

Abstract
Antibiotic use is known to promote the development of antibiotic resistance, but substantial controversy exists about the impact of agricultural antibiotic use (AAU) on the subsequent emergence of antibiotic-resistant bacteria among humans. AAU for animal growth promotion or for treatment or control of animal diseases generates reservoirs of antibiotic-resistant (AR) bacteria that contaminate animal food products. Mathematical models are an important tool for understanding the potential medical consequences of this increased exposure. We have developed a mathematical model to evaluate factors affecting the prevalence of human commensal AR bacteria that cause opportunistic infections (e.g., enterococci). Our analysis suggests that AAU hastens the appearance of AR bacteria in humans. Our model indicates that the greatest impact occurs very early in the emergence of resistance, when AR bacteria are rare, possibly below the detection limits of current surveillance methods.
The development of antibiotic resistance (AR) among pathogenic bacteria has emerged as a major public health concern. The appearance of AR has been directly linked with the use and overuse of antibiotics (1–4). It was reported that as much as 80% of total antibiotic production in the United States is used in agriculture, with a substantial portion of this used for the nontherapeutic purpose of growth promotion (4, 5). AR bacteria have been found in farm animals where antibiotics are heavily used (6–8), in associated food products (9, 10), in environments contaminated by animal waste (11, 12), and in farm workers (13–15). Drugs that are used therapeutically in animals also may generate a reservoir of AR bacteria (16, 17). AR bacteria in food animals threaten the efficacy of human drugs if AR bacteria or AR genes become incorporated into bacteria populations colonizing humans. To provide a basis for public policy discussions about agricultural antibiotic use (AAU), we have developed a mathematical model to quantify the medical consequences.
AAU may cause AR bacterial infections in humans by two different processes. First, AAU increases the frequency of AR in zoonotic pathogens such as Campylobacter or Salmonella. These pathogens are typically acquired through exposure to contaminated animal food products. Human-to-human transmission of zoonotic pathogens is rare, although it may occur in settings where humans are immuno-compromised or where the gut community has been disturbed by heavy medical antibiotic use (MAU; ref. 18). Therefore, the incidence of AR in zoonotic infections of humans is directly related to the prevalence of AR bacteria in food animals. A risk-assessment model examining resistance in a zoonotic pathogen was recently proposed by FDA (see http://www.fda.gov/cvm/antimicrobial/Risk_asses.htm).
Second, AR bacteria from food animals may facilitate the development of AR in human commensal bacteria which ordinarily colonize humans without causing infection. Commensal bacteria typically have long persistence times, frequent human-to-human transmission, and high bacterial loads that are associated with good health but not disease. Commensal bacteria occasionally cause opportunistic infections such as wound or bloodstream infections; the bacteria causing these infections may have originated from the normal flora inhabiting the infected human's gut or from contact with another human. In either case, the risk that a human is infected with commensal AR bacteria increases with the prevalence of AR in the human population. Correctly attributing increases in medical risks associated with AR in commensal bacteria caused by AAU is difficult because infection may be an indirect consequence of exposure.
We have used a mathematical model to evaluate the medical impacts of simultaneously using the same antibiotic in food animals and medicine. The model is based on the ecology of enterococci, a medically important organism for which high-level antibiotic resistance has been linked with AAU (19, 20). The principles also apply to the spread of other commensal AR bacteria.
Mathematical Model
We have developed a model for the population prevalence of human commensal AR bacteria. Commensal bacteria may persist in the gut for a few days or several years depending on whether the population colonizes or, in other words, depending on whether it establishes a persistent population. Exposure to AR bacteria may be quite common but transient, whereas colonization is rare but persistent. Once a population colonizes, population densities may fluctuate over time varying by several orders of magnitude. These fluctuations may be driven, in part, by MAU.
To develop a simple model of commensal bacteria, we have made several simplifying assumptions. We assume that humans are in one of four categories with respect to AR bacteria: unexposed, exposed, colonized, or amplified. Colonized and exposed individuals are assumed to carry relatively low loads. Humans with populations amplified by antibiotic use carry high loads and are highly contagious. We have assumed that human population size is constant; the variables represent the fraction of humans in each state over time: W, X, Y, and Z, respectively. Prevalence is denoted P = 1 − W = X + Y + Z.
Persistence of Bacterial Populations.
Humans are exposed to new strains of AR commensal bacteria from food, water, or contact with other humans. Total prevalence of AR bacteria in a population depends, in part, on the natural turnover rates of AR bacteria in humans. We have assumed that bacteria establish transient populations after exposure, but some populations colonize and persist for much longer. We assume that transient populations are lost at a per capita rate α or colonize at a lower per capita rate θ. Once the population colonizes, it may persist for years. Colonized populations are lost at very low per capita rate σ.
Medical Antibiotic Use.
We assume that MAU alters the community ecology of gut, eliminating competition with antibiotic-sensitive bacteria and allowing the population density of AR bacteria to increase. In some cases, the amplification of population densities could occur by reductions in the population densities of species that were not targeted by the drug treatment. Thus, several antibiotics may influence the emergence of resistance to a focal antibiotic (21). After treatment ends, we assume that the gut eventually returns to its former state, and AR bacteria either recolonize or the populations are lost.
To quantify prescription, amplification, and subsequent reduction of population densities, we let ρ denote the per capita rate that humans are prescribed an antibiotic that amplifies the AR bacteria populations. In those individuals who were colonized or exposed, the AR bacteria increase in density; we call these populations amplified. The rate that AR bacteria are shed into the environment and the contagiousness increase. We assume that amplified populations revert to low-density colonized populations at a per capita rate φ, and they are lost at a per capita rate γ. Because we have assumed that transient bacteria can be amplified by antibiotics, MAU leads to increased colonization.
Horizontal Transmission.
Commensal bacteria are frequently spread among humans by human-to-human contact. To model horizontal transmission, we have assumed populations are well mixed, and horizontal transmission occurs at a higher rate because of contact with a person who has amplified-population densities compared with a person with lower-population densities. The per capita rate of exposure from human-to-human contact is βZ + ηY, where βZ is the contact rate with individuals carrying amplified populations, and ηY is the contact rate with colonized individuals, and β ≫ η.
The Origin of New Resistant Strains.
AAU does not directly select for AR bacteria in humans, except possibly through antibiotic residues on food (5). On the other hand, animal food products often are contaminated with AR bacteria (22). Exposure to AR bacteria may have medical impacts if AR bacteria or AR genes establish in the bacteria of humans as a result of this contamination.
New strains of AR bacteria of humans may evolve in several ways. Human commensal bacteria under selection by MAU may become AR by accumulating multiple-point mutations. This evolutionary process may not account for some clinical isolates with high-level AR; instead, the resistance genes probably originated elsewhere and had been acquired by human commensal bacteria. Genetic elements conferring AR may have been transferred from another bacteria species on mobile genetic elements through bacterial sex. Alternatively, bacteria associated with another host species or environment may have crossed the species barrier and adapted to the human gut. The transfer of high-level AR genes from bacteria in animals to bacteria in humans and the transfer of whole bacteria of animal origin into the human gut are two different ways that new AR bacteria may appear in humans. We have made the simplifying assumption that introducing a new AR strain affects prevalence regardless of whether whole organisms or only genetic material was transferred; thus, our model considers both kinds of evolutionary events.
We assume that new AR strains evolve in humans, with or without AAU. To understand the medical impacts of AAU, it is necessary to estimate how much more frequently these events occur with AAU. To quantify the process, we let μ + λ denote the per capita rate of exposure to new AR strains; μ represents the background rate, and λ represents the increased rate of exposure because of AAU. These rates quantify all evolutionary events that expose humans to new strains with AR from sources other than human-to-human transmission. To estimate the impact, it is not necessary to understand how the strains originate, only how much more often they occur because of AAU.
Equations.
Under these assumptions, the changes in prevalence over time are modeled by a simple set of coupled ordinary differential equations:
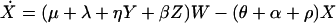 |
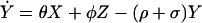 |
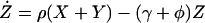 |
1 |
A diagram of the model is found in Fig. 1.
Figure 1.

New strains of high-level resistant bacteria enter the human population because of background processes at rate μW or AAU at rate λW, where the fraction of humans carrying no resistant bacteria is W = 1 − X − Y − Z. Initially, humans are exposed (X), and the populations are transient. Exposed populations colonize (Y) at per capita rate θ or are lost at the per capita rate α. Colonized populations are lost at the much lower per capita rate σ < α. Both transient and persistent populations are assumed to have low-population densities, but antibiotic use in humans generates amplified population densities (Z) at the population rate ρ(X + Y). Once amplified, populations are lost at the per capita rate γ and recolonize at the per capita rate φ. Human-to-human transmission occurs at the rate βZW + ηYW, where the rate is higher after contact with amplified populations than colonized populations (β > η).
Results
Secondary Transmission (R0).
R0 is the number of humans who are exposed by a single exposed human when prevalence in humans is approximately zero. As with other models for the population dynamics of AR bacteria, R0 = 1 is an epidemic threshold; above the threshold, a new AR strain spreads and persists (23, 24). In this model, new strains are introduced from other sources (μ + λ > 0), so AR bacteria are always present in the population, regardless of whether an individual AR strain persists. New strains spread epidemically if each exposed human exposes at least one other human or, in other words, if R0 > 1.
In this model, transient bacterial populations in humans do not expose other humans unless the populations colonize or become amplified by MAU, but a single exposed individual may become colonized, amplified, and recolonized multiple times. In a naive population, a colonized individual would expose η other humans per day for 1/(σ + ρ) days, on average. Amplified humans expose β humans per unit time and stay colonized for 1/(γ + φ) days. Thus, a simple expression for R0 is
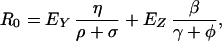 |
2 |
where EY and EZ are the expected number of times that a single exposed individual is colonized or amplified (see Appendix for details).
The two terms, EY and EZ, are functions of the rate of MAU and parameters that determine the natural turnover rates of bacteria in humans after exposure. R0 is also influenced by transmission rates, especially β. Both MAU rates, ρ, and transmission, β, are amenable to control. Equally important, R0 is unaffected by changes to λ, the rate of exposure because of AAU. In other words, AAU increases the rate that new strains are introduced, but it does not affect subsequent transmission.
Equilibrium Prevalence.
Like other models for the prevalence of AR, if R0 > 1, the prevalence of resistance over time follows a sigmoidal curve and asymptotically approaches an equilibrium (Fig. 2A). Although AAU does not influence R0, it does affect the prevalence of AR bacteria in a human population over time. A simple measure of the impact is the difference in human prevalence at the steady state. We define four equilibria under conditions with and without MAU and with and without AAU: the unregulated equilibrium with both MAU and AAU, denoted 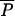 (ρ,λ); the regulated equilibrium with MAU but no AAU, denoted
(ρ,λ); the regulated equilibrium with MAU but no AAU, denoted 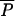 (ρ,0); the agricultural equilibrium with AAU but no MAU, denoted
(ρ,0); the agricultural equilibrium with AAU but no MAU, denoted 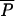 (0,λ); and the pristine equilibrium, with no MAU and no AAU,
(0,λ); and the pristine equilibrium, with no MAU and no AAU, 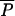 (0,0).
(0,0).
Figure 2.

(A) The projected prevalence of AR over time with AAU (dotted line) and without AAU (solid line). AAU does not change the equilibrium prevalence dramatically, but it does cause AR to invade 3 years sooner. The cumulative excess prevalence is the area between these two curves. The medical consequences of AAU are contrasted with a 50% increase in the rate of MAU (dashed line). Increasing MAU use makes resistance invade earlier and reach a higher equilibrium. The parameter values correspond to Fig. 1. (B) The equilibrium prevalence is very sensitive to changes in the rate of MAU (ρ, solid line) relative to changes in the rate of exposure caused by AAU (λ, dashed line). The equilibrium was computed by multiplying the two parameters by a sensitivity factor, s, ranging from 0.5 to 2.0. At the square, R0 = 1. The parameters at the circle correspond to Fig. 1. (C) The cumulative excess prevalence caused by AAU declines dramatically if exposure resulting from AAU is delayed. The longer AAU is delayed, the lower the impact. The units are scaled to the maximum impact. Each point, t, represents a numerical simulation identical to the top panel in every way except one. For each point, λ = 0 before t.
If a drug is approved for use in agriculture first, and subsequently approved for use in humans, regulation of AAU may not be expected to affect equilibrium prevalence of resistance in the human population, provided that R0 > 1. Eliminating all AAU would be expected to change prevalence slightly by reducing the rate that new strains are introduced, but the reduction would be smaller than the agricultural equilibrium because it can be shown that 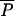 (0,λ) >
(0,λ) > 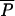 (0,λ) −
(0,λ) − 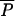 (0,0) >
(0,0) > 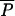 (ρ,λ) −
(ρ,λ) − 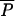 (ρ,0). Intuitively, when R0 > 1, human-to-human transmission is responsible for the high prevalence of AR bacteria near equilibrium. Introducing new AR strains into a population near equilibrium causes fewer new cases than when prevalence is low, because many new contacts are already carrying some AR bacteria from previous human-to-human contact. According to the model, if an AAU had occurred for decades, and the agricultural equilibrium was 1%, regulating AAU after increases in the rate of MAU would not change the equilibrium prevalence by more than 1% compared with the unregulated equilibrium.
(ρ,0). Intuitively, when R0 > 1, human-to-human transmission is responsible for the high prevalence of AR bacteria near equilibrium. Introducing new AR strains into a population near equilibrium causes fewer new cases than when prevalence is low, because many new contacts are already carrying some AR bacteria from previous human-to-human contact. According to the model, if an AAU had occurred for decades, and the agricultural equilibrium was 1%, regulating AAU after increases in the rate of MAU would not change the equilibrium prevalence by more than 1% compared with the unregulated equilibrium.
It is possible that the agricultural equilibrium is relatively high (greater than 5%, for example) only because of high exposure to new strains on contaminated animal food products. The probability of colonizing, θ/(α + θ), the average persistence times of colonized bacteria, 1/σ, and the contact rate with colonized humans, η, affect how long each strain persists in a population in the absence of MAU. High prevalence of AR bacteria is associated with long persistence times, or values of R0 (computed with ρ = 0) that are slightly less than one. Long persistence times may be associated with microbial strains with low biological cost of AR (25). High exposure combined with long persistence times of these strains may explain why the community prevalence of AR bacteria in some areas is as high as 12% despite moderate levels of MAU (14, 26).
Time to Emergence.
AAU may have a large impact on prevalence in humans over time, even if it does not cause large changes in the equilibrium prevalence of AR bacteria. The medical impacts of AAU are assessed by summing the extra prevalence in a human population with AAU compared with a human population without AAU. It is possible that AAU can have a large cumulative impact on the development of AR by introducing high-level AR bacteria into the human population sooner than they would have appeared otherwise.
Assuming that R0 > 1, the equilibrium prevalence is determined largely by the rate of human-to-human transmission. Because AR strains arise from natural processes (μ > 0), the selective pressure provided by MAU guarantees that AR bacteria will eventually appear and approach equilibrium. Although AAU shifts the curve, the basic shape of the sigmoidal approach to equilibrium does not change much regardless of exposure because of AAU (Fig. 2A). The two curves are different in exponentially increasing phase, when prevalence is initially very low.
It may take a very long time for the first AR strain to appear, and AR bacteria may remain rare for years. Epidemic spread is initially exponential, but if prevalence is low, the rate of increase is slow in absolute terms. If λ is large compared with the rate of epidemic spread near the pristine equilibrium, AR bacteria may appear and spread several years earlier than they would without AAU (Fig. 2A). The amount of time “wasted” by AAU is approximately the time it would take the natural epidemic process to reach the agricultural equilibrium starting from the pristine equilibrium. As with other exponential growth processes, the time to increase by some ratio is proportional to the logarithm of the ratio [for example, doubling times are proportional to log(2)]. In this case, the time lost because of AAU is proportional to log[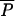 (0,λ)/
(0,λ)/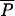 (0,0)] ≈ log(λ/μ). We have not found a simple and intuitive approximation to the amount of time lost involving all of the parameters, but numerical solutions to equations [1] verify that it is approximately proportional to log(λ/μ).
(0,0)] ≈ log(λ/μ). We have not found a simple and intuitive approximation to the amount of time lost involving all of the parameters, but numerical solutions to equations [1] verify that it is approximately proportional to log(λ/μ).
These numerical simulations suggest that the cumulative impact from early invasion of AR bacteria is large provided the agricultural equilibrium is much larger than the pristine equilibrium, and provided that the prevalence of resistance would remain rare for some time after initiating MAU. For this to occur, the pristine equilibrium must remain very low, and R0 must not be much larger than one. In this model, higher values of R0 lead to rapid invasion of resistance without AAU. In short, imprudent use of MAU decreases the efficacy of medical antibiotics, so the potential impact of AAU is much lower.
We conclude that when (i) the pristine equilibrium is very low, (ii) the agricultural equilibrium is high relative to the pristine equilibrium (but both may be low in absolute terms), and (iii) R0 assumes intermediate values, then AAU should have large impacts. Because AR strains must compete with resident antibiotic-sensitive bacteria, values of R0 that are not much higher than one may be typical.
An Illustrative Example
The epidemic spread of AR bacteria is a nonlinear process, and intuition about nonlinear processes is notoriously bad. Mathematical models are a useful tool for guiding intuition, and they provide a basis for heuristic reasoning. To illustrate several important properties of this model, we have estimated the impact for a specific set of parameters that are consistent with current understanding of the relationship between avoparcin use for animal growth promotion, the use of vancomycin in medicine, and the spread of vancomycin-resistant enterococci (VRE) in hospitals. Vancomycin and avoparcin are different names for a glycopeptide antibiotic; bacteria that are resistant to vancomycin also are resistant to avoparcin. Some parameter estimates are poorly supported by data, so the results presented here should be regarded as an illustration of important theories to be tested, not as a risk assessment.
To estimate the impact, we need estimates of exposure rates, λ and μ. These rates could be obtained from direct estimates of exposure at two points in time; once before the use of the avoparcin, and again after avoparcin had been used for a time in agriculture but before vancomycin was heavily used in humans. Alternatively, the difference in rates can be estimated from the difference in the agricultural and pristine equilibria, with some estimates of persistence times. We are not aware of any data of this sort. As a substitute, we have compared the community prevalence of VRE in Europe and the US. Until recently, many European countries used avoparcin in agriculture for growth promotion. Measures of the prevalence of VRE in the human population in Europe after decades of avoparcin use range from 1–12%; the highest prevalence is associated with areas where avoparcin was heavily used (14, 26). In contrast, avoparcin was never approved as a growth promoter in the U.S., and VRE is rarely detected in humans with no recent history of hospitalization (27). Independent lines of circumstantial evidence suggests that the high prevalence of VRE in the European community is due to avoparcin use (19, 28). In both communities, some cases may be related to hospitalization, but the prevalence of VRE is much higher in U.S. hospitals. All else being equal, the higher prevalence of VRE in U.S. hospitals compared with European hospitals leads us to expect more secondary cases associated with hospitalization in the U.S. communities. Based on these arguments, we estimate that λ and μ are both low, but λ is much larger than μ (Table 1).
Some uncertainty exists about the natural turnover of enterococcal strains in the human gut. We assume that, on average, transient populations last 10 days, populations that have colonized last ≈1 year, and high-load populations last 100 days. After exposure, we assume that ≈1% of transient low-load populations colonize (Pr(X→Y) = 0.01), and 30% of high-load populations colonize (Pr(Z→Y) = 0.3). The estimates are consistent with the ecology of VRE in humans (27).
The prescription rate, ρ, and the contact parameter for high-microbial loads, β, were chosen so that R0 and the equilibrium prevalence were consistent with other estimates (29). The amplification of VRE population densities may be affected by vancomycin and other antibiotics (21). We have assumed that low-density populations that have colonized rarely expose others (η is much less than β). The parameter values are reported in Table 1.
To assess the impact for these parameters, we numerically integrated the equations using these parameters comparing resistance with and without avoparcin use (changing λ). To contrast the role played by vancomycin and other medical antibiotics, we numerically integrated the equations without avoparcin use but with higher MAU rates (changing ρ).
Years of Wasted Medical Potential.
For this set of parameters, AAU caused dramatic changes in the prevalence of VRE in humans during the first 3 years, compared with identical populations without AAU (Fig. 2A). Because AR arises from natural sources and is amplified by MAU, VRE would eventually emerge without AAU, but it would reach equilibrium several years later. For this set of parameters, the total excess prevalence of AR integrated over the first 10 years is approximately equal to the excess prevalence generated by a 30% increase in the MAU rates over the same period.
Undetected by Surveillance.
In this case, the change from the pristine equilibrium to the agricultural equilibrium is small, although the ratio of the two is large. The difference between the two equilibria is below detection thresholds; data accumulated through ordinary surveillance would probably not have sufficient statistical power to detect an increase in prevalence caused by AAU. Once an increase was detectable, the spread of AR bacteria would be irreversible. In other words, surveillance on humans would not provide the kind of information that would allow a governing agency to effectively monitor and manage AR bacteria. Small increases in prevalence when AR is rare have dramatic effects, like sparks that start forest fires. The consequences may be due to rare events, which are notoriously difficult to study.
Small Changes in the Equilibrium Prevalence.
Increasing the rate of MAU in humans caused AR to spread faster and reach a higher equilibrium. We performed a sensitivity analysis for the equilibrium prevalence for doubling and halving the parameters (Fig. 2B). The equilibrium prevalence was very sensitive to the contact rate in humans, β, and the rate of MAU, ρ (Fig. 2B). On the other hand, the equilibrium is not sensitive to the rate new strains appear because of AAU (λ). Eliminating all AAU changed the equilibrium prevalence by less than 1%.
Timing.
In this case, the timing of regulation is critical; it matters whether AAU precedes MAU. The impact of AAU is most severe when AAU precedes MAU, but it may be negligible if AAU is delayed until after MAU has been used for a time and AR is near equilibrium. To show this, we numerically integrated the equations [1] with λ varying to simulate delayed AAU. The longer AAU was delayed, the lower the impact (Fig. 2C).
Discussion
The rate of MAU (ρ) and exposure from AAU (λ) have fundamentally different consequences on the emergence of AR. Our model, like others (23, 24), shows that the equilibrium prevalence of AR will be strongly affected by the rate of MAU. In contrast, AAU plays a role that is similar to mutation. Because AAU does not amplify population densities of AR bacteria in humans, it does not have a strong impact on equilibrium prevalence when MAU rates are high.
AAU may be responsible for high community prevalence in humans, but a potentially more serious problem is the effect on the time when AR appears in humans. AAU can have impacts that are equivalent to large increases in MAU by introducing new AR strains earlier than they would appear otherwise. Such impacts may occur even if changes in prevalence caused by AAU are too low to be detected by surveillance. The large impacts occur, because the prevalence of AR increases at a time when AR bacteria would be virtually absent otherwise.
AAU hastens the onset of AR, but quantitative estimates of the number of years lost are sensitive to parameter estimates and model assumptions. Quantitative assessments of the total impact for each drug-bug combination may depend on specific details of the ecology of the bacteria and the mechanisms of AR. On the other hand, most policy is made in the absence of extensive quantitative understanding of microbial population ecology. Without a sound understanding of the ecology, the existing information is subject to misinterpretation.
Stochasticity.
The time to emergence of AR bacteria may depend on stochastic events when AR bacteria are initially rare. After exposure, a sequence of events must occur for the strain to spread; at each stage, the AR bacteria may go extinct because of stochastic fade-out. One shortcoming of the deterministic model we have formulated is that it may not correctly reflect the underlying stochasticity. Detailed stochastic models may provide different estimates of impact.
Increased Exposure from Agricultural Antibiotic Use.
Substantial controversy exists about the rate that bacteria from food animals colonize humans or how frequently AR genes move from bacteria populations of animals into the bacteria of humans. Evidence suggests that AR bacteria or genes move among hosts or habitats. The basic structures of many antibiotics are modified from chemicals that are naturally produced by bacteria (30); genes that confer resistance to these antibiotics also may have an ancient origin in these same bacteria (e.g., in soils). The fact that these high-level AR genes are found in humans strongly implies that μ + λ is higher than zero. On the other hand, to measure impact, it is necessary to compare what happened to what would have happened otherwise. Both rates may be very small, but the absolute value of λ + μ is not as important as their values relative to one another, λ/(λ + μ). In other words, AAU must substantially increase the rate that new AR strains appear in humans.
The more critical issue for assessing impact is whether the rate of exposure because of AAU is high compared with background rates. There is strong evidence that animal food products are often contaminated with AR bacteria (22), leading to dramatically increased exposure to AR bacteria or resistance genes of animal origin. Whether this exposure leads to dramatic increases in the rate these strains colonize humans or the rate resistance genes are transferred is poorly understood from a quantitative perspective.
Are Prevalence and Impact Related?
The prevalence of AR bacteria in humans after decades of AAU is not, by itself, informative about the impact of AAU. The impact of AAU may be low even if the rate of exposure to resistant bacteria on food products is considered high. Alternatively, the impact of AAU may be high, even if the prevalence of AR bacteria in humans remains low after decades of AAU.
The sensitivity of the equilibrium prevalence to minor changes in MAU rates and the insensitivity to major changes in AAU implies that minor differences in MAU may mask major differences in the history of AAU (Fig. 2B). Studies that compare the prevalence of AR in communities with similar patterns of MAU but different patterns of AAU might fail to detect an impact because AAU does not cause large changes in the prevalence of AR bacteria at equilibrium. Once AR is common, the critical question is, “when would AR have invaded otherwise?”
Why Regulate Agricultural Antibiotic Use?
After AR is common in humans, infection control and prudent MAU are more likely to reduce the prevalence of AR in hospitals than eliminating AAU. When AR bacteria spread epidemically (R0 > 1), MAU is largely responsible for human-to-human transmission and the high prevalence of AR. Under these circumstances, eliminating AAU to prevent the introduction of new AR strains from animals has minor effects on the equilibrium prevalence of AR in hospitals (Fig. 2 B and C).
On the other hand, public health benefits may accumulate from restricting AAU before AR bacteria emerge. Restricting AAU in new resistance classes would likely maximize the time when AR in humans is rare (Fig. 2C), suggesting that the best time to regulate AAU is before AR appears. The principle also applies to the evolution of multidrug resistance. If heavy AAU and MAU are concurrent, multidrug resistance may evolve in animal populations and move into human populations. Early invasion of multidrug resistance has profound medical consequences, and might be managed by the regulating AAU as well as prudent MAU.
Quantitative vs. High-Level Resistance.
The rate that new AR human commensal bacteria appear in humans depends on the genetics of AR mechanisms and the population biology of the AR bacteria in different host species. The evolution of AR through the accumulation of point mutations in animal bacteria poses a threat to humans only if these bacteria can colonize humans. If high-level AR genes exist, and if they can be transmitted on mobile genetic elements, the medical risks associated with AAU are much greater. Because genes may be transferred among unrelated species, any microbial species carrying an AR gene could introduce it into human commensal bacteria. Furthermore, AAU may provide strong selection leading to the evolution of increased mobility of genetic elements or linkage to genes conferring resistance to other antibiotics.
Species Barriers.
Evolutionary host-shifting may be rare because of natural species barriers created by ecological differences among different hosts; thus, bacteria may be host- or habitat-specific. Concurrent AAU and MAU may facilitate host shifts by providing a temporary advantage to AR bacteria of animal origin in humans, or vice versa. The concept of a species barrier is controversial, and data are limited in both quantity and quality. A few studies have isolated AR bacteria from humans and food animals with identical fingerprints (31). There are also a few reports indicating that strains from humans and animals form separate clusters and can be identified by molecular typing (32). A simple explanation is that circulation of commensal bacteria between different host species occurs but is infrequent, allowing development of genetically distinct clusters.
Conclusions
AAU and MAU play similar roles in the communities where they are used; hospitals and farms with high rates of antibiotic use are evolutionary “incubators” where high-level AR bacteria and multidrug-resistant bacteria thrive. In such environments, strong selection also favors the evolution of genetic mechanisms that increase the mobility of genes. AAU may introduce new AR strains into the human population; this introduction threatens the public health when important evolutionary events occur first in bacteria populations associated with animals and then move into bacteria populations associated with humans. Some medical impacts may occur as a result of heavy veterinary therapeutic use, not just for animal growth promotion; antibiotic use selects for AR regardless of why it is used.
Restricting AAU is most effective when AR bacteria remain rare. One solution might be to regulate AAU before AR becomes a problem in medicine and then allow prudent AAU once clinically significant resistance has already developed. This solution represents the opposite conclusion from a proposed regulatory concept in which drug use would be allowed until AR exceeds a threshold (see http://www.fda.gov/cvm/antimicrobial/threshold21.pdf). This threshold concept may be an ineffective tool for managing commensal AR bacteria because once AR bacteria are detected, much of the damage has been done. Thresholds may be useful for organisms such as Campylobacter and Salmonella but not AR resistance in enterococci. In commensal bacteria with the potential for epidemic spread, small increases in prevalence when AR is extremely rare can initiate epidemics which may have large consequences. We are skeptical that a threshold in humans could be set low enough to effectively prevent the adverse effects of AAU.
The regulations on animal growth promoters are undergoing serious revision. Before the EU regulated growth promoters, many antibiotics used as animal growth promoters selected for resistance to drugs that were used or were eventually developed for use in humans (8). The situation is similar in the U.S. In addition, many front-line antibiotics are heavily used in animals therapeutically. Our analysis demonstrates that AAU may hasten the appearance of AR and decrease the efficacy of the antibiotic in humans; prudent AAU after the development of AR in humans may have few medical impacts. Regulating early AAU would likely extend the period that a drug can be used effectively in humans and reduce the demands for new antibiotics that must undergo an expensive discovery and approval process. We conclude that agricultural use of antibiotics in new resistance classes should be delayed until the period of maximum medical utility has passed.
Acknowledgments
The authors thank the reviewers for excellent suggestions that substantially improved this paper. We acknowledge financial assistance from a grant by Pfizer Corporation.
Abbreviations
- AR
antibiotic-resistant or antibiotic resistance
- AAU
animal antibiotic use
- MAU
medical antibiotic use
- VRE
vancomycin-resistant enterococci
Appendix
Bacterial populations in humans do not lead to new exposure unless they colonize or become amplified by antibiotics; the probabilities these events occur are denoted Pr(X→Y) = σ/(θ + α + ρ), and Pr(X→Z) = ρ/(θ + α + ρ), respectively. Once a transient population colonizes or becomes amplified, the expected number of humans that would be exposed rises dramatically. We let R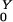 denote the number of humans exposed after colonization and R
denote the number of humans exposed after colonization and R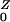 denote the number after amplification. It follows that R0 = Pr(X→Y)R
denote the number after amplification. It follows that R0 = Pr(X→Y)R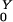 + Pr(X→Z)R
+ Pr(X→Z)R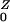 .
.
A single colonized individual in a population otherwise unexposed would expose η/(σ + ρ) other humans on average. If drugs are used, the population is amplified, and R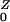 cases are generated. The probability a human receives a drug that amplifies the population density is Pr(Y→Z) = ρ/(σ + ρ). An amplified human exposes β/(γ + φ) new humans, on average. After amplification, they revert to being colonized by low-density populations with probability Pr(Z→Y) = φ/(γ + φ). Thus, R
cases are generated. The probability a human receives a drug that amplifies the population density is Pr(Y→Z) = ρ/(σ + ρ). An amplified human exposes β/(γ + φ) new humans, on average. After amplification, they revert to being colonized by low-density populations with probability Pr(Z→Y) = φ/(γ + φ). Thus, R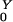 = η/(σ + ρ) + Pr(Y→Z)R
= η/(σ + ρ) + Pr(Y→Z)R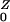 , and R
, and R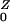 = β/(γ + φ) + Pr(Z→Y)R
= β/(γ + φ) + Pr(Z→Y)R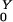 . The expressions for EY and EXare found by solving these last two equations for R
. The expressions for EY and EXare found by solving these last two equations for R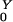 and R
and R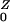 :
:
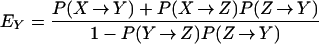 |
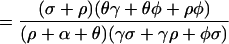 |
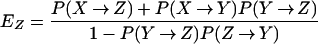 |
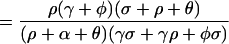 |
[3]
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 5752.
References
- 1.McGowan J E J. Rev Infect Dis. 1983;5:1033–1048. doi: 10.1093/clinids/5.6.1033. [DOI] [PubMed] [Google Scholar]
- 2.Tenover F C, McGowan J E J. Am J Med Sci. 1996;311:9–16. doi: 10.1097/00000441-199601000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Shlaes D M, Gerding D N, John J F J, Craig W A, Bornstein D L, Duncan R A. Clin Infect Dis. 1997;25:584–599. doi: 10.1086/513766. [DOI] [PubMed] [Google Scholar]
- 4.Mellon M, Benbrook C, Benbrook K L. Hogging It: Estimates of Antimicrobial Abuse in Livestock. Cambridge, MA: Union of Concerned Scientists; 2001. [Google Scholar]
- 5.National Research Council, Committee on Drug Use in Food Animals. The Use of Drugs in Food Animals: Benefits and Risks. Washington, DC: Natl. Acad. Press; 1999. [Google Scholar]
- 6.Aarestrup F M. Microb Drug Res. 1995;1:255–257. doi: 10.1089/mdr.1995.1.255. [DOI] [PubMed] [Google Scholar]
- 7.Aarestrup F M. Int J Antimicrob Agents. 1999;12:279–285. doi: 10.1016/s0924-8579(99)90059-6. [DOI] [PubMed] [Google Scholar]
- 8.Aarestrup F M. Microb Drug Res. 1998;4:137–141. doi: 10.1089/mdr.1998.4.137. [DOI] [PubMed] [Google Scholar]
- 9.Chadwick P R, Woodford N, Kaczmarski E B, Gray S, Barrell R A, Oppenheim B A. J Antimicrob Chemother. 1996;38:908–909. doi: 10.1093/jac/38.5.908. [DOI] [PubMed] [Google Scholar]
- 10.Bates J, Jordens J Z, Griffiths D T. J Antimicrob Chemother. 1994;34:507–516. doi: 10.1093/jac/34.4.507. [DOI] [PubMed] [Google Scholar]
- 11.Chee-Sanford J C, Aminov R I, Krapac I J, Garrigues-Jeanjean N, Mackie R I. Appl Environ Microbiol. 2001;201:1494–1502. doi: 10.1128/AEM.67.4.1494-1502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linton A H. Schriftenr Ver Wasser Boden Lufthyg. 1988;78:197–224. [PubMed] [Google Scholar]
- 13.van den Bogaard A E, Jensen L B, Stobberingh E E. N Engl J Med. 1997;40:454–456. doi: 10.1056/NEJM199711203372117. [DOI] [PubMed] [Google Scholar]
- 14.Simonsen G S, Haaheim H, Dahl K H, Cruse H, Lovseth A, Olsvik O, Sundsfjord A. Microb Drug Res. 1998;4:313–318. doi: 10.1089/mdr.1998.4.313. [DOI] [PubMed] [Google Scholar]
- 15.Levy S B, FitzGerald G B, Macone A B. N Engl J Med. 1976;295:583–588. doi: 10.1056/NEJM197609092951103. [DOI] [PubMed] [Google Scholar]
- 16.Smith K E, Besser J M, Hedberg C W, Leano F T, Bender J B, Wicklund J H, Johnson B P, Moore K A, Osterholm M T The Investigation Team. N Engl J Med. 1999;340:1525–1532. doi: 10.1056/NEJM199905203402001. [DOI] [PubMed] [Google Scholar]
- 17.Endtz H P, Ruijs G J, van Klingeren B, Jansen W J, van der Reyden T, Mouton R P. J Antimicrob Chemo. 1991;27:199–208. doi: 10.1093/jac/27.2.199. [DOI] [PubMed] [Google Scholar]
- 18.Olsen S J, DeBess E E, McGivern T E, Marano N, Eby T, Mauvais S, Balan V K, Zirnstein G, Cieslack P R, Angulo F J. N Engl J Med. 2001;344:1572–1579. doi: 10.1056/NEJM200105243442102. [DOI] [PubMed] [Google Scholar]
- 19.Wegener H C, Aarestrup F M, Jensen L B, Mammerum A M, Bager F. Emerg Infect Dis. 1999;5:329–335. doi: 10.3201/eid0503.990303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray B E. N Engl J Med. 2000;342:710–721. doi: 10.1056/NEJM200003093421007. [DOI] [PubMed] [Google Scholar]
- 21.Rice L B. Emerg Infect Dis. 2001;7:183–187. doi: 10.3201/eid0702.010205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teuber M. Cell Mol Life Sci. 1999;56:755–763. doi: 10.1007/s000180050022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Austin D J, Kristinsson K G, Anderson R M. Proc Natl Acad Sci USA. 1999;96:1152–1156. doi: 10.1073/pnas.96.3.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Austin D J, Kakehashi M, Anderson R M. Proc R Soc London Ser B. 1997;264:1629–1638. doi: 10.1098/rspb.1997.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersson D I, Levin B R. Curr Opin Microb. 1999;2:489–493. doi: 10.1016/s1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 26.van der Auwera P, Pensart N, Korten V, Murray B E, Leclercq R. J Infect Dis. 1996;173:1129–1136. doi: 10.1093/infdis/173.5.1129. [DOI] [PubMed] [Google Scholar]
- 27.Roghmann M C, Qaiyumi S, Schwalbe R, Morris J G., Jr Infect Cont Hosp Epidemiol. 1997;18:679–680. doi: 10.1086/647510. [DOI] [PubMed] [Google Scholar]
- 28.Klare I, Badstubner D, Konstabel C, Bohme G, Claus H, Witte W. Microb Drug Res. 1999;5:45–52. doi: 10.1089/mdr.1999.5.45. [DOI] [PubMed] [Google Scholar]
- 29.Austin D J, Bonten M J M, Weinstein R A, Slaughter S, Anderson R M. Proc Natl Acad Sci USA. 1999;96:6908–6913. doi: 10.1073/pnas.96.12.6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabay J E. Science. 1994;264:373–374. doi: 10.1126/science.8153623. [DOI] [PubMed] [Google Scholar]
- 31.Aarestrup F M, Ahrens P, Madsen M, Pallesen L V, Poulsen R L, Westh H. Antimicrobial Agents and Chemotherapy. 1996;40:1938–1940. doi: 10.1128/aac.40.8.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson J, Qaiyumi S, English L, Hayes J, White D, Joseph S, Wagner D. Comparison of Streptogramin-Resistant Enterococcus Faecium from Poultry and Humans. 2000. AVMA Annual Convention, Salt Lake City, UT). [Google Scholar]