal to ΔQ divided by the charge of a Ca ion (3.2⋅10−19 C)] to ΔFLtotal. Under the experimental conditions and assumptions (see below) used for this study, for a 10-ms-exposure frame, the value for the conversion factor is 8.59 ± 0.65 Ca ions per fluorescence unit from the charge-coupled device camera readout, or 1.72 ± 0.13 Ca ions per detected photon (8 transients from 4 cells). Using this value, ΔCa2+ can be obtained from ΔFLtotal, when the standard pipette solution (containing 2 mM Ca2+) is used. The fraction of the current carried by Ca2+ can then be calculated from 3.2⋅10−19 ΔCa2+/ΔQ. All data are expressed as the mean ± SEM.
To apply the conversion factor obtained by using the 90 mM Ca2+ pipette solution to cells where we used the 2 mM Ca2+ pipette solution, the loading of fluo-3 should be similar for both groups of cells. As a way of ensuring this condition, we measured the background-corrected resting fluorescence (normalized to exposure time, FLn) from an assumed cylindrical section of each cell and calculated the resting fluo-3 fluorescence per unit volume (FLn/V). Assuming that the resting [Ca2+] was similar in all of the cells, we selected from our recordings using the 2 mM Ca2+ pipette solution, those having an FLn/V within 2.5 SD from the mean value of the recordings using the 90 mM Ca2+ pipette solution.
The data gathered from the cells where we used the 90 mM Ca2+ pipette solution were obtained with the patches held 100 mV more negative than the resting potential. There was no significant difference between the ΔFLtotal for the two groups of cells, using the 2 mM or 90 mM Ca2+ pipette solutions.
Solutions.
For most of the experiments described here, cells were bathed in one of two solutions: a standard solution containing (in mM): NaCl 127, KCl 3, CaCl2 2, MgCl2 1, Hepes 10, pH 7.4, or a Ca2+-free solution containing (in mM): NaCl 127, KCl 3, MgCl2 1, Hepes 10, Na4BAPTA 0.2, pH 7.4 [BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate]. A high [Ca2+] solution, used as the cell-attached patch pipette solution for calibration purposes, contained 90 mM CaCl2 and 10 mM Hepes, with a pH of 7.4 [adjusted using Ca(OH)2].
When experiments required removing any possible contributions from intracellular Ca2+ stores, thapsigargin (1 μM), and sometimes ryanodine (100 μM), were added to the bathing solutions. In these experiments, caffeine (20 mM in the standard solution) was applied to the cells by pressure ejection by using a Picospritzer (General Valve, Fairfield, NJ) before sealing onto the cell (12). ZhuGe et al. (13) have reported that treatments with thapsigargin and caffeine will lead to a permanent decrease in [Ca2+] in the intracellular stores and, therefore, eliminate the store contribution to cytosolic Ca2+ rise.
Results
Localized Increases in Fluorescence Are Observed When SACs Open.
To demonstrate that we could record the fluorescence associated with Ca2+ passing through SACs, SACs were opened with the effects of intracellular Ca2+ stores eliminated. The fluorescence increased near the location of the pipette tip and could spread to adjacent regions (Fig. 2). The general amplitude and time course of the fluorescence change were similar to those of the averaged current passing through the SACs. Sometimes, when there was Ca2+ present in the bathing solution, a delayed global increase in Ca2+ also occurred (see next section).
Figure 2.

Opening of SACs in a solution containing physiological levels of Ca2+ causes localized fluorescence increases even when the effects of intracellular Ca2+ stores are removed. The bright-field microscope image of the cell and the patch pipette are shown in the leftmost frame of the image set. To its right are the fluorescence ratio images of the cell acquired at the time points indicated by the correspondingly numbered arrows. The fluorescence changes at the pipette tip (location a), and another area away from it (location b), were plotted along with the current. The gray dotted line is the scaled current trace averaged over 200 ms. Thapsigargin (1 μM) was added to the bath solution, and caffeine (20 mM) had been applied to the cell. Inset indicates the conditions under which the recording was carried out. Patch membrane potential is the same as the cell membrane potential which is not clamped (see Methods). Fluorescence images were acquired every 50 ms with a 10-ms exposure time. The calibration bar in the bright-field microscope image is 5 μm. The bottom trace indicates the level of negative pressure (suction). The format used in this figure is also used for Figs. 3, 4, and 5. The bath solution was the standard solution with 1 μM thapsigargin. The pipette solution was the standard solution.
Such localized fluorescence transients were observed from cells subjected to different treatments designed to eliminate the effects of intracellular Ca2+ stores. These treatments included the following: cells bathed in the standard solution with the addition of 1 μM thapsigargin (24 recordings from 13 cells) see, e.g., Fig. 2; cells bathed in the standard solution with the addition of 1 μM thapsigargin and 100 μM ryanodine (6 recordings from 4 cells); cells bathed in the Ca2+-free solution without thapsigargin (9 recordings from 4 cells); and cells bathed in the Ca2+-free solution with 1 μM thapsigargin (38 recordings from 17 cells). Caffeine (20 mM) was applied to all of these cells before forming the cell-attached patch to empty intracellular Ca2+ stores. These experiments were carried out with the patch membrane potential set at or more negative than the cell membrane potential.
Because Kirber et al. (8) did not detect a fluorescence increase with fura-2 when SAC openings occurred without amplification from Ca2+ stores, experiments were also carried out under the conditions used by these authors, but with fluo-3 as the Ca2+ indicator. Cells were bathed in the Ca2+-free solution, with the addition of 0.5 mM caffeine and 100 μM ryanodine, for longer than 30 min before beginning the experiments. Like the results reported above, when SACs opened, localized fluorescence transients were evident and their time course matched the current profile (8 recordings from 3 cells, not shown).
Taken together, all of these results suggest that these localized fluorescence increases were due solely to Ca2+ entry through SACs.
Ca2+ Entry Through SACs Contributes to Global Increases in Intracellular Ca2+.
To examine the effect of SAC opening under more physiological conditions, negative pressure was applied to cell-attached patches at the resting membrane potential, in the absence of any agents affecting intracellular Ca2+ stores. Fig. 3A shows an example when the standard solution (containing 2 mM Ca2+) was used as both the pipette solution and the bath solution. When SACs opened, the fluorescence increase initially appeared locally around the pipette tip, and then usually spread to the adjacent regions of the cell (as also indicated in Fig. 2). As more SAC openings occurred, a fluorescence increase occurred everywhere, almost simultaneously, due to Ca2+ entry through VGCCs caused by the depolarization of the cell membrane (11 recordings from 4 cells; also see ref. 8). The near-simultaneous opening of plasma membrane VGCCs would be expected because the high membrane resistance usually found in these cells causes the membrane to behave as an isopotential surface (8, 16). There are other recordings (4 recordings from 3 cells), however, where opening of SACs did not cause an obvious global Ca2+ increase. Whether cell membrane depolarization and global increases in Ca2+ occur when SACs are opened will depend on a number of factors: the number of SACs that open, the size of the cell, the activity of other plasma membrane ion channels, and the membrane potential before applying suction.
Figure 3.

Openings of SACs can lead to membrane depolarization and global Ca2+ increases. (A) Both local and global fluorescence increases were observed when SACs opened with Ca2+ present in the pipette solution. Localized fluorescence transients at the tip of the pipette (location c, see images) appeared as soon as the SACs opened (trace c). This was followed by a delayed, near-simultaneous increase in fluorescence throughout the cell (see traces from locations a, b, and d). The latter most likely occurred from the opening of VGCCs in response to the membrane depolarization (indicated by the decrease in the unitary current amplitude) caused by the inward SAC currents. The fluorescence transient at the pipette tip remained higher than the rest of the cell, presumably because of the additional Ca2+ entry through SACs. (B) Only global, not local, fluorescence increases were observed when SACs opened in the absence of Ca2+ in the pipette solution. In contrast to when Ca2+ was present in the pipette solution, there was no fluorescence increase at the tip of the pipette when SAC openings occurred. However, a simultaneous global fluorescence increase could still be observed, suggesting the occurrence of membrane depolarization and Ca2+ entry through VGCC. The brief transient at location d toward the end of the recording may be due to a spontaneous release of Ca2+ from intracellular Ca2+ stores (13), which are presumably intact under these experimental conditions; or it may be due to Ca2+ influx through a spontaneous opening of a Ca2+-permeable channel. Patch membrane potential is the same as the cell membrane potential for A and 100 mV more negative for B. Fluorescence images were acquired every 30 ms with a 6-ms exposure time for A and 5 ms for B. The calibration bar in the bright-field microscope image is 5 μm. The bath solution for both experiments was the standard solution, whereas the pipette solution was the standard solution for A and the Ca2+-free solution for B. There were no pharmacological agents present affecting intracellular Ca2+ stores.
As a control, experiments were also carried out with the Ca2+-free solution (containing 200 μM BAPTA and no Ca2+) in the patch pipette to eliminate Ca2+ entry through the patch membrane. When SAC openings occurred, there was, as expected, no fluorescence increase observed (7 recordings from 3 cells). With other recordings from the same three cells, sometimes an almost simultaneous global fluorescence increase was observed (Fig. 3B, 8 recordings) after the initial SAC openings. However, the localized fluorescence transients at the pipette tip observed with Ca2+ in the pipette solution were absent. These results are in agreement with the idea that without Ca2+ entry, the cation (mainly Na+) current through SAC openings could cause depolarization of the cell membrane and bring Ca2+ into the cell through VGCCs throughout the cell membrane. Results similar to those shown in Fig. 3 A and B were also reported by Kirber et al. (8) with a much smaller image set and much lower time resolution, so that the time delay between the focal and global fluorescence increases was not evident.
A Localized Fluorescence Transient Caused by Ca2+ Entry Through a Single SAC Opening Can Be Detected.
Discrete localized increases in fluorescence were observed around the pipette tip, but not elsewhere, during single openings of a SAC in the cell-attached patch (Fig. 4, and also Fig. 1). These experiments were carried out with cells bathed in the Ca2+-free solution and treated with thapsigargin and caffeine to eliminate effects of Ca2+ stores. Therefore, the only source of Ca2+ was the patch pipette solution containing 2 mM Ca2+. Each fluorescence transient corresponds to a SAC opening, suggesting these transients are single channel Ca2+ fluorescence transients [SCCaFTs (12)] caused by Ca2+ entry through SACs. Similar results were also obtained in other Ca2+-store-eliminating solutions: (i) Ca2+-free bathing solution in the absence of thapsigargin, (ii) standard bathing solution with 1 μM thapsigargin, and (iii) standard bathing solution with 1 μM thapsigargin and 100 μM ryanodine. The SCCaFTs were seen in at least 20 recordings from 12 cells, including recordings without the patch membrane potential being more negative than the cell membrane potential.
Figure 4.

Single channel Ca2+ fluorescence transients (SCCaFTs) from openings of SACs. When discrete SAC openings occurred, localized fluorescence transients appeared at the pipette tip. The time course of the rising phase of the transients corresponds to the duration of the channel openings (see ref. 12). Patch membrane potential was held 100 mV more negative than the cell membrane potential to enhance Ca2+ entry. Fluorescence images were acquired continuously with a 10-ms exposure time. The calibration bar in the bright-field microscope image is 5 μm. The Ca2+-free solution containing 1 μM thapsigargin was used in the bath solution, and caffeine had been applied to the cell. The pipette solution was the standard solution.
Normally, the pattern for fluorescence increases was somewhat spatially symmetric, suggesting isotropic movement away from a point source (i.e., a single SAC). The full spatial width at half maximal amplitude (FWHM), measured parallel to the long axis of the cell at about 15 ms after the channel opened, was 2.22 ± 0.14 μm (6 transients from 3 cells). The FWHM for these SAC SCCaFTs is very close to what we found for the caffeine-activated channel SCCaFTs (see caption to figure 5 in ref. 12).
Estimating the Fraction of the Total SAC Current Carried by Ca2+.
To quantitatively determine whether significant Ca2+ enters the cell during SAC openings, the fraction of the SAC current carried by Ca2+ was calculated based on the measurements of total fluorescence as described in Methods (and Fig. 1). The conversion factor, 1.72 Ca ions per detected photon (determined by using the 90 mM Ca2+ pipette solution), was applied to cells where we used the standard pipette solution (containing 2 mM Ca2+), assuming the loading of fluo-3, the buffering capacity, and the resting intracellular Ca2+ levels were similar for the two groups.
On average, we found that 17.8 ± 2.0% (9 transients from 5 cells) of the SAC current was carried by Ca2+. Based on a unitary SAC current of about 2 pA at a resting membrane potential of near −56 mV (17), the unitary SAC Ca2+ current is at least 0.35 pA (see Discussion). This current is larger than, but close to, the value (near 0.2 pA) determined by Rubart et al. (18) for L-type Ca2+ channels at approximately the same potential with 2 mM Ca2+ in the pipette solution.
Because the membrane patch was not voltage clamped, the percent of the SAC current carried by Ca2+ was determined at different patch membrane potentials (50, 100, or 150 mV more negative than the cell membrane potential). However, there was no correlation found between the fraction of the Ca2+ current obtained from different transients and the amplitude of the unitary SAC current (R2 = 0.014). Therefore, over the range of patch membrane potentials that occurred during our experiments, the fraction of the SAC current that is carried by Ca2+ appears to be relatively independent of this parameter.
Ca2+ Influx Through SACs Opens BKCa Channels in the Same Patch.
Ca2+ entering the cell through SACs is sufficient to cause a substantial local increase in the free [Ca2+] (see discussion), as indicated by large-conductance Ca2+-activated K+ (BKCa) channel (19) openings in the same patch (Fig. 5). Application of suction opened SACs, causing an increase in [Ca2+], which in turn led to openings of BKCa channels. These results (19 recordings from 6 cells) were obtained in a Ca2+-free high [K+] bathing solution with the patch membrane potential held at −55 mV, and with the effect of intracellular Ca2+ stores eliminated. These conditions were used to remove all sources of Ca2+, except that which was in the pipette solution, and to hold the patch potential constant near the cell resting potential to prevent possible voltage activation of BKCa channels. To observe BKCa channel openings at −55 mV and make them distinguishable from SAC openings, Na+ was replaced with K+ in the pipette solution. BKCa channels were not activated when similar experiments were carried out with a Ca2+-free (high-[K+]) pipette solution (9 recordings in 3 cells). Therefore, BKCa channels in toad stomach smooth muscle cells do not appear to be directly stretch-activated, at least under the above experimental conditions, which is in agreement with the findings in this preparation by Kirber et al. (4).
Figure 5.

Opening of SACs causes sufficient Ca2+ influx to open BKCa channels in the same patch. The application of negative pressure first caused the opening of SACs (2.2-pA unitary currents), which then raised the [Ca2+] sufficiently to open BKCa channels [identified by the 8.9 pA unitary current, open times, and Ca2+ sensitivity (see text and ref. 19)]. As the Ca2+ fluorescence declined with the subsequent cessation of suction (and closing of SACs), the BKCa channel activity also ceased. A section of the current trace is enlarged to more clearly display the sequential openings of SACs and BKCa channels. The Ca2+-free bath solution (with 1 μM thapsigargin) was used here, but with K+ replacing Na+ to zero the cell membrane potential and thereby effectively clamp the patch membrane potential to −55 mV. Na+ was replaced by K+ in the pipette solution so that BKCa channel activity could be observed at this potential. Under these conditions, the unitary BKCa channel current was much larger than the unitary SAC current. Images were acquired every 30 ms with a 10-ms exposure time. The calibration bar in the bright-field microscope image is 5 μm.
Discussion
We report here the first recording of SCCaFTs attributable to openings of single SACs, demonstrating that significant amounts of Ca2+ can enter the cytoplasm through SACs with physiological solutions. We also provide direct evidence that the opening of SACs, in addition to causing depolarization of the cell membrane to open VGCCs and increase global Ca2+, contributes to the local Ca2+ increase by directly passing Ca2+ into the cytoplasm. This local increase in Ca2+ can be clearly visualized, at high temporal and spatial resolution, before or in the absence of the simultaneous increase in global Ca2+. It can also serve as a signal for carrying out important physiological functions, such as activating BKCa channels (Fig. 5) and triggering Ca2+ release from intracellular stores in smooth muscle (8). The fraction of current carried by Ca2+ through SACs and the Ca2+ current at the resting membrane potential are also estimated.
Imaging Ca2+ Entry Through a Single Opening of SACs – SCCaFTs.
As a fundamental event in Ca2+-signaling, the SCCaFT or Ca2+ entry through a single-channel opening has its unique significance for studying Ca2+ handling in microdomains (20). So far, there have been two other reports of SCCaFTs: SCCaFTs from caffeine-activated cation channels (12) and, by using confocal microscopy, L-type VGCCs (21). The latter study (21) used the term “sparklet” for the SCCaFT recordings, and used 20 mM Ca2+ in the pipette solution, much larger than the physiological levels of Ca2+ (2 mM) used for caffeine-activated channels and SACs.
Estimating Ca2+ Entry Through SACs.
For our estimate of the fraction of the SAC current carried by Ca2+, the most crucial assumption was that the fluo-3 concentration was similar in all of the cells. This was important if we were to capture approximately the same fraction of the Ca2+ that passed through the SAC channels for each cell. Even though the same fluo-3 acetoxymethyl ester loading conditions were used for our studies, based on our estimates of the FLn/V, there was a certain amount of variation in the fluo-3 concentration (see Methods). Some of this variation may be due to our estimates of the volume of the cylindrical section of the cells. Instead of being a cylinder, some cells could have had a somewhat ellipsoidal cross section. Also, the resting [Ca2+] may not have been the same in all of the cells. The value of 17.8 ± 2.0% of the SAC current being carried by Ca2+ was obtained when we chose data where the estimates of FLn/V were within the range indicated in Methods. If the fluo-3 loading was much less variable than that suggested by our measurements of FLn/V, then we could have used 19 transients from 11 cells (instead of 9 transients from 5 cells). The fraction of the SAC current carried by Ca2+ would then be 24.2 ± 2.8%. Although this value is significantly different from 17.8%, both sets of data indicate a much larger fraction of the SAC current being carried by Ca2+ than has been heretofore estimated in the literature.
Based on the unitary SAC current of about 2 pA at a resting membrane potential of near −56 mV, the unitary SAC Ca2+ current is at least 0.35 pA. This is larger than that estimated by Sachs and his colleagues, 0.02 pA at −60 mV in oocytes (9, 22). The value for the percent of the current carried by Ca2+ (≈18%) is also larger than the estimate of 5% in bladder smooth muscle cells by Wellner and Isenberg (6), based on constant field equations. These investigators did not measure the Ca2+ influx for their estimates. Our value is in the upper range for other nonselective cation channels where Ca2+ fluxes were measured (23–25) but lower than for some cyclic-nucleotide-gated channels (25). The fraction of the current carried by Ca2+ at the resting membrane potential might be expected to be somewhat larger than what we obtained at more negative potentials (making the estimated Ca2+ current at the resting potential somewhat larger as well), because the reversal potential for the Ca2+ current would be more positive than that for SAC cation current as a whole.
From the pattern of fluorescence increase when a SAC opens, the diffusion of Ca2+-bound fluo-3 appears as expected for diffusion from a point source or single channel. To determine what happens to the local [Ca2+] in the absence of fluo-3, we used computer simulations of the underlying events with a 0.356-pA Ca2+ current and a 230 μM fixed buffer (for details see ref. 12). The results revealed that the free [Ca2+] at 50 nm from the channel rises to about 20 μM in 1 ms, and about 23 μM in 20 ms. At a distance of 150 nm from the channel, it rises to about 2.7 μM in 1 ms and 5 μM in 20 ms. If we included in the simulation a 10 μM mobile buffer [with an on-rate of 80 (μM⋅s)−1, an off-rate of 90 s−1, and a diffusion constant of 2.2⋅10−7 cm2⋅s−1], the Ca2+ concentrations were diminished by, at most, 6%. This analysis shows that a significant [Ca2+] increase occurs quickly in a relatively extended neighborhood of the channel.
Physiological Significance for Ca2+ Entry Through SACs.
The present study gives experimental support to the suggestion made by Kirber et al. (8) that Ca2+ entry through SACs is strongly amplified by ryanodine-sensitive Ca2+ release, the amplification being greater than for VGCC in the same cell type. Because Ca2+ increases in the absence of intracellular Ca2+ stores were not detected by using fura-2, the observed local Ca2+ increase was attributed mainly to Ca2+ release from intracellular stores triggered by Ca2+ entry through SACs. This Ca2+ entry has now been visualized in this study, and it occurred as soon as the SACs opened, independent of the status of the Ca2+ stores (Figs. 1, 2, and 4).
When using a pipette solution containing normal [Ca2+], but a higher [K+], openings of BKCa channels were observed after the SAC openings and the accompanying increase in fluo-3 fluorescence (see Fig. 5). These results suggest that Ca2+ entry through SACs can activate BKCa channels in the same patch. Therefore, the localized [Ca2+] in a microdomain might play a significant modulating role on adjacent effectors.
Without monitoring intracellular Ca2+, other investigators (26–30) have reported a similar relationship between SACs and BKCa channels in various cell types. However, the contribution of intracellular Ca2+ stores was not eliminated in those studies. Therefore, the activation of BKCa in those studies cannot be explicitly attributed only to Ca2+ entry through SACs. In addition, higher concentrations of extracellular Ca2+ were used in some of those studies.
The sequential activation of SACs and BKCa channels can regulate smooth muscle function by either suppressing or enhancing smooth muscle contraction. The activation of SACs increases the local [Ca2+] and causes membrane depolarization, which can activate VGCCs, leading to contraction. The activation of BKCa channels, however, causes membrane hyperpolarization, which can cause relaxation by deactivating VGCC; but hyperpolarization with maintained membrane stretch could enhance Ca2+ influx through open SACs, favoring contraction.
In conclusion, because of our imaging capabilities, we were able to record the Ca2+ influx associated with a single opening of a SAC, and to determine that there is much more Ca2+ passing through these channels than was estimated previously from ion replacement studies. Moreover, we have been able to clearly demonstrate the delayed, near-synchronous increase in global Ca2+ because of membrane depolarization induced by just a few SAC openings.
Acknowledgments
We thank Michael T. Kirber for his help and participation in some of the initial experiments for this study; Jeff Carmichael, Rebecca Mckinney, and Paul Tilander for their excellent technical assistance; and Karl Bellvé for the customized software. We also thank Robert Drummond, Agustín Guerrero-Hernández, Michael Kirber, and Michael Sanderson for their comments on earlier versions of this manuscript. This work was supported by National Institutes of Health Grants AR47067, DK31620, and HL47530.
Abbreviations
- SAC
stretch-activated channel
- VGCC
voltage-gated Ca2+ channel
- SCCaFT
single-channel Ca2+ fluorescence transient
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate
- BKCa channel
large-conductance Ca2+-activated K+ channel
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Zou, H., Lifshitz, L. M., Kirber, M. T., Tuft, R. A., Fogarty, K. E. & Singer, J. J. (2001) Biophys. J. 80, 112a (abstr.).
References
- 1.Morris C E. J Membr Biol. 1990;113:93–107. doi: 10.1007/BF01872883. [DOI] [PubMed] [Google Scholar]
- 2.Sachs F. Soc Gen Physiol Ser. 1992;47:241–260. [PubMed] [Google Scholar]
- 3.Hamill O P, Martinac B. Physiol Rev. 2001;81:685–740. doi: 10.1152/physrev.2001.81.2.685. [DOI] [PubMed] [Google Scholar]
- 4.Kirber M T, Walsh J V, Jr, Singer J J. Pflügers Arch. 1988;412:339–345. doi: 10.1007/BF01907549. [DOI] [PubMed] [Google Scholar]
- 5.Davis M J, Donovitz J A, Hood J D. Am J Physiol. 1992;262:C1083–C1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- 6.Wellner M C, Isenberg G. J Physiol. 1993;466:213–227. [PMC free article] [PubMed] [Google Scholar]
- 7.Meininger G A, Davis M J. Am J Physiol. 1992;263:H647–H659. doi: 10.1152/ajpheart.1992.263.3.H647. [DOI] [PubMed] [Google Scholar]
- 8.Kirber M T, Guerrero-Hernandez A, Bowman D S, Fogarty K E, Tuft R A, Singer J J, Fay F S. J Physiol. 2000;524:3–17. doi: 10.1111/j.1469-7793.2000.t01-4-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sigurdson W, Ruknudin A, Sachs F. Am J Physiol. 1992;262:H1110–H1115. doi: 10.1152/ajpheart.1992.262.4.H1110. [DOI] [PubMed] [Google Scholar]
- 10.Lassignal N L, Singer J J, Walsh J V., Jr Am J Physiol. 1986;250:C792–C798. doi: 10.1152/ajpcell.1986.250.5.C792. [DOI] [PubMed] [Google Scholar]
- 11.Fay F S, Hoffmann R, Leclair S, Merriam P. Methods Enzymol. 1982;85:284–292. doi: 10.1016/0076-6879(82)85027-1. [DOI] [PubMed] [Google Scholar]
- 12.Zou H, Lifshitz L M, Tuft R A, Fogarty K E, Singer J J. J Gen Physiol. 1999;114:575–588. doi: 10.1085/jgp.114.4.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ZhuGe R, Tuft R A, Fogarty K E, Bellve K, Fay F S, Walsh J V., Jr J Gen Physiol. 1999;113:215–228. doi: 10.1085/jgp.113.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun X P, Callamaras N, Marchant J S, Parker I. J Physiol. 1998;509:67–80. doi: 10.1111/j.1469-7793.1998.067bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ZhuGe R, Fogarty K E, Tuft R A, Lifshitz L M, Sayar K, Walsh J V., Jr J Gen Physiol. 2000;116:845–864. doi: 10.1085/jgp.116.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singer J J, Walsh J V., Jr Am J Physiol. 1980;239:C153–C161. doi: 10.1152/ajpcell.1980.239.5.C153. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi H, Honeyman T W, Fay F S. Am J Physiol. 1988;254:C423–C431. doi: 10.1152/ajpcell.1988.254.3.C423. [DOI] [PubMed] [Google Scholar]
- 18.Rubart M, Patlak J B, Nelson M T. J Gen Physiol. 1996;107:459–472. doi: 10.1085/jgp.107.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singer J J, Walsh J V., Jr Pflügers Arch. 1987;408:98–111. doi: 10.1007/BF00581337. [DOI] [PubMed] [Google Scholar]
- 20.Berridge M J. J Physiol. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S Q, Song L S, Lakatta E G, Cheng H. Nature (London) 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 22.Yang X C, Sachs F. Science. 1989;243:1068–1071. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]
- 23.Neher E. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 24.Burnashev N. Cell Calcium. 1998;24:325–332. doi: 10.1016/s0143-4160(98)90056-2. [DOI] [PubMed] [Google Scholar]
- 25.Ohyama T, Hackos D H, Frings S, Hagen V, Kaupp U B, Korenbrot J I. J Gen Physiol. 2000;116:735–753. doi: 10.1085/jgp.116.6.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen O. Nature (London) 1987;330:66–68. doi: 10.1038/330066a0. [DOI] [PubMed] [Google Scholar]
- 27.Filipovic D, Sackin H. Am J Physiol. 1991;260:F119–F129. doi: 10.1152/ajprenal.1991.260.1.F119. [DOI] [PubMed] [Google Scholar]
- 28.Erxleben C F. NeuroReport. 1993;4:616–618. doi: 10.1097/00001756-199306000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Hoyer J, Distler A, Haase W, Gogelein H. Proc Natl Acad Sci USA. 1994;91:2367–2371. doi: 10.1073/pnas.91.6.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin K S, Park J Y, Ha D B, Chung C H, Kang M S. Dev Biol. 1996;175:14–23. doi: 10.1006/dbio.1996.0091. [DOI] [PubMed] [Google Scholar]