

Abstract
Immunotherapy has revolutionized cancer management, but many tumors, particularly immunologically “cold” tumors, remain resistant. Combination of conventional systemic immunotherapies with locoregional interventional radiology approaches is being explored to transform these “cold” tumors into immunologically active “hot” ones. The present article uses the example of chromophobe renal cell carcinoma (ChRCC), a renal cell carcinoma subtype resistant to current systemic immunotherapies, to address practical and conceptual challenges that have to date prevented the activation of clinical trials specifically designed for this malignancy. The practical framework discussed herein can help overcome logistical and funding limitations and facilitate the development of biology-informed clinical trials tailored to specific rare diseases such as ChRCC.
Introduction
Immunotherapy has transformed the management of patients with cancer (1, 2). However, many tumors still do not show objective responses in a majority of patients treated with immunotherapy. Resistance to immunotherapies can occur through numerous mechanisms, potentially disrupting any phase of the cancer immunity cycle (3). For example, immunologically “cold” tumors are poorly infiltrated by cytotoxic T cells and are therefore less likely to demonstrate clinical responses to conventional systemic immunotherapies (4). Therefore, combinations of conventional systemic immunotherapies with novel approaches are being explored to convert “cold” tumors into immunologically “hot” tumors (5, 6). Locoregional interventional radiology strategies are accordingly being developed to intratumorally deliver immune-activating agents such as small molecules, antibodies, oncolytic viruses, and gene therapies alone or in combination with systemic immunotherapies (7, 8). Nevertheless, challenges such as tumor heterogeneity as well as logistical and funding limitations impede the clinical development of these strategies. The present article aims to provide practical suggestions on how to efficiently address these hurdles using renal cell carcinoma (RCC) as the motivating example.
RCCs are a heterogeneous group of tumors of which clear cell RCC (ccRCC) is the most prevalent subtype, accounting for approximately 75% of RCC cases. The remaining 25% fall under the broad categorization of non-clear cell RCCs, also known as variant histology RCCs (9, 10). The effectiveness of treatments can significantly differ among non-clear cell RCCs because each subtype has distinct clinical, biological, and immune hallmarks (9–12). For this reason, it is crucial to develop prospective clinical trials tailored to each specific non-clear cell RCC subtype (13). Such clinical trials have been activated for common non-clear cell RCCs such as papillary RCC (PRCC)(13, 14) and rare subtypes such as renal medullary carcinoma (RMC) (15, 16). However, to date there have no clinical trials developed specifically for chromophobe RCC (ChRCC), a subtype that is particularly resistant to all currently available systemic therapies and may thus benefit from the testing of locoregional immunomodulatory strategies (9, 10). To address this unmet need, it is important to first consider whether and how reliable evidence can be generated from clinical trials in rare diseases such as metastatic ChRCC.
Methodological considerations
Despite the large number of systemic therapies currently available for the treatment of RCC, their regulatory approvals were predominantly based on randomized controlled trials (RCTs) in ccRCC (9, 10, 17). The methodological framework that can inform and justify the development of trials for more rare non-clear cell RCC subtypes such as RMC and ChRCC relies on the relevance-robustness trade-off (Figure 1), which is a clinical recasting of the more general bias-variance trade-off observed in all data science. Technical elaboration of this framework and its clinical implications across diseases has been provided elsewhere (18–20).
Figure 1.
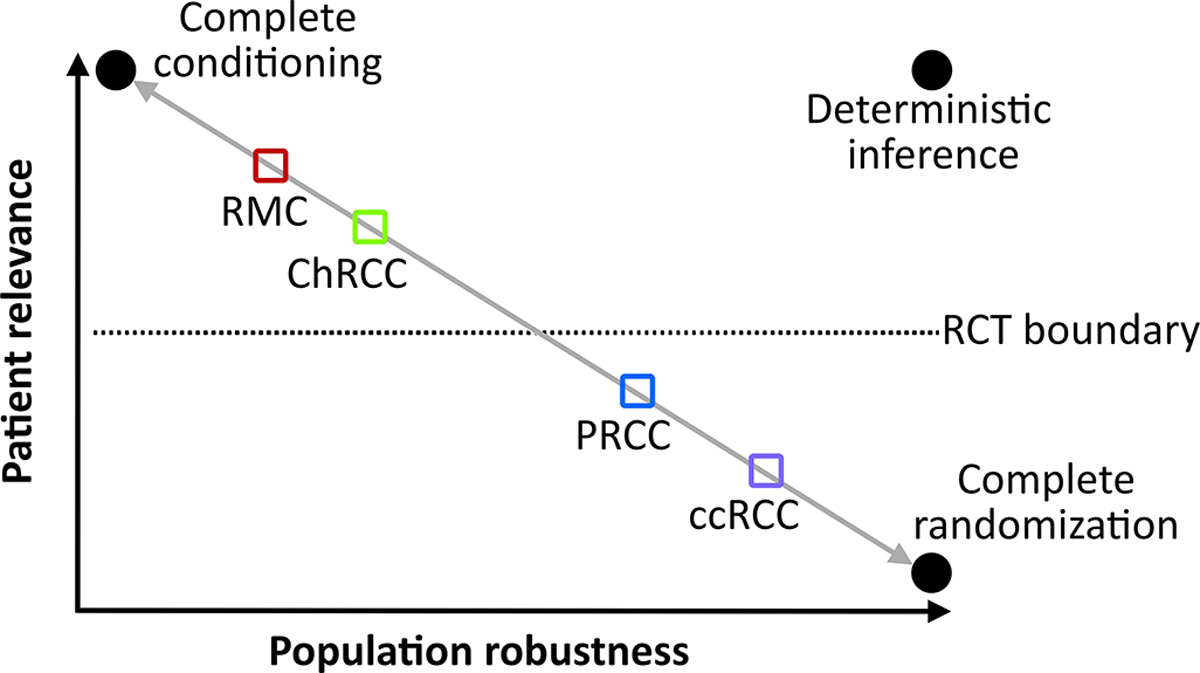
Renal cell carcinoma subtypes mapped according to their patient relevance and population robustness coordinates. The gray diagonal line is the feasibility diagonal that optimally balances relevance and robustness. Moving above the diagonal requires additional assumptions or sacrifices for each renal cell carcinoma subtype. Moving below the feasibility diagonal is a suboptimal position that does not account for important ancillary features of the available data for each histology. Randomization allows the valid estimation of robustness and is therefore necessary for more heterogeneous populations such as PRCC and ccRCC. The more homogeneous histologies above the dotted line corresponding to the “RCT boundary” can be studied by highly relevant biology-driven trials that do not require the large sample sizes used in RCTs. Abbreviations: ccRCC, clear cell renal cell carcinoma; ChRCC, chromophobe renal cell carcinoma; PRCC, papillary renal cell carcinoma; RCT, randomized controlled trial; RMC, renal medullary carcinoma.
Assume we are tasked with choosing between two treatment options, such as immunotherapy or the multi-receptor tyrosine kinase inhibitor (TKI) cabozantinib, for a patient with PRCC. To achieve this, we will have to predict the potential outcomes of using cabozantinib or immunotherapy in exact copies of this particular patient (21, 22). “Conditioning” is the process of accounting for the characteristics defining our patient, as opposed to simply choosing therapies that work “on average” (18–20). Studies performed in other patients who share common characteristics with our patient are “relevant”. Accordingly, studies that perform “apples-to-apples” comparisons between patients that are highly balanced for key PRCC characteristics are relevant to our patient. “Robustness” is the ability of our inferences to withstand perturbations in patient characteristics (23). Therefore, the higher the robustness of a study, the more applicable its results will be on average across heterogeneous patient populations (19, 20). The random allocation of treatments in RCTs by its very design does not yield “apples-to-apples” comparisons. This is because randomization is expected to produce random imbalances in relevant characteristics between groups. But the brilliant, albeit counterintuitive, insight obtained by randomization is that it physically justifies the use of statistical measures of uncertainty such as p-values and confidence intervals to reliably quantify the robustness of RCTs (19, 24).
Figure 1 illustrates how patient relevance and population robustness can be used to design RCC research that can inform histology-specific clinical practice. If we knew perfectly how perturbations in RCC characteristics influence outcomes, then we would achieve deterministic inferences shown on the upper right coordinates of the relevance-robustness plane (Figure 1). However, the complexity of human diseases such as RCC necessitates the use of stochastic rather than deterministic inferences. Perfect relevance in stochastic inference is achieved by complete conditioning if we know every source of outcome heterogeneity for the treatment options being considered for a specific patient. This is practically impossible in all but the most well-characterized diseases. Perfect robustness in stochastic inference is achieved by complete randomization inference via both random allocation and random sampling. This allows reliable, physically justifiable, and fully generalizable estimation of the average treatment effect in a patient population (25, 26). While all medical RCTs use random allocation, they enroll convenience samples of patients who are eligible, available, and willing to participate. No oncology RCT to date has enforced participation from the general population via random sampling. Therefore, there will always be a trade-off between patient relevance and robustness alongside a feasibility continuum illustrated by the gray diagonal line in Figure 1. For example, an RCT that compared cabozantinib with immunotherapy in patients with RCC will be relevant for our patient with PRCC only if it conditioned for PRCC histology.
Conditioning is practically feasible for more homogeneous diseases such as RMC driven by specific biological drivers (Figure 1) (11). Carefully conducted biology-driven small trials and mechanistic studies can meaningfully inform patient care in these situations. Conversely, more heterogeneous histologies such as ccRCC and PRCC require robust inferences obtained ideally by RCTs (Figure 1). Furthermore, emerging insights may allow splitting of ccRCC and PRCC into more homogeneous clinical and molecular subgroups to increase patient relevance. Clinical risk models such as the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) score can be used for this purpose (27, 28). For example, molecular insights have additionally allowed the distinction of fumarate hydratase-deficient RCC from PRCC and the development of tailored clinical trials and therapeutic strategies targeting oncogenic pathways induced by FH deficiency (12, 29, 30). As relevance increases by splitting RCC subtypes into more homogeneous subgroups, it becomes progressively more difficult to enroll adequate sample sizes that can yield robust inferences. This is why there is always a trade-off between relevance and robustness (18–20).
Focusing on patient relevance requires very careful considerations informed by deep understanding of clinical and biological context as well as use of efficient trial designs. As the statistician and physicist Edwin Jaynes wrote: “Whenever there is a randomized way of doing something, then there is a nonrandomized way that delivers better performance but requires more thought” (31). Therefore, to design trials testing the use of locoregional therapies in ChRCC it is critical to very clearly delineate the biological and clinical parameters that will facilitate relevance for this histology.
Clinical and biological considerations
ChRCC is the third most common RCC, consisting of ~5% of cases (9, 10, 32), with approximately 4,090 new cases expected to occur in the United States in 2023 (33). Only 5–10% of these patients with ChRCC will develop metastatic disease. However, the currently available systemic immunotherapies and TKIs were developed primarily for the treatment of ccRCC and demonstrate limited efficacy against ChRCC (9, 10). This highlights the need to develop tailored therapies for metastatic ChRCC. Emerging areas of interest focus on addressing ferroptosis as a metabolic vulnerability of ChRCC (34), as well as modulation of the interleukin-15 (IL-15) pathway via locoregional or systemic interventions to enhance anti-tumor immunity (35). Such strategies may be explored using the trial design considerations explored herein.
In contrast to other RCC subtypes like ccRCC and PRCC, the mutation profile of metastatic CHRCC uniquely consists of a significant number of TP53 mutations (found in 58% of cases), PTEN mutations (24% of cases), and uneven chromosome duplications (36). This mutation profile is more diverse than that of other RCCs such as RMC which is invariably driven by the loss of the tumor suppressor SMARCB1 resulting in a uniformly aggressive malignancy (11). Furthermore, ChRCC tumors are immunologically “cold” and harbor substantially less immune cell infiltrates than ccRCC, PRCC, or RMC (11, 12). As a result, ChRCC is highly refractory to immune checkpoint therapies (ICTs) such as the combination of the programmed cell death protein 1 (PD-1) inhibitor nivolumab with the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitor ipilimumab. A recent meta-analysis of 13 clinical studies revealed an objective response rate (ORR) of only 6.3% [95% confidence interval (CI) 1.9% to 12.4%] to systemic ICT alone or in combinations (10). There was notably low between-study heterogeneity as measured by the Cochran Q statistic (X2) = 8.22 (p=0.77). There was also no evidence of clinically relevant between-study variance (τ2 = 0) or total variability due to between-study heterogeneity (I2 = 0%) (10). These metrics all suggest that ChRCC consistently demonstrates poor responses to systemic ICTs across all 13 studies reported to date. Therefore, an intervention that improves this ORR to > 10% would be clinically relevant.
Notably, ChRCC is almost twice more likely to metastasize to the liver than ccRCC (34% versus 18% of cases) (37). This allows the potential use of liver-directed locoregional therapies to reprogram the immunologically cold state into an ICT responsive state (7, 38, 39). Thus, if there is biological rationale motivating the clinical investigation of an interventional radiology (IR) locoregional immunomodulatory approach exclusively to the liver, then this would be feasible if the trial aims to enroll a sample size of up to 30 patients. This is the maximum sample size used for all RMC-specific clinical trials to date (NCT03274258, NCT03587662, and NCT05347212 at clinicaltrials.gov) and would be realistic for liver-directed metastatic ChRCC trials given that approximately 70 new patients in the United States are expected to be diagnosed with ChRCC metastatic to the liver within 2023 alone (33). Accrual rates can be even higher if the locoregional therapy can instead be administered to other sites where ChRCC commonly metastasizes such as the lymph nodes (approximately 51% of cases) or the bone (approximately 33% of cases) (37).
Trial design
Practical limitations prohibit the enrollment of large sample sizes of patients with metastatic ChRCC in dedicated trials for this histology. However, one biology-driven trial (NCT03587662 at clinicaltrials.gov) have successfully completed enrollment of the projected 30 patients with metastatic RMC within less than 4 years from activation at a single center in the United States despite delays posed by the concurrent COVID-19 pandemic. The incidence of metastatic RMC is lower than that of metastatic ChRCC (9, 10). Therefore a maximum sample size of up to 30 patients would be very realistic for this RCC subtype. The trial design can follow the same Bayesian optimal phase II (BOP2) approach successfully used in the RMC trial (40). BOP2 allows any number of interim analyses for futility and toxicity, and can accommodate both binary and complicated efficacy endpoints while controlling the type I error rate. Furthermore, a suite of easy to use web-based BOP2 designs are freely available at http://www.trialdesign.org. This website is accessible to all stakeholders and provides an intergrated platform for developing clinical trials reproducibly using phase I designs such as the Bayesian optimal interval (BOIN) approach (41), phase II designs such as BOP2, and phase I-II designs such as the Bayesian Optimal Interval Phase I-II (BOIN12) dose-finding method (42).
Say we want to design a trial testing whether the addition of IR-based locoregional therapy to liver metastases can activate immune responses to ICT with nivolumab plus ipilimumab in ChRCC. For simplicity, we will assume in this hypothetical example that phase I testing of this locoregional therapy in combination with nivolumab plus ipilimumab has already been completed with no prohibitory safety signals observed. Therefore, phase II testing of the hypothetical combination can now be conducted in cancers such as ChRCC. Our phase II trial will enroll up to 30 patients with ChRCC metastatic to the liver. We will also conduct interim analyses for futility with every additional 10 patients enrolled. This will allow us to discontinue the trial and allocate resources towards other regimens if insufficient efficacy is observed after n = 10 or n = 20 patients have been enrolled. The primary endpoint will be ORR defined as the proportion of patients achieving a best response of either complete response or partial response according to the Response Evaluation Criteria In Solid Tumors (RECIST) 1.1 criteria recorded from the first day of the study until either the date of objectively documented progression per RECIST 1.1 or the date when subsequent anti-cancer treatment is initiated, whichever occurs first.
The null hypothesis for the trial can be ORR = 10%, which is more conservative than the point estimate of ORR = 6.3% observed in the most recent meta-analysis of ICT efficacy in metastatic ChRCC (10). Our chosen null hypothesis is in the upper range of the 95% CI for this estimate and thus lowers the likelihood of false positive signals. To further reduce the chances of false positive assertions, we will choose an alternative hypothesis of ORR = 30%, strictly control the two-sided type I error rate at 5% (i.e., the probability of falsely rejecting the null hypothesis when the null hypothesis is correct will be 5%), and use the default BOP2 vague prior probability corresponding to effective sample size = 1 (41). This produces the stopping boundaries listed in Table 1 that yield a statistical power of ~83% under the alternative hypothesis. Accodring to these stopping rules, if no objective responses are noted after enrolling the first 10 patients, or only 2 patients demonstrate objective response after enrolling 20 patients, then the trial should not continue enrolling further. Furthermore, the trial will meet its primary endpoint only if at least 7 out of 30 patients (23.3%) have demonstrated an objective response. Indeed, a standard frequentist 1-sample proportion test with Yates continuity correction (43), code in R V 4.3.0: prop.test(x=7, n=30, conf.level=0.95, correct = TRUE), reveals that the 95% CI for this ORR = 23.3% will be 10.6% to 42.7% and will therefore not include the null hypothesis of ORR = 10%. Therefore, the proposed design demonstrates good operating characteristics while being realistic and feasible within a reasonable timeframe and logistical constraints.
Table 1.
Stopping citeria for insufficient objective responses during the proposed trial of a logoregional immunomodulatory therapy in combination with nivolumab plus ipilimumab.
| Number of patients treated | Stop if there are this many patients (or fewer) with an objective response |
|---|---|
| 10 | 0 |
| 20 | 2 |
| 30 | 6 |
Emphasis should be placed on gaining information that will move the field forward and prepare the next steps regardless of whether the trial’s primary endpoint is “positive” or “negative”. Towards this goal, the trial should ideally longitudinally collect correlative blood and tissue samples that can be used for basic and translational studies to identify therapeutic vulnerabilities as well as biomarkers and other potential sources of heterogeneity (20, 44). The ability to collect tumor samples during treatment is an advantage of IR-based locoregional approaches versus other metastasis-directed strategies such as radiation therapy (45).
Conclusion
ChRCC is an immunologically “cold” tumor that spreads to areas, such as the liver, that are amenable to IR-based locoregional therapies. The standard systemic ICT regimens are ineffective creating an unmet need and opportunity for biology-driven clinical trials focused on this RCC subtype. The focus of these trials can be to reprogram the tumor immune microenvironment towards an immunologically active state that is responsive to ICT. Such trials can be realistically designed and conducted with reasonable operating characteristics at reasonable samples sizes of N = 30 patients. Biological and clinical knowledge is key to enhancing patient relevance and can be augmented by high-quality prospective clinical trial data that rigorously collect patient specimens to support correlative research irrespective of the trial meeting its clinical efficacy endpoints.
References
- 1.Sharma P, Allison JP. Dissecting the mechanisms of immune checkpoint therapy. Nat Rev Immunol 2020; 20:75–6. [DOI] [PubMed] [Google Scholar]
- 2.Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol 2020; 27:S87–S97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017; 168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria JC. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov 2021; 11:874–99. [DOI] [PubMed] [Google Scholar]
- 5.Kirchhammer N, Trefny MP, Auf der Maur P, Laubli H, Zippelius A. Combination cancer immunotherapies: Emerging treatment strategies adapted to the tumor microenvironment. Sci Transl Med 2022; 14:eabo3605. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Huang D, Saw PE, Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol 2022; 43:523–45. [DOI] [PubMed] [Google Scholar]
- 7.Brito-Orama S, Sheth RA. The Contemporary Landscape and Future Directions of Intratumoral Immunotherapy. J Immunother Precis Oncol 2023; 6:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheth RA, Murthy R, Hong DS, et al. Assessment of Image-Guided Intratumoral Delivery of Immunotherapeutics in Patients With Cancer. JAMA Netw Open 2020; 3:e207911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zoumpourlis P, Genovese G, Tannir NM, Msaouel P. Systemic Therapies for the Management of Non-Clear Cell Renal Cell Carcinoma: What Works, What Doesn’t, and What the Future Holds. Clin Genitourin Cancer 2021; 19:103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Msaouel P, Genovese G, Tannir NM. Renal Cell Carcinoma of Variant Histology: Biology and Therapies. Hematol Oncol Clin North Am 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Msaouel P, Malouf GG, Su X, et al. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020; 37:720–34 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linehan WM, Ricketts CJ. The Cancer Genome Atlas of renal cell carcinoma: findings and clinical implications. Nat Rev Urol 2019; 16:539–52. [DOI] [PubMed] [Google Scholar]
- 13.Maughan BL, Sirohi D. Papillary Renal Cell Carcinoma: A Review of Prospective Clinical Trials. Curr Treat Options Oncol 2023. [DOI] [PubMed] [Google Scholar]
- 14.Pal SK, Tangen C, Thompson IM Jr., et al. A comparison of sunitinib with cabozantinib, crizotinib, and savolitinib for treatment of advanced papillary renal cell carcinoma: a randomised, open-label, phase 2 trial. Lancet (London, England) 2021; 397:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Msaouel P, Hong AL, Mullen EA, et al. Updated Recommendations on the Diagnosis, Management, and Clinical Trial Eligibility Criteria for Patients With Renal Medullary Carcinoma. Clin Genitourin Cancer 2019; 17:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Msaouel P, Walker CL, Genovese G, Tannir NM. Molecular hallmarks of renal medullary carcinoma: more to c-MYC than meets the eye. Mol Cell Oncol 2020; 7:1777060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adashek JJ, Genovese G, Tannir NM, Msaouel P. Recent advancements in the treatment of metastatic clear cell renal cell carcinoma: A review of the evidence using second-generation p-values. Cancer Treat Res Commun 2020; 23:100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craiu RV, Gong R, Meng X-L. Six Statistical Senses. Annual Review of Statistics and Its Application 2023; 10:null. [Google Scholar]
- 19.Liu K, Meng X-L. There Is Individualized Treatment. Why Not Individualized Inference? Annual Review of Statistics and Its Application 2016; 3:79–111. [Google Scholar]
- 20.Msaouel P The Big Data Paradox in Clinical Practice. Cancer Invest 2022; 40:567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubin DB. Causal Inference Using Potential Outcomes. Journal of the American Statistical Association 2005; 100:322–31. [Google Scholar]
- 22.Msaouel P, Lee J, Karam JA, Thall PF. A Causal Framework for Making Individualized Treatment Decisions in Oncology. Cancers (Basel) 2022; 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaelin WG Jr. Common pitfalls in preclinical cancer target validation. Nat Rev Cancer 2017; 17:425–40. [DOI] [PubMed] [Google Scholar]
- 24.Senn S Seven myths of randomisation in clinical trials. Stat Med 2013; 32:1439–50. [DOI] [PubMed] [Google Scholar]
- 25.Senn S Controversies concerning randomization and additivity in clinical trials. Stat Med 2004; 23:3729–53. [DOI] [PubMed] [Google Scholar]
- 26.Degtiar I, Rose S. A Review of Generalizability and Transportability. Annual Review of Statistics and Its Application 2023; 10:null. [Google Scholar]
- 27.Heng DY, Xie W, Regan MM, et al. External validation and comparison with other models of the International Metastatic Renal-Cell Carcinoma Database Consortium prognostic model: a population-based study. Lancet Oncol 2013; 14:141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connor Wells J, Donskov F, Fraccon AP, et al. Characterizing the outcomes of metastatic papillary renal cell carcinoma. Cancer Med 2017; 6:902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linehan WM, Rouault TA. Molecular pathways: Fumarate hydratase-deficient kidney cancer--targeting the Warburg effect in cancer. Clin Cancer Res 2013; 19:3345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skala SL, Dhanasekaran SM, Mehra R. Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome (HLRCC): A Contemporary Review and Practical Discussion of the Differential Diagnosis for HLRCC-Associated Renal Cell Carcinoma. Arch Pathol Lab Med 2018; 142:1202–15. [DOI] [PubMed] [Google Scholar]
- 31.Jaynes ET, Bretthorst GL. Probability theory : the logic of science. Cambridge, UK ; New York, NY: Cambridge University Press, 2003. [Google Scholar]
- 32.Garje R, Elhag D, Yasin HA, Acharya L, Vaena D, Dahmoush L. Comprehensive review of chromophobe renal cell carcinoma. Crit Rev Oncol Hematol 2021; 160:103287. [DOI] [PubMed] [Google Scholar]
- 33.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023; 73:17–48. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Hobeika CS, Khabibullin D, et al. Hypersensitivity to ferroptosis in chromophobe RCC is mediated by a glutathione metabolic dependency and cystine import via solute carrier family 7 member 11. Proc Natl Acad Sci U S A 2022; 119:e2122840119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kansler ER, Dadi S, Krishna C, et al. Cytotoxic innate lymphoid cells sense cancer cell-expressed interleukin-15 to suppress human and murine malignancies. Nat Immunol 2022; 23:904–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casuscelli J, Becerra MF, Seier K, et al. Chromophobe Renal Cell Carcinoma: Results From a Large Single-Institution Series. Clin Genitourin Cancer 2019; 17:373–9 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudani S, de Velasco G, Wells JC, et al. Evaluation of Clear Cell, Papillary, and Chromophobe Renal Cell Carcinoma Metastasis Sites and Association With Survival. JAMA Netw Open 2021; 4:e2021869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehta A, Oklu R, Sheth RA. Thermal Ablative Therapies and Immune Checkpoint Modulation: Can Locoregional Approaches Effect a Systemic Response? Gastroenterol Res Pract 2016; 2016:9251375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erinjeri JP, Fine GC, Adema GJ, et al. Immunotherapy and the Interventional Oncologist: Challenges and Opportunities-A Society of Interventional Oncology White Paper. Radiology 2019; 292:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou H, Lee JJ, Yuan Y. BOP2: Bayesian optimal design for phase II clinical trials with simple and complex endpoints. Stat Med 2017; 36:3302–14. [DOI] [PubMed] [Google Scholar]
- 41.Yuan Y, Hess KR, Hilsenbeck SG, Gilbert MR. Bayesian Optimal Interval Design: A Simple and Well-Performing Design for Phase I Oncology Trials. Clin Cancer Res 2016; 22:4291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin R, Zhou Y, Yan F, Li D, Yuan Y. BOIN12: Bayesian Optimal Interval Phase I/II Trial Design for Utility-Based Dose Finding in Immunotherapy and Targeted Therapies. JCO Precision Oncology 2020:1393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newcombe RG. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med 1998; 17:857–72. [DOI] [PubMed] [Google Scholar]
- 44.Logothetis CJ, Gallick GE, Maity SN, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov 2013; 3:849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang C, Msaouel P, Hara K, et al. Definitive radiotherapy in lieu of systemic therapy for oligometastatic renal cell carcinoma: a single-arm, single-centre, feasibility, phase 2 trial. Lancet Oncol 2021; 22:1732–9. [DOI] [PMC free article] [PubMed] [Google Scholar]