

Abstract
Oceans general circulation models predict that global warming may cause a decrease in the oceanic O2 inventory and an associated O2 outgassing. An independent argument is presented here in support of this prediction based on observational evidence of the ocean's biogeochemical response to natural warming. On time scales from seasonal to centennial, natural O2 flux/heat flux ratios are shown to occur in a range of 2 to 10 nmol of O2 per joule of warming, with larger ratios typically occurring at higher latitudes and over longer time scales. The ratios are several times larger than would be expected solely from the effect of heating on the O2 solubility, indicating that most of the O2 exchange is biologically mediated through links between heating and stratification. The change in oceanic O2 inventory through the 1990s is estimated to be 0.3 ± 0.4 × 1014 mol of O2 per year based on scaling the observed anomalous long-term ocean warming by natural O2 flux/heating ratios and allowing for uncertainty due to decadal variability. Implications are discussed for carbon budgets based on observed changes in atmospheric O2/N2 ratio and based on observed changes in ocean dissolved inorganic carbon.
Repeated hydrographic surveys indicate that the upper 3 km of the oceans have warmed (1) and intermediate waters of high-latitude origin have freshened (2) over the past few decades. Model studies indicate that upper-ocean warming, high-latitude freshening, and an associated increase in the density stratification of the upper ocean are expected consequences of the changes in atmospheric radiative forcing caused by fossil-fuel burning and other human activities (3–5).
Repeated hydrographic surveys also indicate that small detectable changes have occurred in oceanic dissolved O2 concentrations. As summarized in Table 1, detectable decreases in O2 have been found in intermediate waters in the North Pacific, North Atlantic, South Pacific, and South Indian oceans, while small increases have possibly been found in deeper waters in the North Pacific and South Indian Oceans. What caused these O2 changes is unclear, and different mechanisms, including changes in ocean circulation rates (6–9), changes in preformed values (10), changing Redfield ratios (11), and changes in biological production (8) have been offered as possible explanations in different regions. While the changes may partly reflect natural decadal variability, the clearest O2 changes, found at intermediate depths, are in the direction of decreasing O2 concentrations. A global reduction in dissolved O2 is predicted by ocean general circulation models (OGCMs) driven by increasing greenhouse gases (12–15). In the model simulations, most of the O2 decrease is attributed to enhanced stratification.
Table 1.
Significant recent changes in dissolved O2 concentrations
| Study | Location | Time span | Depth range, m | O2 change, μmol kg−1 |
|---|---|---|---|---|
| Garcia et al. (10) | Atlantic 24°N section, averaged over full section | 1981–1992 | 800–1,900 | −3 to −7 |
| Pahlow and Riebesell (11) | North Pacific, basin average | 1950s –1990s | Unreported | ∼−5 |
| Keller et al. (9)* | North Pacific, basin average | 1970s –1990s | 300–500 | −1 to −2 |
| 1,000–1,600 | +2 to +3 | |||
| Shaffer et al. (7) | Pacific, 28°S section, averaged from 52°–88°W | 1967–1995 | 800–1,200 | −5 to −8 |
| Bindoff and McDougal (6) | Indian Ocean, 32°S section, averaged from 30°–117°E | 1962–1997 | 300–800 | −7 to −8 |
| 2,500–4,000 | +3 | |||
| Matear et al. (13) | Southern Ocean, 110°–170°E, 50°–60°S | 1965–1995 | >400 | −5 to −15 |
| Emerson et al. (8)* | North Pacific, 154°W section, averaged from 22°–44°N | 1980–1997 | 100–600 | −9 to −20 |
Report change in O2 − O , rather than O2 concentration, where O
, rather than O2 concentration, where O is the solubility.
is the solubility.
Stratification has two competing effects on subsurface oxygen concentrations. First, it reduces the upwelling of nutrients from deeper waters into surface waters, thus decreasing photosynthetic production and the associated flux of organic detritus into the ocean interior. This flux is often referred to as the “biological pump” (16), and reducing the rate of this pump increases subsurface O2 concentrations by reducing subsurface O2 utilization rates. Second, stratification limits the downward transport from O2 from well-oxygenated surface waters into the ocean interior, which serves to reduce subsurface O2 concentrations.
In the modeling studies cited above, the effect of stratification on O2 transport exceeds the effect on subsurface O2 utilization, leading to a net O2 decrease. This result is expected, considering that stratification allows for more complete biological utilization of nutrients in surface waters, thus lowering the “preformed” (i.e., initial) nitrate and phosphate content of waters sinking into the oceans' interior. To conserve the total ocean nutrient inventory, an increase must occur in the inventory of nonpreformed nutrients—i.e., those that accumulate in subsurface waters from oxidative decomposition of organic detritus. Since O2 is consumed by organic decomposition in proportion to the amount of nitrate or phosphate released (17), subsurface O2 inventories can be expected to decrease in response to increased stratification. In general, the competing effects of the biological pump and vertical mixing on O2 concentrations can be assessed based on their net impact on the vertical nutrient distributions. This net impact we refer to as the “efficiency” (as opposed to “rate”) of the biological pump.
Stratification can also be expected to induce a net release of O2 from the ocean to the atmosphere. The oceanic O2 inventory (I, mol) and sea-to-air O2 flux (Z, mol yr−1) are linked according to
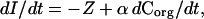 |
1 |
where Corg is the oceanic inventory of organic carbon, including both particulate and dissolved forms, and α is the O2:C oxidative ratio for destruction or production of marine organic matter, and where small terms related to river and sediment transports have been neglected. The term αdCorg/dt effectively accounts for the column-integrated net production of O2 by marine photosynthesis and respiration. This term is presumably small, although not necessarily negligible (18), because the main effect of marine photosynthesis and respiration on time scales of years to centuries is to redistribute inorganic materials within the ocean, rather than to cause accumulation or destruction of organic carbon (see also ref. 14). On these time scales, we therefore expect that changes in the oceanic O2 inventory, due to stratification or other causes, should be roughly balanced by O2 exchanges with the atmosphere (dI/dt ≈ −Z).
Estimates based on OGCMs for oceanic O2 outgassing due to global warming lie in the range of 0.2 to 0.7 × 1014 mol yr−1 for the past few decades, with predictions of 1.0 to 1.6 × 1014 mol yr−1 for late 21st century (12–15). According to these estimates, roughly a quarter of the predicted outgassing is attributable to the direct effect of warming on the O2 solubility, while the remainder is due to increased stratification. The predicted changes for the past few decades amount to an average decrease in oceanic O2 concentrations over a 20-year time frame of between 1 and 5 μmol kg−1 if the changes are confined to the top 1,000 m of the oceans. Larger changes are predicted for the Southern Oceans due to reductions in deep convection (12, 13). It is hard to test these predictions with existing hydrographic data, given the sparse coverage and the lack of comprehensive syntheses.
There are several reasons why changes in the oceanic O2 inventory could be important. Dissolved O2 concentration is a useful diagnostic of ocean circulation and biological activity which can provide constraints on models of physical and biogeochemical response to climate change (13). Small changes in O2 content could influence extent of hypoxic regions in coastal seas, in sediments, or in the open ocean, with consequences for the cycling of nitrogen and other redox-sensitive elements and for the distribution of many marine organisms (19). Changes in O2 are diagnostic of changes in the efficiency of the marine biological pump, which may influence the rate at which the oceans absorb anthropogenic CO2 (12). Finally, at least two approaches for estimating sinks of anthropogenic carbon dioxide require corrections for changes in oceanic O2 inventory. The purpose of this paper is to provide an independent estimate of the plausible O2 inventory changes associated with recent global warming and to discuss the implications for global carbon budgeting.
Natural Warming and O2 Outgassing
It is easily established that a close association exists between ocean warming and O2 outgassing over a range of space and time scales in the open ocean. At middle and high latitudes during the spring and summer, when the upper ocean is heated by the atmosphere, the oceans are a source of O2 to the atmosphere, while in the fall and winter, when the upper ocean is cooled, the oceans are a sink of O2 from the atmosphere. These seasonal air–sea O2 exchanges are driven partly by biological exchanges, linked to seasonal stratification, nutrient supply, and irradiance, and partly by effects of heating and cooling on O2 solubility (20).
We have estimated the ratio of seasonal O2 outgassing to seasonal heating from global archived measurements of dissolved O2 in surface waters, climatological winds, and climatological air–sea heat fluxes (21). The O2 flux/heating ratio varies between 1.5 nmol of O2 per joule at lower latitudes to 4 or 5 nmol of O2 per joule in the 40° to 60° latitude bands, as shown in Fig. 1. Consistent with Najjar and Keeling (20), we find ratios that are larger than expected from the effect of warming on the O2 solubility by factors between 1.5 and 2.5. We further have shown, based on comparisons with atmospheric O2/N2 data (21), that the component of the O2 flux that correlates with heating dominates large-scale seasonal O2 exchange. These results indicate that seasonal heating, through its effect on stratification, biological productivity, and O2 solubility, is a major driver of the exchange, and not just coincidentally correlated with the exchange.
Figure 1.

Ratio of the seasonal O2 outgassing to the seasonal upper ocean warming by 10° latitude bands. Dashed line indicates the ratios expected based on changes in O2 solubility alone. Ratios from 20°N to 20°S are poorly defined due to weak seasonal forcing. The seasonal O2 outgassing here accounts for the anomalous flux after the mean annual outgassing or ingassing is subtracted.
Linkages between heat fluxes and oxygen fluxes are also evident on longer time scales, as revealed from a plot of the tracer O*2 = O2 + 175PO4 versus potential temperature (Θ), as shown in Fig. 2. The tracer O*2, which is identical to Broecker's tracer PO*4 (25) but expressed in O2 rather than PO4 units, is a measure of the O2 gained or lost by a water parcel through air–sea gas exchange (26). O*2 is largely conserved below the sea surface, where photosynthesis and respiration produce compensating effects on O2 and PO4. O*2 keeps track of air–sea O2 exchanges driven by both solubility changes and the processes controlling the efficiency of the biological pump. Assuming rapid air–sea equilibration, the solubility component is simply given by the O2 solubility (O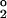 ), so that the remaining biological pump component is proportional to the (apparent) preformed phosphate content (PO4 − (O
), so that the remaining biological pump component is proportional to the (apparent) preformed phosphate content (PO4 − (O − O2)/175) of the water (26).
− O2)/175) of the water (26).
Figure 2.

Scatter plot of the tracer O = O2 + 175PO4 versus potential temperature for data collected on the Geochemical Ocean Section Study (22–24). The data through the main thermocline of the Indian and Pacific oceans are seen to scatter around a line with a slope of approximately −22 μmol kg−1 °C−1. Changes in O
= O2 + 175PO4 versus potential temperature for data collected on the Geochemical Ocean Section Study (22–24). The data through the main thermocline of the Indian and Pacific oceans are seen to scatter around a line with a slope of approximately −22 μmol kg−1 °C−1. Changes in O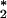 are generally larger than expected from changes in the O2 solubility (dashed line). Antarctic surface water data are from ref. 25. NADW, North Atlantic deep water.
are generally larger than expected from changes in the O2 solubility (dashed line). Antarctic surface water data are from ref. 25. NADW, North Atlantic deep water.
A prominent feature in Fig. 2 is the strong association between O*2 and Θ in waters of the main thermocline between 6° and 18°C. Waters around 18°C consistently have lower O*2 than waters around 6°C, indicating that conversion of cooler water to warmer water, which occurs mostly at low latitudes, leads to outgassing of O2, while the conversion of warmer water back to cooler water, which occurs mostly at higher latitudes, leads to uptake of O2. Outside the North Atlantic, the O*2/Θ slope of ∼22 μmol kg−1 °C−1 in waters between 6° and 18°C, is equivalent to ∼5 nmol of O2 per joule of warming, as derived by multiplying by seawater density and dividing by heat capacity. The slope is several times larger than expected from solubility changes alone, indicating that the O2 exchanges are mainly controlled by variations in the efficiency of the marine biological pump. A generally weaker and but more variable O2/heat relationship is indicated for waters warmer than about 18°C, which is consistent with the lower nutrient content of these waters and a reduction of influence of biological relative to solubility effects.
A less steep O*2/Θ trend is seen in Fig. 2 for the thermocline of the North Atlantic Ocean compared with other oceans, possibly owing to the lower nutrient content of the North Atlantic. The North Atlantic trend of ∼13 μmol kg−1 °C−1 (equivalent to ∼3 nmol J−1) connects low-latitude surface waters in the Atlantic with North Atlantic Deep Water (NADW), which lies below the O*2/Θ trend of the other oceans. A steeper O*2/Θ ratio of around 30 to 40 μmol kg−1 °C−1 (equivalent to 7.5–10 nmol J−1) is found for the deep Antarctic sequence (25), which is driven by ventilation of deep waters around Antarctica. Here stratification induced by warming and freshening in the summer months inhibits the uptake of O2 by deeper waters, while the breakdown of stratification induced by wintertime cooling and brine rejection from sea ice enhances O2 uptake and deepwater formation (27). The air–sea exchanges and water-mass-mixing around Antarctica effectively convert circumpolar deep water, which derives largely from NADW, into colder Antarctic surface waters. By this conversion, the oceans around Antarctica release heat to the atmosphere and take up O2.
Due to sparse coverage, Fig. 2 does not resolve well water masses in the Equatorial Pacific. Here, however a very different relationship between heating and O2 flux is known to exist. Equatorial upwelling raises cool, oxygen-deficient waters to the surface, where a net uptake of O2 from the atmosphere (20) and a net heating of the water occurs. The O2 flux/heating ratio in the Equatorial Pacific is thus opposite in sign to the cases considered above. This feature is a result of the upwelled waters having been exposed to the surface for a very brief period of weeks or days, which is insufficient to allow O2 to equilibrate with the atmosphere and for biological production to remove the nutrients. As the upwelled waters spread laterally away from the Equator, the net effect of warming and nutrient withdrawal leads to an overall O2 release to the atmosphere (20, 28). Integrated over a wider latitude band, the net effect of Equatorial upwelling on heat and O2 exchange is therefore more concordant with the main thermocline trend.
The patterns noted above suggest the following generalizations: Although the changing efficiency of the biological pump dominates the O2 response of the ocean to warming and stratification, the O2 response is nevertheless strongly tied, over a range of space and time scales, to the net air–sea heat flux. For time scales of months to centuries, the O2 flux/heating ratios generally lie in the range of 2 to 10 nmol J−1. Larger ratios are found at higher latitudes, particularly in the Southern Hemisphere and for processes occurring over time scales of decades to centuries (e.g., thermocline ventilation) compared with time scales of months (seasonal exchanges). Some differences exist from ocean to ocean, with the North Atlantic having smaller O2 response per unit heating or cooling than other high-latitude regions. This analysis does not resolve the O2 response of the oceans to heating and cooling on thousand-year and longer time scales.
Anthropogenic Warming and O2 Outgassing
In Table 2 we use the O2 flux/heating relationships found above to formulate an estimate of the global air–sea O2 flux from 1990 to 2000. We consider three ocean regions: (i) North Atlantic (at all depths), (ii) the deep Southern Ocean (>1,000 m), and (iii) the remaining oceans (at all depths). Warming in each region is assumed to produce O2 outgassing proportional to observed steady-state O*2/Θ relationships in these regions. The approach is motivated by the rough universality of O2/heat ratios for processes ranging from warming and cooling on seasonal time scales to steady-state warming/cooling over decades to centuries. The approach effectively adopts the null hypothesis that the ocean's response to transient warming on decadal time scales is governed by similar ratios.
Table 2.
Estimated oceanic O2 outgassing 1990–2000 due to changing radiative forcing
| Ocean region | Heating
|
O2/heat ratio, nmol J−1 | O2 outgassing
|
||
|---|---|---|---|---|---|
| 1022 J yr−1 | % | 1014 mol yr−1 | % | ||
| North Atlantic | 0.18 ± 0.08 | 30 | 3 ± 1.5 | 0.05 ± 0.04 | 18 |
| Deep Southern Ocean | 0.06 ± 0.03 | 10 | 10 ± 5.0 | 0.06 ± 0.04 | 20 |
| Other oceans | 0.36 ± 0.16 | 60 | 5 ± 2.5 | 0.18 ± 0.12 | 61 |
| Global total | 0.6 ± 0.18 | 100 | 4.9 | 0.29 ± 0.13 | 100 |
Heating rates are based on observed warming in each region from 1955 to 1995 (1), scaled to yield total of 0.6 × 1022 J yr−1, the warming attributable to increasing radiative forcing from 1990 to 2000 (4, 5). Error analysis assumes 45% uncertainty in individual heating rates and 50% uncertainty in O2/heat ratios, with all errors uncorrelated.
Because compilations of ocean warming (1) have been completed only through year 1998, and because these compilations do not resolve warming on a yearly basis below 300-m depth, we rely on model simulations (4, 5) as a means of extrapolating the observed long-term (1950s–1990s) warming in each region through the 1990–2000 period. The approach effectively accounts for the warming caused by anthropogenic radiative forcing but not natural variability. The latter we treat as a source of noise. We derive a central estimate of the global O2 outgassing of 0.29 × 1014 mol of O2 yr−1 for 1990–2000, where 18% of the total O2 outgassing is attributable to warming in the North Atlantic, 20% in the deep Southern Ocean, and 61% in the remainder of the upper ocean. Our estimate corresponds to a global average O2 flux heating ratio of 5 nmol J−1.
The uncertainty around our central estimate attributable to uncertainties in regional O*2/Θ ratios and long-term warming rates is ±0.13 × 1014 mol of O2 yr−1. A much larger uncertainty must be allowed for decadal variability (15, 29, 30). One estimate of this can be derived by multiplying the global O2/heat ratio of 5 nmol of O2 J−1 by the decadal variability in global ocean heat storage, which we estimate from heat storage data (1), after removing the long-term trend, to be ±5 × 1022 J, which yields ±2.5 × 1014 mol of O2 variability on a decadal basis. Here we adopt a slightly higher estimate of ±4 × 1014 mol of O2, on the grounds that the heat storage data (1) may underestimate true variability due to spatial and temporal averaging, and given uncertainties in the appropriate O2/heat ratio. Treating the decadal variability as a source of random noise, we derive an estimate of 0.29 ± 0.4 × 1014 mol of O2 yr−1 for the total oceanic O2 outgassing from 1990 to 2000.
From 1990 to 1998, the global upper ocean (<300 m) heat content increased at a rate of ∼0.5 × 1022 J yr−1 (1), which is faster than the rate of ∼0.2 × 1022 J yr−1 that we would estimate from projecting long-term warming rates. If we assume that the additional warming of ∼0.3 × 1022 J yr−1 persisted through year 2000, and we add the difference to our outgassing estimate, then our central estimate increases from 0.29 × 1014 mol yr−1 to 0.44 × 1014 mol yr−1, assuming a scaling of 5 nmol J−1. To apply such a correction is premature, however, because the global 1990–1998 heating trend is heavily influenced by a large anomaly in the North Atlantic in 1998, which was possibly a transient associated with the 1997–1998 El Niño event (1). In any case, the correction would be within our allowed uncertainties for decadal variability of ±0.4 × 1014 mol yr−1. Once heat storage data are compiled through 2000, it may be possible to refine the decadal outgassing estimate and reduce the allowed uncertainties.
Carbon Budgeting
What are the implications of oceanic O2 outgassing for carbon budgeting? A correction for oceanic O2 outgassing is needed to estimate land and ocean carbon sinks based on the global budgets of atmospheric O2 and CO2 (31, 32). These can be written
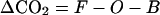 |
2 |
and
 |
3 |
where F is the CO2 source from fossil-fuel burning, O and B are, respectively, the ocean and land sinks, αF and αB are global-average O2:C exchange ratios for fossil-fuel and land biota, and Z is the net source of O2 from the oceans. Eqs. 2 and 3 are solved to yield estimates of O and B, using ΔO2 and ΔCO2 derived, respectively, from observed changes in atmospheric O2/N2 ratio and CO2 mole fraction, F and αF derived from industrial records, and αB ≈ 1.1 (31, 32).
Previously it has been assumed that the ocean outgassing term Z is zero to within the uncertainties (31, 32), or allowance has alternately been made for O2 outgassing based on the solubility effect alone (33). In Table 3, we correct the estimate of Manning (33), as cited in the recent Intergovernmental Panel on Climate Change (34) report, based on our above estimate of Z = 0.29 ± 0.4 mol of O2 per yr, where this estimate implicitly allows for the effect of anthropogenic warming on both solubility and stratification. The correction increases the oceanic sink by 0.18 Pg of C yr−1 and decreases the land sink by the same amount relative to the Manning (33) estimate. Although the change is small relative to other uncertainties, it nevertheless helps to reconcile the estimated oceanic sink with recent model estimates (35).
Table 3.
O2-based global carbon budget 1990–2000
| Budget item | Pg of C yr−1
|
|
|---|---|---|
| Manning (33) | Revised budget | |
| Fossil-fuel burning | 6.33 ± 0.4 | 6.33 ± 0.4 |
| Atmospheric CO2 increase | 3.21 ± 0.1 | 3.21 ± 0.1 |
| Total ocean CO2 sink | 1.68 ± 0.5 | 1.86 ± 0.6 |
| Total land CO2 sink | 1.44 ± 0.7 | 1.26 ± 0.8 |
Budgets are based on the decadal trend in atmospheric O2/N2 ratio at two northern hemisphere stations in the Scripps O2/N2 network. Manning (33) assumes Z = 0.16 ± 0.16 × 1014 mol of O2 yr−1; revised budget assumes 0.29 ± 0.4 × 1014 mol of O2 yr−1 (see text). Budgets differ in assumed ocean warming rate, and in assumed oceanic O2 outgassing per unit warming. Both budgets allow for outgassing of N2 (which affects the observed O2/N2 ratio) at a rate based on N2 solubility of 2.2 nmol of N2 per J of warming.
A correction for oceanic O2 is also needed for carbon budgets based on direct measurement of changes in dissolved inorganic carbon (DIC) in the ocean. Detecting the input of anthropogenic CO2 into the oceans is difficult due to large natural variability in DIC caused by ocean biology. This difficulty is commonly overcome by normalizing to a constant O2 concentration to filter out the variability due to ocean biology (36–40). In effect, what is reported is not the change DIC, but rather the change in the quantity DIC + O2/α, where α ≈ 1.3 (36, 37). By design, this approach neglects changes in DIC caused by variations in the efficiency of the biological pump. If the dissolved O2 inventory decreases globally due to stratification, the approach will underestimate oceanic CO2 uptake by an amount given approximately by (dI/dt)/α, where dI/dt is the change in oceanic O2 inventory. The correction is the same sign and a similar magnitude to that required based on atmospheric O2 and CO2 budgets. Taking the observed ocean warming of ∼2 × 1023 J between the middle 1950s and 1990s (1), and assuming the O2 inventory decreases by 5 nmol of O2 J−1, yields an upwards correction of ∼9 Pg of C (1 Pg = 1015 g) over the 1955–1995 period. In comparison, the total oceanic uptake from preindustrial times through 1990 is estimated to be 107 ± 27 Pg of C yr−1 (34).
Recent estimates of change in oceanic inventories of DIC (38–40) have not allowed for changing O2 inventory, and therefore are presumably biased low, although more work is needed to establish reliable corrections in the individual ocean basins and globally. If the DIC data are additionally normalized based on regressions against Θ and alkalinity, then additional corrections may be needed for changing ocean heat content (independent of the effect on O2) and alkalinity inventory.
Discussion
Our estimate of 0.29 × 1014 mol of O2 yr−1 for recent oceanic O2 outgassing caused by changing radiative forcing lies within the range estimated based on recent OGCMs (12–15). The relatively good agreement between these independent estimates strengthens the case that a long-term ocean outgassing of this magnitude may actually be occurring. Nevertheless, the similarity in these estimates undoubtedly results partly from similar assumptions. For example, the estimates commonly assume that Redfield P/O2 ratios are constant with time and space, and that phosphate is the ultimate limiting nutrient. Also, while our estimate assumes that the O2 response to transient warming is similar to the O2 response to steady-state warming and cooling, the OGCMs similarly assume that the response of the ocean biology to transient warming can be understood on the basis of parameters adjusted to duplicate steady-state behavior. It is unclear if these assumptions form a realistic basis for projection.
Our estimate effectively assumes that O*2/Θ relationships—or equivalently, preformed phosphate/Θ relationships—remain constant during transient warming. For example, the O2 flux/heating ratio we adopt for warming of the main thermocline would be obtained if the main effect of warming was to deepen the thermocline without altering the preformed phosphate/Θ relationship across the thermocline, or if the surface isotherms progress polewards during the transient at the same rate as the isolines of surface phosphate. The O2 flux/heating ratio we adopt for the deep Southern Ocean would be obtained if warming caused a reduction in deepwater formation around Antarctica, thus increasing the influence of other source waters on the chemistry of the deep ocean.
Our estimate of the effect of Southern Ocean warming on O2 exchange can be compared with the study Broecker et al. (41), who used PO*4, 14C, and chlorofluorocarbon (CFC) data to support the argument that the ventilation rate of deep waters around Antarctica slowed substantially in the 20th century. According to their scenario, the PO*4 content of the deep ocean should now be decreasing with time, in which case a net O2 outgassing should also be occurring, as required by the links between PO*4 (i.e., O*2) and the air–sea O2 flux. Taking their estimate of the difference in PO*4 content of southern source waters relative to average deep waters and their estimate that the input rate has slowed by 10 × 106 m3 s−1 yields a required outgassing rate of 0.3 × 1014 mol of O2 yr−1, which is 5 times larger than our estimate of 0.06 × 1014 mol of O2 yr−1 for the deep Southern Ocean (Table 2). We defend our smaller estimate on the following grounds: First, the Broecker et al. scenario implies that the deepwater heat content should be increasing by ∼0.4 × 1022 J yr−1, based on the ∼3°C difference in temperature between southern surface waters and average deep waters. A warming rate of this magnitude is inconsistent with the observed temperature trends (1), unless the warming is mostly confined below 3,000 m, the maximum depth considered by Levitus et al. (1). Second, recent work (42) suggests that a large 20th-century slow-down is not necessary to explain the CFC and PO*4 data.
A reliable assessment of the global air–sea O2 flux will ultimately require an approach based on direct observations rather than model studies. Over the next few decades, global systematic decreases in dissolved O2 can be expected at the level of 0.4 × 1014 mol of O2 yr−1 or larger, which corresponds to a change of 0.7 μmol of O2 kg−1 per decade, if spread uniformly over 2,000 m. Resolving these changes against natural variability will require a high measurement density, but this may be feasible with appropriate sensor and platform development and with a concerted long-term observing program. Resolving these changes is needed for carbon budgeting, as a complement to lower-density DIC measurements and atmospheric O2/N2 measurements, and would help to assess the overall impact of climate change on the biogeochemistry and biodiversity of the oceans. Another source of uncertainty in carbon budgets involves change in oceanic organic carbon (18), which we have assumed is small, but is not well constrained. We suggest that a program to directly monitor oceanic inventories of O2 and organic carbon, along with inorganic carbon and nutrients, should be given some priority in future ocean observing systems.
Acknowledgments
We thank Scott Doney, Ray Najjar, and Corinne Le Quéré for helpful comments. This work was supported by the National Science Foundation under Grant ATM-0000923, National Oceanic and Atmospheric Administration under Grant NA77RJ0453A, and the National Aeronautics and Space Administration under Grant NAG5–6668, and was completed in part while one of us (R.K.) was hosted at the Max Planck Institute for Biogeochemistry in Jena, Germany.
Abbreviations
- OGCMs
ocean general circulation models
- DIC
dissolved inorganic carbon
References
- 1.Levitus S, Antonov J I, Boyer T P, Stephens C. Science. 2000;287:2225–2229. [Google Scholar]
- 2.Wong A P S, Bindoff N L, Church J A. Nature (London) 1999;400:440–443. [Google Scholar]
- 3.Manabe S, Stouffer R J, Spelman M J, Bryan K. J Climate. 1991;4:785–818. [Google Scholar]
- 4.Levitus S, Antonov J I, Wang J, Delworth T L, Dixon K W, Broccoli A J. Science. 2001;292:267–270. doi: 10.1126/science.1058154. [DOI] [PubMed] [Google Scholar]
- 5.Barnett T P, Pierce D W, Schnur R. Science. 2001;292:270–274. doi: 10.1126/science.1058304. [DOI] [PubMed] [Google Scholar]
- 6.Bindoff N L, McDougall T J. J Phys Oceanogr. 2000;30:1207–1222. [Google Scholar]
- 7.Shaffer G, Leth O, Ulloa O, Bendtsen J, Daneri G, Dellarossa V, Hormazabal S, Sehlstedt P-I. Geophys Res Lett. 2000;27:1247–1250. [Google Scholar]
- 8.Emerson S, Mecking S, Abell J. Global Biogeochem Cycles. 2001;15:535–554. [Google Scholar]
- 9.Keller K, Slater R D, Bender M, Key R M. Deep-Sea Res II. 2002;49:345–362. [Google Scholar]
- 10.Garcia H, Cruzado A, Gordon L, Escanez J. J Geophys Res. 1998;103:2817–2830. [Google Scholar]
- 11.Pahlow M, Riebesell U. Science. 2000;287:831–833. doi: 10.1126/science.287.5454.831. [DOI] [PubMed] [Google Scholar]
- 12.Sarmiento J L, Hughes T M C, Stouffer R J, Manabe S. Nature (London) 1998;393:245–249. [Google Scholar]
- 13. Matear, R. J., Hirst, A. C. & McNeil, B. I. (2000) Geochem. Geophys. Geosyst.1, American Geophysical Union paper no. 10.1029/2000GC000086.
- 14.Plattner G-K, Joos F, Stocker T F, Marchal O. Tellus Ser B. 2001;53:564–592. [Google Scholar]
- 15. Bopp, L., Le Quéré, C., Heimann, M., Manning, A. C. & Monfray, P. (2002) Biogeochem. Cycles, in press.
- 16.Volk T, Hoffert M I. The Carbon Cycle and Atmospheric CO2, Archean to Present. 1985. AGU Geophysical Monograph 32, eds. Sundquist, E. T. & Broecker, W. S. (Am. Geophys. Union, Washington, DC), pp. 99–110. [Google Scholar]
- 17.Redfield A C, Ketchum B H, Richards F A. In: The Sea. Hill M N, editor. Vol. 2. New York: Interscience; 1962. pp. 26–77. [Google Scholar]
- 18.Hansell D A, Carlson C A. Deep-Sea Res II. 2001;48:1649–1667. [Google Scholar]
- 19.Rogers A D. Deep-Sea Res II. 2000;47:119–148. [Google Scholar]
- 20.Najjar R G, Keeling R F. Global Biogeochem Cycles. 2000;14:573–584. [Google Scholar]
- 21.Garcia H E, Keeling R F. J Geophys Res. 2001;106:31155–31160. [Google Scholar]
- 22.Bainbridge A E. Hydrographic Data 1972–1973, GEOSECS Atlantic Expedition. Vol. 1. Washington, DC: National Science Foundation; 1981. [Google Scholar]
- 23.Broecker W S, Spenser D W, Craig H. Hydrographic Data 1973–1974, GEOSECS Pacific Expedition. Vol. 3. Washington, DC: National Science Foundation; 1982. [Google Scholar]
- 24.Weiss R F, Broecker W S, Craig H, Spenser D W. Hydrographic Data 1977–1978, GEOSECS Indian Ocean Expedition. Vol. 5. Washington, DC: National Science Foundation; 1983. [Google Scholar]
- 25.Broecker W S, Peacock S L, Walker S, Weiss R, Fahrbach E, Schroeder M, Mikolajewicz U, Heinze C, Key R, Peng T-H, Rubin S. J Geophys Res. 1998;103:15833–15843. [Google Scholar]
- 26.Keeling R F, Peng T-H. Philos Trans R Soc London B. 1995;348:133–142. [Google Scholar]
- 27.Gordon A L, Huber B A. J Geophys Res. 1990;95:11655–11672. [Google Scholar]
- 28.Stephens B B, Keeling R F, Heimann M, Six K D, Murnane R, Caldeira K. Global Biogeochem Cycles. 1998;12:213–230. [Google Scholar]
- 29.Bender M, Ellis T, Tans P, Francey R, Lowe D. Global Biogeochem Cycles. 1996;10:9–21. [Google Scholar]
- 30.McKinley G A, Follows M J, Marshall J. Geophys Res Lett. 2000;27:2933–2936. [Google Scholar]
- 31.Keeling R F, Shertz S R. Nature (London) 1992;358:723–727. [Google Scholar]
- 32.Battle M, Bender M L, Tans P P, White J W C, Ellis J T, Conway T, Francey R J. Science. 2000;287:2467–2470. doi: 10.1126/science.287.5462.2467. [DOI] [PubMed] [Google Scholar]
- 33.Manning A C. Ph.D. thesis. San Diego: Univ. of California; 2001. [Google Scholar]
- 34.Prentice I C, Farquhar G D, Fasham M J R, Goulden M L, Heimann M, Jaramillo V J, Kheshgi H S, Le Quéré C, Scholes R J, Wallace D W R, et al. In: Climate Change 2001, Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change. Pitelka L, Ramirez Rojas A, editors. Cambridge, U.K.: Cambridge Univ. Press; 2001. pp. 183–237. [Google Scholar]
- 35.Orr J C, Maier-Reimer E, Mikolajewicz U, Monfray P, Sarmiento J L, Toggweiler J R, Taylor N K, Palmer J, Gruber N, Sabine C L, et al. Global Biogeochem Cycles. 2001;15:43–60. [Google Scholar]
- 36.Brewer P G. Geophys Res Lett. 1978;5:997–1000. [Google Scholar]
- 37.Chen C-T A, Millero F J. Nature (London) 1979;277:205–206. [Google Scholar]
- 38.Gruber N. Global Biogeochem Cycles. 1998;12:165–191. [Google Scholar]
- 39.Sabine C L, Key R M, Johnson K M, Millero F J, Poisson A, Sarmiento J L, Wallace D W R, Winn C D. Global Biogeochem Cycles. 1999;13:179–198. [Google Scholar]
- 40.Peng T-H, Wanninkkof R, Bullister J L, Feely R A, Takahashi T. Nature (London) 1998;396:560–563. [Google Scholar]
- 41.Broecker W S, Sutherland S, Peng T-H. Science. 1999;286:1132–1135. doi: 10.1126/science.286.5442.1132. [DOI] [PubMed] [Google Scholar]
- 42.Orsi A H, Jacobs S S, Gordon A L, Visbeck M. Geophys Res Lett. 2001;28:2923–2926. [Google Scholar]