

Abstract
Cathepsins B and L are widely expressed cysteine proteases implicated in both intracellular proteolysis and extracellular matrix remodeling. However, specific roles remain to be validated in vivo. Here we show that combined deficiency of cathepsins B and L in mice is lethal during the second to fourth week of life. Cathepsin B−/−/L−/− mice reveal a degree of brain atrophy not previously seen in mice. This is because of massive apoptosis of select neurons in the cerebral cortex and the cerebellar Purkinje and granule cell layers. Neurodegeneration is accompanied by pronounced reactive astrocytosis and is preceded by an accumulation of ultrastructurally and biochemically unique lysosomal bodies in large cortical neurons and by axonal enlargements. Our data demonstrate a pivotal role for cathepsins B and L in maintenance of the central nervous system.
Cathepsins B and L are widely expressed cysteine proteases. In vitro studies using purified enzymes and cysteine protease inhibitors suggested roles for these cathepsins in lysosomal protein turnover at acidic pH. In a number of pathological conditions, both cathepsins are reported to be secreted and involved in degradation of extracellular matrix components (1, 2). More specific functions have recently been postulated in a wide range of physiological and pathological processes. These include antigen presentation (3), prohormone processing (4), turnover of β-amyloid in Alzheimer's disease (5), tumor invasion (6), tumor angiogenesis (7), and apoptosis (8, 9).
An in vivo validation of specific roles for cathepsins B and L proved to be difficult, likely due to overlapping substrate specificities of cysteine-type cathepsins. Cathepsin B null mice show no obvious phenotype (10), but a reduction in premature intrapancreatic trypsinogen activation (11) and increased resistance to tumor necrosis factor-α-mediated hepatocyte apoptosis (12) were observed under experimental conditions. Cathepsin L-deficient mice present only with a reduction in CD4+ T cells (13) and recurrent hair loss (14). In contrast, cathepsin K deficiency causes pyknodysostosis in mice and humans, consistent with its expression in osteoclasts and function in bone resorption (15, 16). In addition, loss-of-function mutations in the human cathepsin C gene have been associated with Papillon–Lefèvre syndrome, an autosomal recessive palmoplantar keratosis with periodontopathia (17).
To assess whether cathepsins B and L compensate for each other and to identify specific roles for these two proteases in vivo, all organs of mice lacking from one to four cathepsin B and L alleles were analyzed at various ages. Despite ubiquitous expression of the two proteases, homozygous double-mutant mice exhibit a selective neuronal vulnerability with rapid loss of cerebral cortical neurons as well as cerebellar Purkinje and granule cells. Dying cells display nuclear condensation, DNA fragmentation, and other features characteristic of apoptosis. As observed in human neurodegeneration and aging, fading of functional neurons is accompanied by an increase of glial tissue and preceded by the occurrence of intraneuronal inclusions. These are, however, ultrastructurally and biochemically distinct from those seen in classical Alzheimer's disease and neuronal ceroid lipofuscinoses (NCLs). Our data demonstrate that cathepsins B and L are required for integrity of the postnatal central nervous system and clarify previous assumptions concerning the biology of cathepsins B and L. The observed phenotype is, to our knowledge, without precedent in mice and provides an early-onset mouse model of brain atrophy.
Materials and Methods
Genotyping.
Mice examined in this study were from a mixed genetic background (C57BL/6 × 129/sv). The experiments were performed in compliance with the regulations of the Harvard Medical School Standing Committee on Animals. The neo-expression cassettes had been inserted into exon 4 of the cathepsin B and L genes upstream of the coding region for the active site of the enzymes (see ref. 10; AstraZeneca, London). PCR analyses were performed with primers CTSLex3F (common oligonucleotide; 5′-CATGGAGATGAACGCCTTTGG-3′), CTSLex4R (wild-type oligonucleotide; 5′-ATAGCCATTCACCACCTGCC-3′), CTSL-PGK-neo (mutant oligonucleotide; 5′-ATCGCCTTCTTGACGAGTTCTTC-3′), CTSBex4F (common oligonucleotide; 5′-GGTTGCGTTCGGTGAGG-3′), CTSBex4R (wild-type oligonucleotide; 5′-AACAAGAGCCGCAGGAGC-3′), and CTSB-pMC1-neo (mutant oligonucleotide; 5′-CGATCCCATATTGGCTGC-3′).
Histology and Immunohistochemistry.
Fifty double-mutant mice and their littermates were analyzed morphologically. Affected animals at postnatal day 10.5 (P10.5) and older (n = 19) were generally killed when they were close to death as measured by weight loss of up to one-fourth of their body weight and reduced activity. Of these, 11 cathepsin B−/−/L−/− mice represented the period in which double-mutants died if litters were not reduced (P10.5–17.5). Eight cathepsin B−/−/L−/− mice between P21.5 and P50.5 were derived from breedings with reduced litters representing the stages in which brain shrinkage became visible. To determine the age of onset of each of the phenotypes, 31 double-mutant mice and their littermates between E17.5 and P9.5 were studied. For initial histological analyses, tissues were fixed in Bouin's fixative (Sigma). Multiple coronal brain sections were taken of each animal. Several cathepsin B−/−/L−/− brains were serial sectioned. Samples of all tissues were analyzed from six cathepsin B−/−/L−/− animals (P12.5 twice, P17.5, P24.5, and P50.5 twice). In addition, brains of 5-, 8-, and 12-month-old cathepsin B−/−/L+/−, B+/−/L−/−, B−/−/L+/+, B+/+/L−/−, and B+/+/L+/+ mice were examined. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E), the periodic acid/Schiff reagent method counterstained with hematoxylin, and luxol fast blue in combination with cresylecht violet. Glial fibrillary acidic protein (GFAP)-immunohistochemistry was performed on P17.5, P21.5, P24.5, and P50.5 paraffin sections with polyclonal rabbit anti-cow GFAP (1:200, Dako) using the Vectastain Elite ABC kit and the diaminobenzidene substrate kit from Vector Laboratories. The antibody against subunit c of the mitochondrial F1F0-ATP synthase was kindly provided by E. Kominami (Department of Biochemistry, Juntendo University, Tokyo, Japan) and used as described on P17.5 brain sections (18).
In Situ Labeling of Fragmented DNA.
P16.5, P17.5, P23.5, and P24.5 double-mutant and control brains were frozen in OCT embedding medium (Tissue-Tek, Sakura Finetek Europe, B.V., Zoeterwoude, The Netherlands), serial sectioned, and thaw-mounted on Polysine slides (Menzel Gläser, Braunschweig, Germany). Apoptotic cells were identified with the terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) method by using the ApopTag Plus peroxidase In Situ apoptosis detection kit from Intergen (Oxford, U.K.) on 8-μm frozen sections representative of all major brain regions.
Electron Microscopy.
Brains of P7.5, P12.5, and P24.5 cathepsin B−/−/L−/− mice were fixed by intracardiac perfusion of Karnovsky's fixative. One cerebral hemisphere was processed for paraffin embedding and histopathological analysis. Semithin Epon-embedded sections of the other hemisphere were stained with toluidine blue for orientation. Ultrathin sections were stained with uranyl acetate and lead citrate and examined with a 1200EX JEOL electron microscope.
Biochemical Analyses.
Brains and spleens from P10.5, P12.5, P13.5, P17.5, and P18.5 representative mice of all nine genotypes were snap frozen and kept at −80°C. Tissues were homogenized in ice-cold PBS (10 ml/g of tissue) with five passes of a Teflon/glass Dounce homogenizer. The tissue homogenates were washed twice with PBS and pellets were kept at −80°C.
Preparation of the 125I-radiolabeled active-site-directed probes LHVS-PhOH and JPM-565, lysis of brain tissue homogenates, and labeling with the radioactive probes were performed as reported (19). Palmitoyl-protein-thioesterase- and tripeptidyl-peptidase-I enzyme activities were determined by using the fluorogenic substrates MU-6S-palm-βGlc (4-methylumbelliferyl-6-thiopalmitoyl β-d-galactopyranoside) (Moscerdam, Rotterdam, The Netherlands) and AAF-AMC (Ala-Ala-Phe-7-amido-4-methylcoumarin) (Sigma), respectively, and the assays were performed twice in triplicate as described (20–22).
Subcellular fractionations, preparations of enriched fractions of dense bodies, and preparations of proteolipids were performed as described (23–25). The anti-cathepsin D antibody used for Western blot analysis was supplied by Santa Cruz Biotechnology.
Results
Cathepsin B−/−/L−/− Mice Die in Infancy.
Mice homozygous for cathepsin B and L deficiency were generated by intercrossing cathepsin B+/−/L+/− mice and genotyped by PCR (Fig. 1a). Labeling of tissue homogenates with the thiol protease-specific active site probes 125I-LHVS-PhOH and 125I-JPM-565 confirmed that these proteins were absent in null mice (Fig. 1b). The numbers of offspring of each of the nine possible genotypes were in accord with Mendelian segregation at birth (5 double-mutants of 77 genotyped newborn animals). Newborn mice of all genotypes were indistinguishable visibly. However, cathepsin B−/−/L−/− mice gained weight only slowly after day 5, and their body weight started to decline during the second week of life. Most homozygous double-mutant mice died around day 12 in an emaciated state (Fig. 1c). No animal lacking all four cathepsin B and L alleles was found among 307 mice genotyped at day 21, whereas the expected proportions of all other genotypes were obtained.
Figure 1.

Early death in cathepsin B−/−/L−/− mice. (a) Genotyping of wild-type (lanes 1), cathepsin B−/−/L−/− (lanes 2), and cathepsin B+/−/L+/− (lanes 3) mice by PCR. (b) 125I-LHVS-PhOH and 125I-JPM565 active-site labeling of brain homogenates from cathepsin B+/−/L+/+ (lane 1), cathepsin B+/−/L+/− (lane 2), cathepsin B+/−/L−/− (lane 3), cathepsin B+/+/L+/− (lane 4), cathepsin B+/−/L+/− (lane 5), and cathepsin B−/−/L+/− (lane 6) mice. Cathepsins B and L (CTSB and CTSL) and their corresponding proforms were identified according to previous studies (19, 24). (c) Mortality diagram of wild-type (n = 20, dashed line) and cathepsin B−/−/L−/− mice (n = 14, solid line). Generally, the lifetime could be extended by only a few days through reduction of litter size to 3 to 5 pups at day 7 in further breedings (II) (n = 34). Importantly however, these breedings yielded 8 mice aged 21 to 50 days in which brain shrinkage became evident.
The lifespan of double-mutant mice increased by several days when healthy littermates were removed at day 7 (Fig. 1c). At this stage, growth impairment, combined with a delay in fur development, enabled the identification of the cathepsin B−/−/L−/− phenotype. Four cathepsin B−/−/L−/− mice survived the weaning period but depended on continuous nursing with wetted food twice daily. These mice demonstrated hesitating voluntary movements, a mild unusual tremor, and subtle rear limb spasticity. Characteristically, they swayed backwards while grooming and used their tail for balance. Animals of both sexes were killed at day 50, weighing less than half of the expected body weight. Thus, reduction of competitors in the cage and special animal care enabled us to study the pathological changes from late embryonal stages (E17.5/E18.5) through P50.5.
Cerebral and Cerebellar Atrophy in Cathepsin B−/−/L−/− Mice.
Besides hyperproliferation of keratinocytes as described for cathepsin L-deficient mice (14) and an unspecific involution of the thymus in terminally ill animals, histology of all organs revealed pathological features only in the brains of cathepsin B−/−/L−/− mice.
Fifty-day-old mice exhibited a pronounced cerebral and cerebellar atrophy. The total thickness of the cerebral cortex was significantly reduced when compared with that of wild-type or younger double-mutant mice (compare Fig. 2c with Fig. 2 a and b). On cut surfaces, the cerebral white matter could hardly be distinguished from gray matter and proved to be thinned in Luxol fast blue (LFB)-stained sections (Fig. 2c). Furthermore, the cerebellar molecular and internal granule cell layers were strikingly reduced (compare Fig. 2f with Fig. 2 d and e), and the Purkinje-cell layer had disappeared (compare Fig. 2j with Fig. 2h). These changes were the result of massive neuronal death in the cerebellar Purkinje- and granule-cell layers (Fig. 2i) and the cerebral cortex (Fig. 2g) during the third and fourth week of life.
Figure 2.

Cerebral and cerebellar atrophy in cathepsin B−/−/L−/− mice. (a–c) Luxol fast blue (LFB)-stained coronal sections of P50.5 littermate control (a), P17.5 (b), and P50.5 (c) cathepsin B−/−/L−/− cerebrum demonstrating cortical shrinkage accompanied by white-matter (wm) reduction between days 17 and 50 in double-mutants (cx = cortex; scale bar in a = 1 mm). The deep cerebral white matter is not yet myelinated in 17-day mutants (b). (d–f) LFB-stain of P50.5 wild-type (d), P17.5 (e), and P50.5 (f) cathepsin B−/−/L−/− cerebellum (m = molecular layer, g = granular layer; scale bar = 1 mm). (h–j) H&E stain of the above samples at ×100. Note the dying bright red Purkinje cell in i, the complete absence of the Purkinje cell layer (p), and the striking reduction in cellular density in the granule cell layer (g) in j. (g) Nuclear pyknosis, karyorrhexis, and brightly red-stained angular cytoplasm in dying cortical neurons at P24.5. Healthy neurons exhibit a lightly stained nucleus and a darkly stained nucleolus as seen in the bottom part of g. (k) Lipid-laden macrophages in the corpus callosum at P50.5 (×1250 in g–k).
To assess whether the numerous pyknotic and karyorrhectic nuclei seen in H&E stained neurons (Fig. 2 g and i) have fragmented DNA indicative of an apoptotic mechanism of cell death, TUNEL (26) was performed. Widespread TUNEL-positive staining was found in the serial sectioned P23.5–24.5 cathepsin B−/−/L−/− cerebral cortex and the cerebellar granule-cell layer (Fig. 3 b, c, and e), with only an occasional TUNEL-positive cell in the cerebellar molecular cell layer (data not shown). This staining was absent from wild-type control sections (Fig. 3 a and d). While intact up to postnatal day 16, the Purkinje-cell monolayer had largely vanished in P21.5–24.5 double-mutant mice. However, P17.5 cathepsin B−/−/L−/− brain sections revealed some TUNEL-positive cells in the Purkinje-cell monolayer (data not shown). At this stage, significantly fewer TUNEL-positive granule cells were observed when compared with P23.5–24.5 double-mutant sections. This observation suggests that Purkinje cells are the more likely first target.
Figure 3.

Positive TUNEL-labeling indicative of apoptotic cell death and positive GFAP immunostaining demonstrating reactive gliosis in response to neuron death in cathepsin B−/−/L−/− mice. (a–c) Control (a) and cathepsin B−/−/L−/− P24.5 (b and c) cryosections of the cerebral cortex showing small TUNEL-positive globules in the center of a shrunken cathepsin B−/−/L−/− neuron in b and division into apoptotic cell fragments in c. (d and e) Control (d) and cathepsin B−/−/L−/− (e) P24.5 granule layer of the cerebellar central lobe shows TUNEL-positive material around the periphery of double-mutant nuclei, suggesting chromatin margination. (f–i) GFAP-immunostaining of 6-μm coronal paraffin sections reveals abundant star-shaped hypertrophic astrocytes in the 50-day-old double-mutant cerebral cortex (g) not present in the control cortex (f). Control (h) and double-mutant (i) 50-day-old cerebellum shows the picture of an isomorphic gliosis in the latter. The processes of the Bergmann glia are in parallel orientation. Note the tiny healthy area in i with a residual Purkinje cell (arrow). [Scale bars = 7 μm (a–e), and 0.1 mm (f–i).]
Neuronal loss was paralleled by an increasing occurrence of hypertrophic astrocytes and Bergmann glia as demonstrated by immunostaining with an antibody against GFAP. Fifty-day-old double-mutant mice showed the most striking reactive gliosis in the cerebral cortex (Fig. 3g). Consistent with the almost complete loss of Purkinje cells, the P24.5 and P50.5 cathepsin B−/−/L−/− cerebellar cortex showed the picture of an isomorphic gliosis (Fig. 3i).
Accumulation of Membranous Compartments in Large Neurons and Axons.
Before TUNEL reactivity and cell death, cell bodies of about 50% of neurons in the cerebral cortex accumulated eosinophilic, periodic acid Schiff-, toluidine blue-, and LFB-positive material. The amount of inclusion material increased rapidly between P11.5 and P15.5 (Fig. 4 a and b). Already at P7.5, a large subset of Purkinje cells showed a haze of LFB stain and some granules (data not shown). Apart from occasional inclusions in the CA3 region, the hippocampus was spared. Similarly, only a few larger neurons in the thalamus and basal ganglia and select nuclei in the brainstem revealed some granules. The spinal cord and peripheral ganglia were largely unaffected. There was no motoneuron loss or neurogenic muscular atrophy, consistent with the observation that cathepsin B−/−/L−/− mice do not experience flaccid paralysis.
Figure 4.

Accumulation of amorphous and membranous material in neuronal perikarya, dendrites, and axons in cathepsin B−/−/L−/− mice. (a and b) LFB stain of P11.5 and P15.5 double-mutant cortical neurons. At P11.5 (a), only a few inclusions are observed in some neuronal perikarya (arrow). The arrowhead points to a normal neuron. Only a few days later, at P15.5 (b), the perikarya are expanded and filled with LFB-positive inclusions. (c–e) Electron micrographs of inclusions in P12.5 (c and d) and P24.5 (e) cathepsin B−/−/L−/− cortical neurons. (d) Higher magnification of the inclusion indicated by an arrow in c. (f) Sagittal section of the P11.5 double-knockout brain shown in a revealing a dense accumulation of eosinophilic globules in cerebral white matter (H&E stain). (g and h) Magnification of the area surrounding the asterisk shown in f stained with H&E and LFB, respectively. Normal white matter is seen in the upper right corner. All LFB stainings were performed simultaneously. (i) Electron micrograph of membranous inclusions in a swollen axon in the corpus callosum of a P12.5 cathepsin B−/−/L−/− mouse. [Scale bars = 15 μm (a, b, g, h), 2 μm (c), 100 nm (d), 1.6 μm (e), 0.15 mm (f), and 500 nm (i).]
Ultrastructurally, intraneuronal inclusions of 12-day-old cathepsin B−/−/L−/− mice consisted of electron-dense, often membrane-bound bodies of varying size and shape occasionally containing lamellae (Fig. 4 c and d), suggesting lysosomal residual bodies. These inclusions formed circumscribed, but loosely arranged, groups within the neuronal perikarya. Later, they occupied almost the entire perikaryon, displacing and replacing other organelles such as mitochondria (P24.5, Fig. 4e). The remaining neurons in 50-day-old mice were largely devoid of LFB-positive inclusions.
A hallmark of double-mutant mice, observed as early as day 3 and preceding all aforementioned changes, was a dense accumulation of eosinophilic and LFB-positive material appearing as round globules of less than 20 μm in axon-rich areas such as the corpus callosum, fornix, rostral commissure, fimbriae hippocampi, and deep cerebellar white matter (Fig. 4 f–h). Electron microscopy of the 12-day-old cathepsin B−/−/L−/− corpus callosum revealed expanded axons that contained a large number of dense bodies often replete with lamellae and mitochondria. Preexisting microtubules were displaced to the periphery of the cell process (Fig. 4i). While unique in location and dense packing, the profiles resembled axonal inclusions seen in other lysosomal diseases (27, 28).
Cathepsin B−/−/L−/− mice also showed a hippocampal lamination defect. Whereas the Ammon's horn of the hippocampus usually presents as a morphologically distinct C-shaped structure, the CA3 region of the stratum pyramidale was broadened and split in double-mutant mice older than P3.5 (U.F., R.T.B, and B.R.O, unpublished data). Variable phenotypes in cathepsin B−/−/L−/− mice included white matter hypodensities, a hydrocephalus ex vacuo, and a cavity forming between the corpus callosum and the ventral commissure of the fornix and extending rostrally to divide the septal nuclei (data not shown). The brains of 5-, 8-, and 12-month-old cathepsin B−/−/L+/−, B+/−/L−/−, B−/−/L+/+, B+/+/L−/−, and B+/+/L+/+ mice showed neither neuronal loss, nor intraneuronal LFB-positive inclusions, nor a hippocampal split, indicating that the observed phenotype requires the absence of both cathepsin B and L activity.
The Cathepsin B−/−/L−/− Phenotype Is Distinct from Classical NCLs.
The early and severe brain atrophy is highly reminiscent of NCLs, the most common group of autosomal-recessively inherited neurodegenerative disorders in children and young adults (29). However, lipopigment accumulation in NCLs is found in retina, skin, myocardium, and visceral organs which were inconspicuous in LFB-stained cathepsin B−/−/L−/− sections up to day 50. In addition, the ultrastructural morphology of the accumulated material in cathepsin B−/−/L−/− neurons did not unequivocally resemble the granular, curvilinear, or fingerprint profiles seen in NCLs (30). Furthermore, the activities of palmitoyl protein thioesterase (31) and tripeptidyl peptidase I (32), the only two enzymes known to be mutated in NCLs, were normal in 12-day-old and elevated in older cathepsin B−/−/L−/− brain lysates (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). Immunohistochemistry with an antibody against subunit c of the mitochondrial F1F0-ATP synthase (18), a standard marker for NCLs, showed no relevant staining in the cerebral and cerebellar cortex of cathepsin B−/−/L−/− mice (data not shown). Accordingly, dense-body preparations (25) from wild-type, cathepsin B−/−/L+/+, B+/+/L−/−, and B−/−/L−/− mice separated on SDS and Tris-Tricine polyacrylamide gels, proteolipid fractions prepared from dense bodies (25), and subcellular fractions of total brain homogenates (23) did not show any specific accumulation of polypeptides such as the subunit c of the mitochondrial F1F0-ATP synthase (data not shown).
Procathepsin D and Cathepsin D Are Increased in Cathepsin B−/−/L−/− Brains.
Accumulation of ceroid-lipofuscinous material was observed in sheep with an enzymatically inactive form of the aspartic protease cathepsin D (33) and in mice with a truncated cathepsin D polypeptide (34, 35). The accumulating inclusions in cathepsin B−/−/L−/− mice have a significantly earlier onset and a much more rapid progression resulting in apoptotic neuronal death and brain shrinkage not reported for cathepsin D-deficient mice. Because in vitro studies suggested, however, that the conversion of a 47-kDa cathepsin D intermediate into the mature 31-kDa enzyme might be accelerated by cysteine proteases after its transfer into dense lysosomes (36), we assessed whether such processing is impaired in brains lacking cathepsins B and L. Western-blot analyses showed that processing of procathepsin D is not blocked in cathepsin B−/−/L−/− brains (Fig. 5). Since the intermediate and mature forms of cathepsin D serve as organelle markers for prelysosomal and lysosomal compartments, respectively, the observed proportional increase of both forms in cathepsin B−/−/L−/− brains suggests that endosomal and lysosomal compartments accumulate with time.
Figure 5.
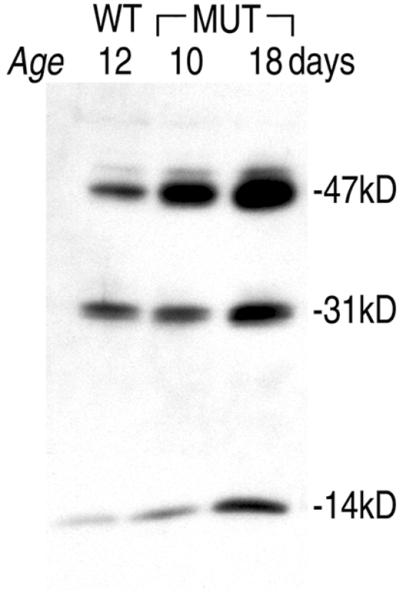
Endosomal and lysosomal compartments accumulate proportionally in null mice because processing of the short-lived 53-kDa precursor of cathepsin D into the 47-kDa intermediate form and the mature enzyme composed of a 31-kDa and a 14-kDa fragment is not altered in cathepsin B−/−/L−/− brain lysates. Ten micrograms of brain homogenates from wild-type (WT), 10-, and 18-day-old cathepsin B−/−/L−/− mice (MUT) were separated on 7.5–17.5% SDS/PAGE gels, electroblotted onto nitrocellulose, and probed with an anti-cathepsin D antibody.
Discussion
This study demonstrates that cathepsins B and L are essential for maturation and integrity of the postnatal central nervous system (CNS) and that the two proteases compensate for each other in vivo. Consistent with an increase of cathepsin B and L mRNA expression from normal newborn to 5-day-old mouse brains (37) and cathepsin B and L mRNA coexpression in rat brain neurons (38), cathepsin B−/−/L−/− mice exhibit histopathological alterations in the CNS that change rapidly as a function of time. The dramatic course of neurodegeneration in infant cathepsin B−/−/L−/− knockouts is unlike neuronal loss in, e.g., Munc18–1-deficient mice, which show a failure in prenatal development (39). It is also distinct from known mouse models of human NCLs, which have late disease onset and do not reveal cytological changes indicative of neuronal cell death and brain atrophy (40).
Cysteine proteases whose active-site sulfhydryl groups are oxidation-sensitive have been implicated in NCL etiology as well as in decreased protein turnover in aging brains. Intraventricular infusions of the broad-spectrum serine and cysteine protease inhibitor leupeptin and the cysteine protease inhibitor E-64c resulted in a general accumulation of dense granular aggregates throughout rodent brains, as well as in the retina and internal organs, thereby partially reproducing features of NCLs, Alzheimer's disease, and aging (41). Several in vitro studies using cultured hippocampal and cortical slices (42, 43) or cardiac myocytes (44) appeared to reproduce the in vivo experiments. In contrast, cathepsin B−/−/L−/− mice show no general pleiotropic effects in membrane turnover within their lifetime. One possible explanation for the selective neuronal vulnerability in cathepsin B−/−/L−/− knockouts is that the inhibitors also affect additional enzymes.
An initial biochemical characterization of cathepsin B−/−/L−/− mice revealed no obvious metabolic substrate accumulating in cathepsin B−/−/L−/− brains. It remains to be clarified whether the earliest and pathognomonic finding, the accumulation of membranous compartments in cathepsin B−/−/L−/− axons throughout the corpus callosum far distant from neuronal perikarya, as well as the perikaryal inclusions in large neurons, is causally related to neurodegeneration or result from the cell's protective response to earlier pathogenetic mechanisms. Taking into account that the pH range of cathepsins B and L should allow activity in less acidic endosomal compartments (1, 2), it is conceivable that these two cathepsins are more widely involved in the homeostasis of vesicle trafficking and formation of axons and synaptic connections during early postnatal life. The early onset and rapid progression of neurodegeneration should make cathepsin B−/−/L−/− mice ideal for further studies into the neurobiology of these enzymes and for evaluation of therapeutic strategies designed to counteract apoptosis or to replace dying or lost neurons.
Supplementary Material
Acknowledgments
We thank C. Peters and J. Deussing for providing cathepsin B−/− mice, AstraZeneca for cathepsin L−/− mice, E. Kominami for the antibody against subunit c of the mitochondrial F1F0-ATP synthase, and N. Rüttling, L. Zhang, and M. Ericsson for expert assistance with the histotechnology and electron microscopy. This work was supported by Deutsche Forschungsgemeinschaft Grant Fe 432/6-1 (to U.F.), the Human Science Frontier Program Organisation (B.K.), Hoffman La Roche (B.K.), Entremed, Inc. (B.R.O.), and the National Institutes of Health (H.L.P. and B.R.O.).
Abbreviations
- GFAP
glial fibrillary acidic protein
- H&E
hematoxylin/eosin
- LFB
luxol fast blue
- NCL
neuronal ceroid lipofuscinosis
- Pn
postnatal day n
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mort J S. In: Handbook of Proteolytic Enzymes. Barrett A J, Rawlings N D, Woessner J F, editors. San Diego: Academic; 1998. pp. 609–617. [Google Scholar]
- 2.Kirschke H. In: Handbook of Proteolytic Enzymes. Barrett A J, Rawlings N D, Woessner J F, editors. San Diego: Academic; 1998. pp. 617–621. [Google Scholar]
- 3.Blum J S, Cresswell P. Proc Natl Acad Sci USA. 1988;85:3975–3979. doi: 10.1073/pnas.85.11.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shinagawa T, Do Y S, Baxter J D, Carilli C, Schilling J, Hsueh W A. Proc Natl Acad Sci USA. 1990;87:1927–1931. doi: 10.1073/pnas.87.5.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein H-G, Kirschke H, Wiederanders B, Pollak K-H, Zipress A, Rinne A. Mol Chem Neuropathol. 1996;27:225–247. doi: 10.1007/BF02815106. [DOI] [PubMed] [Google Scholar]
- 6.Berquin I M, Sloane B F. Adv Exp Med Biol. 1996;389:281–294. doi: 10.1007/978-1-4613-0335-0_35. [DOI] [PubMed] [Google Scholar]
- 7.Felbor U, Dreier L, Bryant R A, Ploegh H L, Olsen B R, Mothes W. EMBO J. 2000;19:1187–1194. doi: 10.1093/emboj/19.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isahara K, Ohsawa Y, Kanamori S, Shibata M, Waguri S, Sato N, Gotow T, Watanabe T, Momoi T, Urase K, et al. Neuroscience. 1999;91:233–249. doi: 10.1016/s0306-4522(98)00566-1. [DOI] [PubMed] [Google Scholar]
- 9.Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, Elling F, Leist M, Jäättelä M. J Cell Biol. 2001;153:999–1009. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deussing J, Roth W, Saftig P, Peters C, Ploegh H L, Villadangos J A. Proc Natl Acad Sci USA. 1998;95:4516–4521. doi: 10.1073/pnas.95.8.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halangk W, Lerch M M, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. J Clin Invest. 2000;106:773–781. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guicciardi M E, Deussing J, Miyoshi H, Bronk S F, Svingen P A, Peters C, Kaufmann S H, Gores G J. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa T, Roth W, Wong P, Nelson A, Farr A, Deussing J, Villadangos J A, Ploegh H, Peters C, Rudensky A Y. Science. 1998;280:450–453. doi: 10.1126/science.280.5362.450. [DOI] [PubMed] [Google Scholar]
- 14.Roth W, Deussing J, Botchkarev V A, Pauly-Evers M, Saftig P, Hafner A, Schmidt P, Schmahl W, Scherer J, Anton-Lamprecht I, et al. FASEB J. 2000;14:2075–2086. doi: 10.1096/fj.99-0970com. [DOI] [PubMed] [Google Scholar]
- 15.Gelb B D, Shi G-P, Chapman H A, Desnick R J. Science. 1996;273:1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- 16.Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz J D, Schu P, von Figura K. Proc Natl Acad Sci USA. 1998;95:13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toomes C, James J, Wood A J, Wu C L, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods C G, et al. Nat Genet. 1999;23:421–424. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- 18.Kominami E, Ezaki J, Muno D, Ishido K, Ueno T, Wolfe L S. J Biochem (Tokyo) 1992;111:278–282. doi: 10.1093/oxfordjournals.jbchem.a123749. [DOI] [PubMed] [Google Scholar]
- 19.Bogyo M, Verhelst S, Bellingard-Dubouchaud V, Toba S, Greenbaum D. Chem Biol. 2000;7:27–38. doi: 10.1016/s1074-5521(00)00061-2. [DOI] [PubMed] [Google Scholar]
- 20.Sohar I, Lin L, Lobel P. Clin Chem. 2000;46:1005–1008. [PubMed] [Google Scholar]
- 21.van Diggelen O P, Keulemans J L M, Winchester B, Hofman I L, Vanhanen S L, Santavuori P, Voznyi Y V. Mol Genet Metab. 1999;66:240–244. doi: 10.1006/mgme.1999.2809. [DOI] [PubMed] [Google Scholar]
- 22.Vines D J, Warburton M J. FEBS Lett. 1999;443:131–135. doi: 10.1016/s0014-5793(98)01683-4. [DOI] [PubMed] [Google Scholar]
- 23.Castellino F, Germain R N. Immunity. 1995;2:73–88. doi: 10.1016/1074-7613(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 24.Driessen C, Bryant R A, Lennon-Dumenil A M, Villadangos J A, Bryant P W, Shi G-P, Chapman H A, Ploegh H L. J Cell Biol. 1999;147:775–790. doi: 10.1083/jcb.147.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faust J R, Rodman J S, Daniel P F, Dice J F, Bronson R T. J Biol Chem. 1994;269:10150–10155. [PubMed] [Google Scholar]
- 26.Gavrieli Y, Sherman Y, Ben-Sasson S A. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolman C L, MacLeod P M, Chang E. J Neurol Neurosurg Psychiatry. 1977;40:588–594. doi: 10.1136/jnnp.40.6.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter S, Goebel H H. Acta Neuropathol. 1988;76:489–495. doi: 10.1007/BF00686388. [DOI] [PubMed] [Google Scholar]
- 29.Rider J A, Rider D L. Mol Genet Metab. 1999;66:231–233. doi: 10.1006/mgme.1999.2827. [DOI] [PubMed] [Google Scholar]
- 30.Elleder M, Lake B D, Goebel H H, Rapola J, Haltia M, Carpenter S. In: The Neuronal Ceroid Lipofuscinoses (Batten Disease) Goebel H H, Mole S E, Lake B D, editors. Amsterdam: IOS Press; 1999. pp. 5–15. [Google Scholar]
- 31.Vesa J, Hellsten E, Verkruyse L A, Camp L A, Rapola J, Santavuori P, Hofmann S L, Peltonen L. Nature (London) 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 32.Sleat D E, Donnelly R J, Lackland H, Liu C-G, Sohar I, Pullarkat R K, Lobel P. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 33.Tyynelä J, Sohar I, Sleat D E, Gin R M, Donnelly R J, Baumann M, Haltia M, Lobel P. EMBO J. 2000;19:2786–2792. doi: 10.1093/emboj/19.12.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H, Koster A, Hess B, Evers M, von Figura K, et al. EMBO J. 1995;14:3599–3608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, et al. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gieselmann V, Hasilik A, von Figura K. J Biol Chem. 1985;260:3215–3220. [PubMed] [Google Scholar]
- 37.Söderström M, Salminen H, Glumoff V, Kirschke H, Aro H, Vuorio E. Biochim Biophys Acta. 1999;1446:35–46. doi: 10.1016/s0167-4781(99)00068-8. [DOI] [PubMed] [Google Scholar]
- 38.Petanceska S, Burke S, Watson S J, Devi L. Neuroscience. 1994;59:729–738. doi: 10.1016/0306-4522(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 39.Verhage M, Maia AS, Plomp J J, Brussaard A B, Heeroma J H, Vermeer H, Toonen R F, Hammer R E, van den Berg T K, Missler M, et al. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 40.Bronson R T, Donahue L R, Johnson K R, Tanner A, Lane P W, Faust J R. Am J Med Genet. 1998;77:289–297. doi: 10.1002/(sici)1096-8628(19980526)77:4<289::aid-ajmg8>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 41.Ivy G O, Schottler F, Wenzel J, Baudry M, Lynch G. Science. 1984;226:985–987. doi: 10.1126/science.6505679. [DOI] [PubMed] [Google Scholar]
- 42.Bednarski E, Ribak C E, Lynch G. J Neurosci. 1997;17:4006–4021. doi: 10.1523/JNEUROSCI.17-11-04006.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yong A P, Bednarski E, Gall C M, Lynch G, Ribak C E. Exp Neurol. 1999;157:150–160. doi: 10.1006/exnr.1999.7038. [DOI] [PubMed] [Google Scholar]
- 44.Terman A, Brunk U T. Mech Ageing Dev. 1998;100:145–156. doi: 10.1016/s0047-6374(97)00129-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}