

Abstract
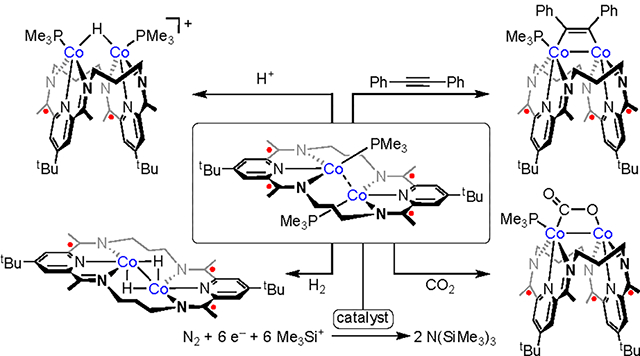
In this article, we discuss the synthesis, characterization, and reactivity of a neutral dicobalt complex, [Co2(PMe3)2]0, supported by a highly reduced, redox-active macrocyclic ligand. The reducing equivalents stored within the ligand framework proved capable of activating a wide scope of small molecule substrates. The addition of a weak acid, lutidinium triflate, led to the clean formation of a bridging hydride that was found to undergo fluxional geometric changes in solution, while exposure of [Co2(PMe3)2]0 to H2 resulted in a rare example of net binuclear oxidative addition to give two bridging hydrides. The addition of diphenylacetylene to [Co2(PMe3)2]0 gave an unusual metallacyclobutane configuration for the coordinated alkyne, a geometry that was mirrored by the product obtained on activation of CO2 by [Co2(PMe3)2]0. Dinitrogen was not observed to bind to [Co2(PMe3)2]0, but the use of catalytic quantities of [Co2(PMe3)2]0 in mixtures of KC8 and Me3SiCl under 1 atm of N2 yielded N(SiMe3)3, which was quantified by conversion into NH4Cl. A screen of related dicobalt complexes revealed a dependence on the identity of the pre-catalyst, with yields of fixed nitrogen ranging up to 50(6) %.
Graphical Abstract
INTRODUCTION
Inspired by metalloenzymes and heterogeneous catalysts, cooperative bond activation using two metal centers in a binuclear ligand scaffold is an increasingly well-studied area. The primary motivation for this chemistry is based on the notion that two metals may access reactivity not available to their mononuclear counterparts.1, 2 While numerous inroads have been made in the design of binucleating ligands, a subset has developed around redox-active binucleating frameworks that have the potential to cooperatively make use of metal-metal and metal-ligand bonding.3 As an example, the Uyeda group has used this principle to great effect in the development of dinickel complexes that can perform a variety of cycloaddition, C–C activation, and C–H functionalization reactions. Their system exploits the redox cycling of naphthyridyldiimine ligands while maintaining consistent metal oxidation states and bonds.4
While the Uyeda system is rigid and has fixed M–M distances, many other binucleating frameworks are geometrically flexible and can accommodate a variety of M–M distances. A common motif for building binuclear scaffolds is tethering together two known chelating ligands.5 The choice of this tether is key to determining the intermetal distance and the flexibility of the complex, and it can have profound effects on catalysis. Rigid aromatic groups are popular tethers, with xanthyl and naphthyl groups often used to link together well-studied ligands such as β-diketiminates or pyridyldiimines (PDI).6, 7 Depending on the tether, these complexes often have substantial vertical flexibility while still maintaining a cofacial arrangement between the metal chelates.8 An example of this for cobalt, which will be the continued focus herein, comes from the Groysman group, who have tethered PDI moieties together with a wide assortment of linkers.9, 10
We have recently sought to marry the two design principles discussed above by using a bis-pyridyldiimine (3PDI2) scaffold to create redox-active and geometrically flexible dinucleating macrocycles. Prior studies have shown that these 3PDI2 macrocycles adopt a variety of geometries11, 12 and ligand reduction states.13 Though the macrocyclic ligand can adopt multiple conformations, small molecule-based ligands that bridge between the two metals are often held in constrained positions. This leads to unusual geometries and electronic structures.14 Furthermore, the two metals in proximity have the potential to cooperatively activate small molecule substrates, especially when aided by the reducing capacity of the PDI ligand. The combination of broad substrate activation and the strained configurations of the resulting activated species may lead to unexplored and interesting reactivity domains. Herein, we describe the synthesis, characterization, and small-molecule reactivity of a highly reduced, macrocycle-supported dicobalt complex that is poised to allow for dual metal-metal and metal-ligand cooperativity.
RESULTS AND DISCUSSION
We previously reported a redox series of dicobalt complexes supported by the 3PDI2 macrocycle. The starting species was a tricationic dicobalt complex bound by two coordinating PMe3 groups. Upon addition of stoichiometric amounts of KC8, the isostructural one- and two-electron reduction products were isolated and characterized (Scheme 1).13
Scheme 1.

With an interest in developing the chemistry at the bridging position, we reasoned that the μ-Cl in the more electron-rich complex, [Co2Cl]+, would be most amenable to substitution, but repeated attempts at salt metathesis of the bridging halide proved difficult. Combined spectroscopic, crystallographic, and computational investigations into the electronic structures of the [Co2Cl]n+ electron transfer series revealed that, while the M-M bond order decreased across the series, electron density also accumulated in the π* manifold of the 3PDI2 ligand. The two-electron reduced product [Co2Cl]+ is thus best described as a (3PDI2)3−:Co(II)/Co(II) system, similar to the (3PDI2)−:Co(II)/Co(II) description assigned to [Co2Cl]3+. The similarity in the Co-based oxidation states across the series likely plays a role in the difficulty encountered when attempting to substitute the bridging chloride for alternative X-type ligands. Subsequently, we became interested in exploring further reduction of the [Co2Cl]+ complex to free the bimetallic core from a bridging halide. We envisioned that doing so would poise the dicobalt center for receiving substrates and transferring to them the reducing equivalents stored on the ligand.
While the one electron-reduction of [Co2Cl]+ was previously discussed in the presence of acetonitrile to give [Co2CN]+ and [Co2CH3]+,12 performing the reduction in the absence of exogenous substrates gave a mixture of products in low-yield that likely resulted from H-atom abstraction of the ligand (vide infra). In contrast, upon addition of two equivalents of KC8 to [Co2Cl]+ (Figure 1) a single, symmetric diamagnetic product was observed by 1H and 31P{1H} NMR spectroscopy. Crystallization from a saturated diethyl ether solution at low temperature yielded dark green blocks that were identified as (3PDI2)Co2(PMe3)2 ([Co2(PMe3)2]0) in low to moderate yields.
Figure 1.
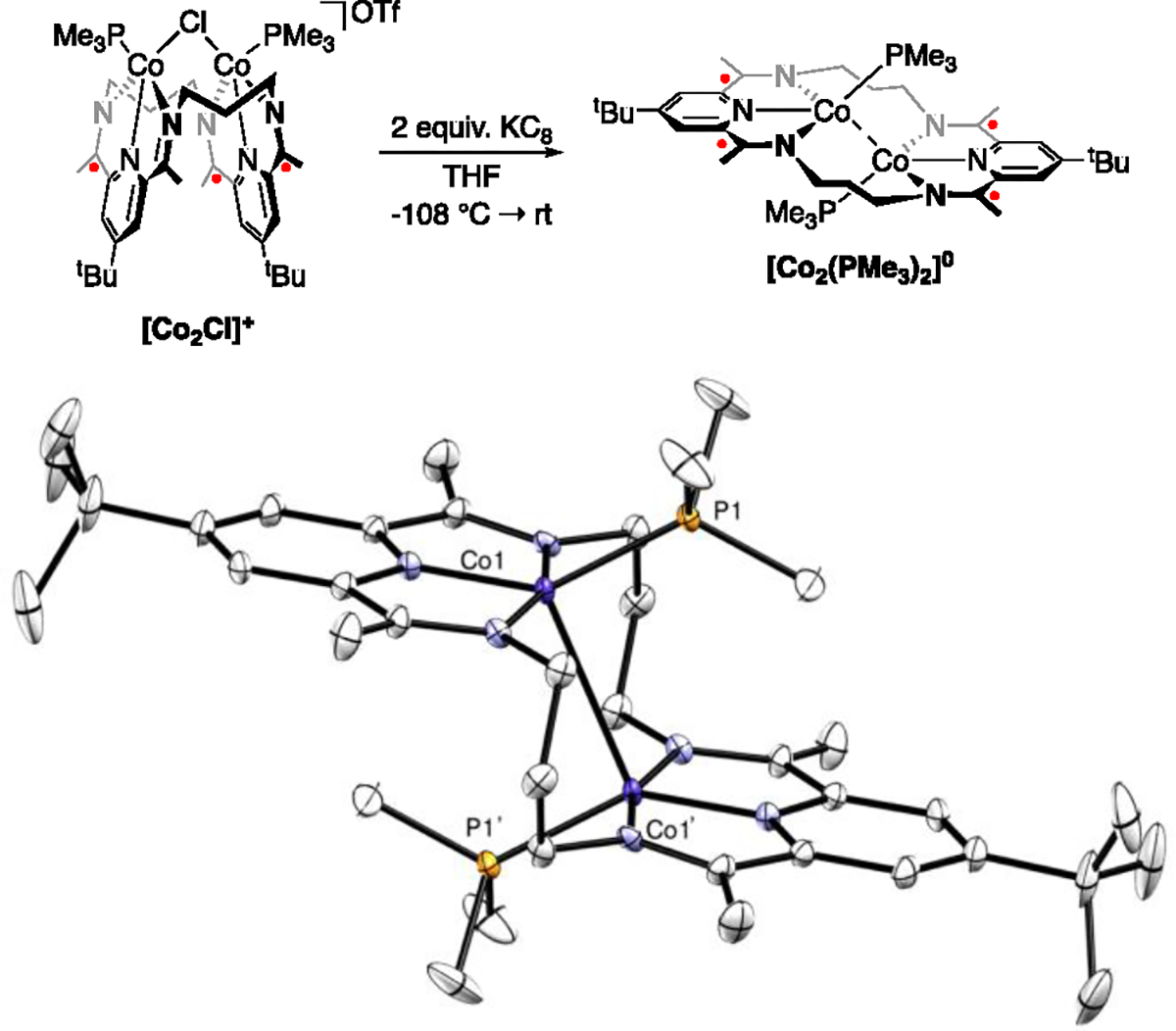
Top: Scheme showing the initial procedure used to make [Co2(PMe3)2]0. Bottom: Thermal ellipsoid plot (50 % probability level) from a crystal structure of [Co2(PMe3)2]0. All hydrogen atoms were removed for clarity. Symmetry generated atoms are labeled with (‘). Symmetry code: 1-X, +Y, 1-Z. Selected bond lengths: Co1–Co1’ = 2.957(1) Å, Co1–P1 = 2.231(1) Å, Co1–N2(py) = 1.823(4) Å.
Crystallographic analysis of [Co2(PMe3)2]0 revealed that the ligand had unfolded into a “stair-step” geometry with each cobalt coordinated by a PDI unit and one PMe3 ligand. Including a potential Co–Co interaction, the metal atoms have a highly distorted square pyramidal geometry with a τ5 value of 0.325 (with 0 being an idealized square pyramidal geometry). The metal-metal distance of 2.957(1) Å is long.15, 16 The formal shortness ratio (FSR) can also be used as a tool to help define the extent of metal-metal bonding. It is defined as the M-M distance divided by the product of the ionic radii of the metals, and a value of 1 represents a single bond, with higher numbers indicative of less bonding. Consistent with a weak metal-metal interaction, the FSR of [Co2(PMe3)2]0 is 1.29.
Due to the popularity of PDI as a well-known redox-active ligand, Wieghardt and coworkers empirically developed the Δ parameter17 (defined as the average Cimine–Cpy distance minus the average of the C–N distances) to describe the reduction level of the PDI ligand. In this case, lower values correspond to higher electron density in the ligand’s π* manifold. We have extrapolated the known Δ ranges to our nPDI2 frameworks13 and found that the value for [Co2(PMe3)2]0 (0.052 Å) corresponds very closely to a (3PDI2)4− state-of-charge for the macrocycle, indicating the presence of a Co(II)/Co(II) core. The Co–P distance of 2.231(1) Å is invariant compared to its more oxidized congener [Co2Cl]+; however, the Co–Npy distance is 1.823(4) Å, which is 0.025 Å longer than in [Co2Cl]+ and in opposition to the trend of the distance decreasing upon continued reductions.
The 31P{1H} NMR spectrum for [Co2(PMe3)2]0 revealed a broad singlet at −12 ppm, and the ligand resonances in the complex’s 1H NMR spectrum were consistent with the C2h geometry observed crystallographically. As a result of this symmetry the methylene resonances displayed a 4:4:4 integration pattern, in contrast to the 4:4:2:2 pattern observed for the more common C2v symmetry commonly encountered for the 3PDI2 systems.
Initial synthetic efforts to form [Co2(PMe3)2]0 led to only trace amounts of isolated material in varying purities, as established by NMR spectroscopy. Extensive optimization trials were carried out to increase the yield of the reaction. As described below, two primary challenges emerged from these studies, namely the lability of the phosphine group(s), which led to decomposition of the complex in solution, and the difficulty in avoiding competitive decomposition of the one-electron reduced intermediate during the two-electron reduction process.
The solution-state phosphine lability was established following observation of the disappearance of the signals corresponding to [Co2(PMe3)2]0 in the material’s 1H NMR spectra over the course of several hours at room temperature. The addition of several equivalents of exogenous PMe3 prevented most of this decomposition to the extent that only ca. 20% of the complex had decomposed in solution after 12 h at room temperature. Though phosphine lability appeared to be present, the rate of exchange was slower than the NMR timescale, as the proton resonances appeared sharp and no exchange was seen on a 31P{1H}−31P{1H} EXSY NMR experiment (see Supporting Information). The methyl resonance for the phosphine did, however, display some mild broadness and poor resolution of the splitting, suggesting that phosphine lability may still be chemically meaningful, even if exchange is not observed on the NMR timescale. Consistent with this interpretation, little change was observed upon cooling the sample to 210 K. The primary difference was a sharpening of the PMe3 signal in the 1H NMR and 31P{1H} NMR spectra (Supporting Information), with no decoalescence or substantial changes in integrations occurring.
Numerous phosphines were explored as supporting ligands for the dicobalt core in [Co2(PMe3)2]0 including PR3 (R = Ph, OPh, iPr, nBu, tBu, Cy), PPhMe2, PPh2Me, and the diphosphine ligands bis(dimethylphosphino)methane (dmpm) and bis(diphenylphosphino)ethane (dppe) through independent syntheses. Other donors consisted of CO, pyridine, THF, DME, and dioxane, with the latter three being used as solvents. Out of the above variants, the only analogous complexes to [Co2(PMe3)2]0 that were isolable as crystals suitable for X-ray diffraction were [Co2(PPh2Me)2]0, [Co2(dmpm)2]0, and [Co2(CO)2]0 (see Supporting Information), although their poor yields limited further characterization. As there is a geometric rearrangement of the ligand and a change in relative orientation (cis to trans) of the phosphines that accompanies reduction, the phosphine must be labile enough to dissociate from the more oxidized congener and remain coordinated to the reduced species to prevent decomposition. Empirically, PMe3 appeared to strike this balance most effectively.
Another challenge to a high-yielding synthesis of [Co2(PMe3)2]0 appeared to lie in ensuring that the kinetics of a two-electron reduction were competitive with the decomposition of an intermediate one-electron reduced species, which was found to produce [(3PDI2)Co2(μ-H)(PMe3)2][OTf] ([Co2H]+). This product was likely generated through the transient formation of a cationic complex with no bridging ligand, followed by rapid H-atom abstraction from the ligand, as has been observed previously.18 Additionally, while no other diamagnetic species were visible in the NMR spectra, Co(PMe3)4 was observed in the 1H NMR spectrum as a broad singlet near 30 ppm, suggesting that demetallation is another decomposition pathway. To address these issues, a wide range of reduction conditions was tested. Based on the cyclic voltammogram of [Co2Cl]3+, a strong reductant with a potential of at least −2.2 V vs. Fc0/+ was needed. Oddly enough, the use of two-electron (Mg(anthracene), Mg, [Mg2(Dippnacnac)2], 1,4-bis(trimethylsilyl)dihydropyrazine, Zn) or homogeneous (Na/K naphthalenide, sodium acenaphthylene) reductants did not aid in overcoming the one-electron sink and similar or worse yields of [Co2(PMe3)2]0 were observed. The use of sodium amalgam, Na/NaCl, sodium mirror or other dispersed, high surface area alkali metals led either to comparable results with KC8 or evidence of over-reduced products. Lastly, the use of borohydride reductants often led to mixtures of the desired [Co2(PMe3)2]0 product with [Co2H]+. The ease of use and high performance associated with KC8 led to its use in the optimized synthesis.
The yield of [Co2(PMe3)2]0 was eventually increased to 30% through the development of a one-pot procedure starting with ligand transmetallation from (3PDI2)Sr(OTf)2 ([Sr]+) to two equivalents of CoCl2, followed by the addition of PMe3, then addition of KC8 in two aliquots at low temperature in THF (Scheme 2). Washing with diethyl ether, followed by extraction in benzene and rapid removal of the solvent gave analytically pure powder. A lower yield of analytically pure crystals could be isolated from saturated toluene solutions of [Co2(PMe3)2]0 (with PMe3 added) layered with pentane and stored at low temperature for several days.
Scheme 2. Optimized synthesis of [Co2(PMe3)2]0.

Reactivity with a weak acid.
The monohydride product [Co2H]+ was repeatedly crystallized as a side-product, albeit in low yields and purities, during our investigations into the synthesis of [Co2(PMe3)2]0. We endeavored to synthesize [Co2H]+ independently through the addition of a weak acid to [Co2(PMe3)2]0. Following the addition of 1.0 equiv of lutidinium triflate to [Co2(PMe3)2]0 in THF, a rapid reaction was observed, as evidenced by a color change to a more intense and vibrant shade of green (Figure 2). The product of the reaction was determined through crystallographic and spectroscopic characterization to be [Co2H]+, which was isolated in good yields. Attempts to synthesize [Co2H]+ through treatment of [Co2Cl]+ with various hydride donors (NaBH4, LAH, Vitride, NaH, LiHBEt3, KHBEt3, and Et3SiH) reliably produced [Co2H]+, albeit as mixtures with [Co2(PMe3)2]0 and other unidentified products.
Figure 2.
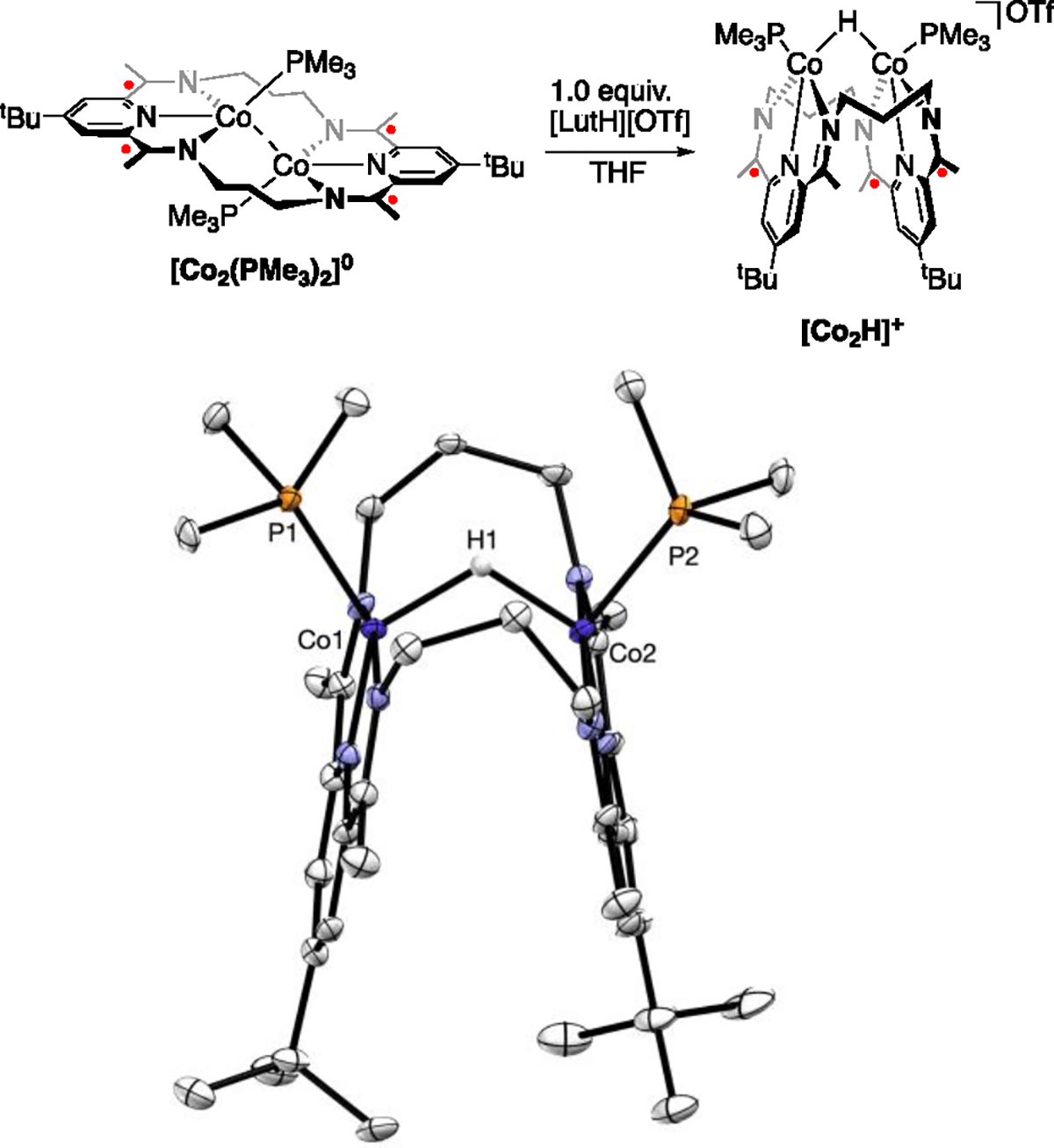
Top: Synthetic scheme for the addition of lutidinium triflate to [Co2(PMe3)2]0 to form a bridging hydride. Bottom: Thermal ellipsoid plot (50 % probability level) of the cationic portion of [Co2H]+ from a crystal structure. Hydrogen atoms (except H1), co-crystallized solvent, and the triflate counterion were removed for clarity. Selected bond lengths: Co1–Co2 = 2.8051(9) Å, Co1–P1 = 2.221(2) Å, Co2–P2 = 2.225(1) Å, Co1–N2 (py) = 1.820(4) Å, Co2–N5 (py) = 1.818(4) Å, Co1–H1 = 1.65(5) Å, Co2–H1 = 1.71(6) Å.
[Co2H]+ adopts a folded configuration in the solid state. The Co–Co distance of 2.8051(9) Å is short relative to its isoelectronic and isomorphic congeners, although a previous analysis of [Co2Cl]+ has shown that complexes at this level of charge in this geometry are unlikely to exhibit M–M bonding. The proximity of the metals in [Co2H]+ appears to be an effect of the small size of the bridging ligand. Also, like its isoelectronic congeners, the Δ value (0.086 Å) is consistent with a (PDI)3− ligand. The cobalt hydride distance is typical at 1.68(6) Å, though these distances are underestimated by X-ray crystallography.
Despite the unremarkable geometry observed crystallographically, the solution-state behavior of [Co2H]+ appeared more complex, with evident dynamic processes occurring. Two resonances (13.9 and 0.9 ppm) were observed in the material’s room temperature 31P{1H} NMR spectrum in a ~1:6 ratio of integrals. Similarly, two species were observed in the 1H NMR spectrum in the same ~1:6 ratio, as evidenced by two distinct sets of 3PDI2 ligand resonances. [Co2H]+ also displayed several features in the hydride region (−10 – −40 ppm) of the material’s 1H NMR spectrum. As seen in Figure 3, the spectrum displayed two broad overlapping features at −20.72 and −21.77 ppm and a sharper singlet more upfield at −26.76 ppm. Integration of the ligand resonances relative to the hydride resonances revealed 4:1 ratios between a) the pyridyl resonance at 7.46 and the overlapping hydride resonances at −21 ppm and b) the pyridyl signal at 8.08 ppm and the hydride resonance at −27 ppm. For clarity, the major product, with the diagnostic pyridyl signal at 7.46 ppm and two broad hydride signals at room temperature, will be referred to as “complex A” and the minor product as “complex B”.
Figure 3.

1H NMR spectrum of [Co2H]+ in THF-d8 at 300 K.
We envisioned three scenarios that could account for our results. First, the minor product, complex B, could be a side reaction product, unrelated to the crystallographic data. However, NMR spectroscopic analysis of several single crystals that were individually determined to contain only the structure of [Co2H]+ described above, gave identical ratios between complexes A and B as in the crude NMR spectra. In addition, the ratio between A and B was observed to be invariant, regardless of synthetic route (either through protonation or reduction of [Co2Cl]+) despite the mechanisms for these reactions being unrelated. The relative ratios of the products do not appear to change with extended time in solution, although it is difficult to distinguish between interconversion and overall decomposition when samples are stored for extended periods in solution without excess PMe3. Aside from increasing the lifetime of the complexes in solution, the addition of excess PMe3 induced no change to the NMR spectra at room temperature; however, we have consistently seen lability of one PMe3 group on all of the isoelectronic complexes and some direct evidence for lability on [Co2H]+ in particular.19 The data thus suggest that it is unlikely that complex B is simply an unrelated diamagnetic hydride impurity, with no relation to the observed crystal structure.
A second potential explanation for the observed solution dynamics was a monomer-dimer equilibrium, akin to that seen in [Co2CN]+.12 In support of this, we observed an 8:1:1 ratio between the pyridyl resonances of the major product and the broad hydride signals at ~ −21 ppm. This would imply that complex B adopts a monomeric configuration, likely with a geometry matching the observed crystal structure. However, a DOSY NMR experiment showed identical diffusion constants for the two species, A and B, and variable temperature analysis of the mixture did not show a change in integration from one species into the other (vide infra). In addition, this explanation would require further phosphine binding equilibria on the proposed dimeric species to account for the continued decoalescence at low temperature. However, this is inconsistent with the splitting patterns of the hydrides observed at low temperature.
Our third potential explanation for the solution-phase data involves interconversion of different geometric isomers of [Co2H]+, taking advantage of the various geometries the ligand is known to adopt and the likely decreased barrier to fluxionality with a small ligand, like hydride, bridging the cobalt atoms. To explore the extent of fluxionality and gain insight into the solution geometries, we performed variable temperature NMR spectroscopy experiments.
At room temperature, two of the expected methylene signals in complex A could not be located, and the ones present were very broad, with no correlations observed in 2D NMR spectroscopy experiments. None of the methylene carbons were visible in the 13C{1H} NMR spectrum, and the other ligand 13C resonances were very broad. Upon cooling to temperatures in the range of 255–240 K, the pyridyl, imine, and tert-butyl resonances assigned to complex A began to broaden and decoalesce. At the same time, some of the associated methylene signals sharpened, though distinct sets of methylene signals attributable to two different species could not be identified.
In the hydride region for complex A, the two broad overlapping signals observed at room temperature coalesced into one broad singlet on warming to temperature above 310 K and became clearly distinguishable from one another on cooling to 285 K. On further cooling to 255 K, one of the signals appeared as a sharp triplet, while the other signal remained broad, but the latter signal began to further decoalesce into two resonances at 240 K. At 225 K, three distinct triplets were observed for what was formerly assigned as complex A (Figure 4). A 1H{31P} NMR spectrum at 225 K (Figure 5) showed full decoupling of the hydride resonances into singlets, consistent with the splitting coming from coordinated phosphines. The J-coupling values (between 23–26 Hz) are also consistent with two-bond phosphorus coupling to a metal hydride.20
Figure 4.

VT 1H NMR spectra (hydride region) of [Co2H]+ in THF-d8 from 215–330 K.
Figure 5.
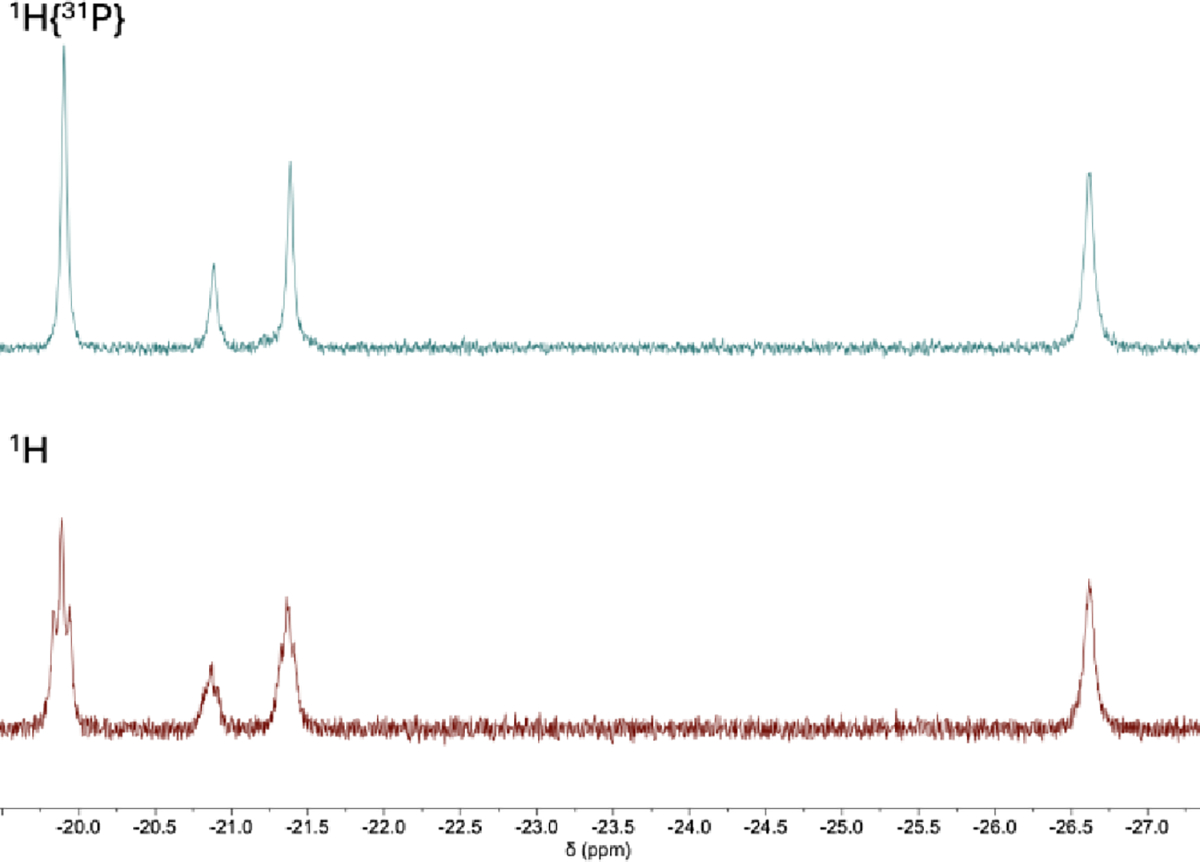
Hydride region of the 1H NMR spectra of [Co2H]+ with phosphorus decoupling (top) and coupling (bottom) at 225 K.
In the variable temperature 31P{1H} spectra, the signal for complex A at ~ 1 ppm similarly decoalesced into two signals on cooling, starting at 285 K. Unlike in the low-temperature hydride region in the 1H NMR spectra, however, the 31P{1H} signals at ~ 0 and 2 ppm did not further separate at 240 K or below, though splitting of the more downfield signal into a doublet (~2 ppm) was observed. Upon adjusting the center of the decoupling parameter to −20 ppm to account for the hydrides, that resonance was converted into an expected singlet. The observed coupling is consistent with the a single hydride coordinated in proximity to that phosphine.
The more upfield hydride singlet, observed at −26.8 ppm at 300 K and corresponding to complex B, was largely unaffected by temperature variations. No decoalescence or splitting of the hydride resonance was observed with temperature variations, though the signal did sharpen somewhat at intermediate temperatures. The corresponding ligand resonances similarly broadened at the extremes of the temperature regime studied. Additionally, the resonances for the methylene protons in complex B appeared in a 4:4:4 pattern, a trend observed only in 3PDI2Co2 complexes adopting a stair-step geometry and C2h symmetry. Relative to an internal standard, the integrations of B over the temperature range studied remained invariant. For complex A, however, the total integrations of the decoalesced signals reversibly changed with temperature. At 215 K, the integration had decreased relative to the starting room temperature value and the spectrum was overall broadened, suggesting the solubility limit of complex A may have been reached. Notably, the two complexes did not appear to interconvert appreciably at low temperatures.
Due to the number of observed species and the breadth of possible geometries it is difficult to assign the solution-state geometries with high confidence with the available data, but we propose an explanation based upon the following key pieces of data. First, though the proton and phosphorus resonances do not show a similar level of deconvolution, likely due to different relaxation rates, the number of hydride resonances at low temperatures clearly indicate the presence of at least four different species present in solution. Second, while the identification of two species being present in solution at room temperature was made based on the phosphorus and pyridyl proton resonances, the broadened hydride signals assigned to complex A that decoalesce upon cooling likely represent an equilibrium between three hydride species – A1, A2, and A3 – that are only fully deconvoluted at low temperature. Third, complex B likely adopts a stair-step geometry based upon the characteristic methylene proton pattern. Fourth, each complex observed likely has two PMe3 molecules coordinated due to the high symmetries observed and, in the case of the A complexes, the triplet splitting pattern of the hydrides. Lastly, the integrations and observed coupling in the 31P{1H} NMR spectrum suggest one hydride ligand per complex.
Taken together, we propose the major species at room temperature to represent an average of three hydride complexes (A1, A2, and A3; Scheme 3) that bind PMe3 ligands on the same side of the macrocycle as one another (cis phosphines). This configuration was observed crystallographically for A1, and the geometries for A2 and A3 are proposed based on data for related M2(μ-E) complexes bound by the 3PDI2 macrocycle.18, 21–23 While crystalline samples of A1 equilibrate with A2, A3, and B at room temperature, the interconversion between “A” and B appears to slow dramatically at lower temperatures. This behavior, along with the methylene 1H NMR spectral pattern for B, is consistent with an unfolded ligand geometry supporting a linear Co–H–Co core and trans phosphines.22 We have previously observed the capacity of the constrained geometry of the nPDI2 framework to enforce unusual bridging ligand configurations; however, in this case, where the bridging ligand is small, the flexibility appears to create varied and dynamic behavior.
Scheme 3. Proposed geometries of [Co2H]+ in solution.

Reactivity with H2.
After optimizing the synthesis of [Co2(PMe3)2]0 and producing high-purity batches separable from [Co2H]+, we sought to use the open coordination sites and reducing potential of the ligand to perform exploratory reactivity with fundamental organometallic ligands. We found that exposure of [Co2(PMe3)2]0 to 1 atm of H2 provided the complex [(3PDI2)Co2(μ-H)2] ([Co2H2]0, Figure 6). This reactivity represents the oxidative addition of one equivalent of H2 by [Co2(PMe3)2]0 to form two bridging hydride ligands with concomitant loss of two equivalents of PMe3. The spectroscopic yield of the reaction was low and some precipitate was observed, suggesting that additional products are formed. However, the only species that could be isolated from the mixture was [Co2H2]0 as dark green blocks. An alternative synthetic procedure for [Co2H2]0 that appears to involve PCET from the medium is presented in the Experimental section. This alternative method is not affected by the presence of H2 or typical H-atom donor reagents and provides the product in very low yields.
Figure 6.

Top: Synthetic scheme for the activation of dihydrogen to form the bridging dihydride complex [Co2H2]0. Bottom: Thermal ellipsoid plot (50 % probability level) from a crystal structure of [Co2H2]0. All hydrogen atoms (except H1) were removed for clarity. Symmetry generated atoms are labeled with (‘). Symmetry code: 1-X, 1-Y, 1-Z. Selected bond lengths: Co1–Co1’= 2.5304(4) Å, Co1–H1 = 1.5898 Å, Co1–H1’ = 1.6048 Å, Co1–N2(py) = 1.818(2) Å.
Unlike the monohydride species, the dihydride complex appeared to remain static in solution on the NMR timescale, with a C2h symmetry that matches the solid-state structure. The complex’s 1H NMR spectrum showed symmetric 3PDI2 resonances with a 4:4:4 ratio for the methylene proton integrations and a broad singlet corresponding to the hydrides at −27.4 ppm that integrated to 2H. Only free PMe3 was observed in the in situ 31P{1H} NMR spectrum following H2 addition to [Co2(PMe3)2]0, with little change upon removal of the volatile materials and redissolution in deuterated solvent. By X-ray crystallography, the molecule was found to lie on a crystallographic center-of-symmetry and the structure showed a stair-step geometry with a dicobalt dihydride core. The two unique Co–H distances are nearly identical at 1.5898 and 1.6048 Å and the Co–Co distance is 2.5304(4) Å. The small bridging groups appear to displace the metal atoms from the PDI planes and this is reflected in the distance between the plane created by the three PDI nitrogen atoms and the metal center, being 0.356 Å in [Co2H2]0 relative to 0.036 Å in [Co2(PMe3)2]0.24 Compared to the related [κ2-(NacNac)Co]2(μ-H)2 complex reported by the Holland group25 and another dicobalt dihydride supported by an enamidophosphinimine ligand,26 the Co–H and Co–Co distances are consistent with literature precedent. Both of those literature examples are paramagnetic, however, and the Holland example reductively eliminates H2 in the presence of dinitrogen. [Co2H2]0 is stable under N2, which could be partly due to mildly more electron-rich metal centers disfavoring reductive elimination. The Δ value of 0.084 Å (corresponding to PDI3−) is consistent with electron density from the ligand facilitating the oxidative addition of H2.
Bimetallic H2 oxidative addition to form bridging dihydrides is a relatively rare phenomenon for first-row transition metals. In 2009, Holland and coworkers examined possible mechanisms for this reaction using a β-diketiminate iron complex to form a high-spin paramagnetic diiron dihydride species.27 Additionally, Limberg reported an analogous reaction with a related BDI-Ni complex.28 Reduction of their metal halide precursor under H2 or exposure of a reduced dimeric complex to H2 both generated the dihydride complex. A dimeric diphosphine (dtbpe) nickel complex with a conjoining benzene moiety also reacted with H2 to make a bridging hydride dinickel species that displayed interesting fluxional behavior in solution.29 Despite numerous examples of cobalt-mediated dihydrogen activation, to our knowledge there are no examples of dicobalt complexes forming dihydride species through H2 oxidative addition. A trinuclear cobalt cluster with a central bridging hydride was reported to react with H2 to form a purported μ3-trihydride complex, though only two hydrides could be located in the Fourier difference map.30 Additionally, a (dippp)Co allyl complex was shown to react with H2 to generate a binuclear cobalt complex with three apparent bridging hydrides and one terminal hydride.31 However, the starting complex in this case is a mononuclear species, which may preclude a bimetallic oxidative addition process.
Reactivity with an alkyne.
Alkyne chemistry has a rich history with reduced cobalt complexes. Both hydrogenation catalysts and cycloaddition reactions, like in the Pauson-Khand reaction, are known to be mediated by low-valent Co catalysts. With this history, along with the potential for atypical, macrocycle-enforced geometric configurations, in mind, we investigated the addition of diphenylacetylene to [Co2(PMe3)2]0.
Upon treatment of a toluene solution of [Co2(PMe3)2]0 with 1.0 equiv of diphenylacetylene, a vibrant deep-green solution immediately formed (Figure 7). A 31P{1H} NMR spectrum of the reaction mixture revealed free PMe3 and a new signal at −11.5 ppm. In the 1H NMR spectrum, a new asymmetric Cs-symmetric diamagnetic product had formed, consistent with the loss of one PMe3 ligand. Following removal of volatile materials, crystallization of the green solid revealed the product to be [(3PDI2)2Co2(PhCCPh)(PMe3)] ([Co2PhCCPh]0, Figure 7).
Figure 7.

Top: Synthetic scheme for the binding and activation of PhCCPh to form [Co2PhCCPh]0. Bottom: Thermal ellipsoid plot (50 % probability level) from a crystal structure of [Co2PhCCPh]0. All hydrogen atoms were removed for clarity. Selected bond lengths: Co1–Co2 = 2.6012(6) Å, Co1–P1 = 2.2041(9) Å, Co1–C36 = 1.992(3) Å, Co2–C37 = 1.948(3) Å, C36–C37 = 1.349(4) Å, Co1–N2(py) = 1.851(2) Å, Co2–N5 = 1.855(3) Å.
A crystallographic study of the product revealed that the 3PDI2 ligand adopts a folded geometry upon loss of one PMe3 ligand and the addition of the alkyne. Dicobalt alkyne complexes are a heavily studied area, but [Co2PhCCPh]0 is unique in that it features an alkyne binding mode that is unknown for isolated dicobalt complexes. Generally speaking, dinuclear transition metal complexes with bridging alkyne ligands exhibit a structural dichotomy between those with the alkynyl C2 unit lying either perpendicular (dimetallatetrahedrane) or parallel (dimetallacyclobutene) to the metal-metal vector.32 Hundreds of crystallographically characterized dicobalt alkyne complexes are known, but the alkynyl C2 units in these species invariably form a dimetallatetrahedrane structure, a geometry that is thought to maximize the cobalt-alkyne interaction through greater π interactions. In [Co2PhCCPh]0, the alkyne adopts a μ2-η1:η1-C2R2 binding motif, with the cobalt and alkynyl carbon atoms forming a four-membered metallacycle.33 This alkyne orientation is known for other first-row transition metals, but for cobalt, it appears to only be proposed as an intermediate geometry during alkyne fluxionality.34 This unusual geometry in [Co2PhCCPh]0 may occur to minimize the steric interactions between the alkyne R groups and the ligand backbone methylenes, as we have seen in the past. Notably, this geometry was recently proposed as a key intermediate along the path toward C(sp)–C(sp2) bond oxidative addition by the diiron analog of [Co2(PMe3)2]0.35
The crystallographic and spectroscopic parameters for [Co2PhCCPh]0 are consistent with a dimetallacyclobutene-type structure of the alkyne. The alkynyl C–C bond length is 1.349(4) Å, which is expanded from 1.212(5) Å in the free ligand36 and similar both to other alkynes coordinated in this fashion and to a typical C=C double bond. The Co–C bond lengths of 1.992(3) Å and 1.948(3) Å are similar to those in the diphenylacetylene adduct of dicobalt hexacarbonyl,37 despite the difference in acetylene coordination mode. In addition, the hybridization around the carbons changed from sp to sp2, as evidenced by the C–C–Ph angles of 122.4(3)° and 126.2(3)°, close to the ideal of 120°. The cobalt-cobalt distance is 2.6012(6) Å with an FSR of 1.12, indicative of potential weak overlap. The Δ value of the ligand increased to 0.090 Å from 0.052 Å in [Co2(PMe3)2]0, suggesting that the reducing equivalents in the ligand were used to aid in alkyne binding and activation through electron density transfer from the 3PDI2 ligand to the alkyne.
In the 1H NMR spectrum of [Co2PhCCPh]0, the alkynyl phenyl groups showed distinct resonances, due to the asymmetry provided by the single coordinated PMe3 ligand. The phenyl groups could not be unambiguously assigned with respect to phosphine proximity. The phenyl protons were in the range 6.84–7.84 ppm, and each appeared in a 2:2:1 pattern as expected, although one of the ortho proton environments gave an unusually broad resonance in comparison to the more defined features on the rest of the aromatic protons, perhaps due to 31P coupling. Assignment of the alkynyl resonances in the complex’s 13C{1H} NMR spectrum were made to signals at 149.7 and 137.5 ppm. These signals are ~50 ppm downfield from the free ligand (90.75 ppm),38 a shift that is consistent with a change to sp2 hybridization for the central carbons.
Reactivity with CO2.
We were interested to find that CO2 binding to the dicobalt core of [Co2(PMe3)2]0 yielded a related binding mode to that observed with diphenylacetylene. Upon treatment of [Co2(PMe3)2]0 with 1 atm of CO2 in benzene, the brown-green solution turned blue-green with formation of small amounts of a dark precipitate (Figure 8). Monitoring the reaction mixture by 31P{1H} NMR spectroscopy allowed for the observation of free PMe3 and the growth of a new broad signal at −5.22 ppm. The reaction mixture’s 1H NMR spectrum revealed near quantitative conversion to an asymmetric complex, consistent with one coordinated PMe3 molecule. The 13C{1H} NMR spectrum for [Co2CO2]0 had a signal at 196.2 ppm, which is within the range of previously characterized C-bound monometallic CO2 complexes. For bimetallic complexes, δ CO2 can appear as far downfield as ~250 ppm,39, 40 but several examples are known that exhibit resonances in the range of 190–210 ppm.39, 41
Figure 8.

Top: Synthetic scheme for the binding and activation of CO2 to form [Co2CO2]0. Bottom: Thermal ellipsoid plot (50 % probability level) from a crystal structure of [Co2CO2]0. All hydrogen atoms were removed for clarity. Selected bond lengths: Co1–Co2 = 2.5539(6) Å, Co1–C33 = 1.952(3) Å, Co2–O2 = 1.921(2) Å, C33–O2 = 1.322(3) Å, C33–O1 = 1.231(3) Å, Co1–P1 =2.1793(7) Å, Co1–N2(py)=1.852(2) Å, Co2–N5(py) = 1.804(2) Å.
A crystallographic analysis of material isolated from the reaction mixture revealed the formation of [(3PDI2)2Co2(CO2)(PMe3)] ([Co2CO2]0, Figure 8), the structure of which is consistent with the NMR spectroscopic data. Like in [Co2PhCCPh]0, one of the C–O linkages bridged along the Co–Co vector between the metal centers to form a dimetallaoxacyclobutane ring. The Co–Co distance of 2.5539(6) Å is slightly shorter than the analogous distance in [Co2PhCCPh]0, which may be due to the decreased steric pressure of the CO2 unit compared to PhCCPh acting on the weak Co–Co interaction. In [Co2CO2]0, the coordinated C–O group has a bond distance of 1.322(3) Å. This distance is significantly longer than the C=O distance of 1.231(3) Å in free CO2, indicative of C=O bond reduction in [Co2CO2]0. The Co–O and Co–C bond lengths of 1.921(2) and 1.952(3) Å, respectively, support this view, as they are in line with known Co–X distances for anionic 2p-element donors bound to Co. The Δ value for [Co2CO2]0 of 0.092 Å, which suggests a 3PDI2 physical oxidation state between (PDI)3− and (PDI)2−, also suggests significant electron density transfer into the CO2 unit. Few examples of crystallographically characterized dicobalt CO2 complexes are known. Fujita has characterized and studied the reaction of CO2 with mononuclear, macrocycle-supported Co complexes that form dinuclear, bridging CO2 complexes on exposure to CO2. However, the crystallographically characterized species is assigned as a −CO2H group.42 Later, the Fujita group isolated an ethylenediamine-supported Co complex with a bridging (CO2)2− ligand that was not synthesized through addition of CO2.43 Recently, they undertook an extensive investigation into the role and advantages that a bimetallic cobalt system confers over the monometallic congener for CO2 reduction.44 They were able to isolate a bridging CO2 intermediate and showed that the kinetic barrier of the elementary steps from CO2 to CO conversion were lowered through the bimetallic interaction.
Catalytic N2 Reduction.
Finally, [Co2(PMe3)2]0 was found to catalyze N2 fixation to form N(SiMe3)3. The first examples of Co-catalyzed N2 silylation were reported in 2015 by Lu45 and Nishibayashi.46 This chemistry has since been extended to other Co systems and is known for a range of transition metals.47 [Co2(PMe3)2]0 was not observed to bind N2; this contrasts with the analogous diiron system, which binds N2 in a cis-μ−1,2 bridging mode within the neutral complex (3PDI2)Fe2(μ-N2)(PR3)2 (R = Me, Ph).14 The dicobalt species, [Co2(PMe3)2]0, with two added valence electrons, may exclude N2 due to the capacity of the cobalt system to form a weak Co–Co bonding interaction. Interestingly, even though the N2 unit in the diiron system was only modestly activated (dNN = 1.14 Å vs. 1.10 Å for free N2), the diiron system was observed to mediate N2 fixation catalysis (see below). This suggested that further activation of N2, likely via in situ reduction, was needed to access a stage of N2 reduction that would be suitable for N-based reactivity with an electrophile. We posited that [Co2(PMe3)2]0 may bind N2 transiently or, more likely, on further reduction to form a species capable of N2 reduction chemistry.
Treatment of [Co2(PMe3)2]0 with 500 equiv of KC8 and 500 equiv of TMSCl (0.2 mol % Co2 complex) in THF at room temperature for 24 h resulted in a 12 % yield of NH4Cl following workup with HCl (Figure 9), corresponding to a turnover number (TON) of 10. The formation of N(SiMe3)3 as an intermediate in this process was confirmed by NMR spectroscopic analysis of the reaction mixture prior to HCl addition. The diiron complex [Fe2N2]0 performed similarly under identical reaction conditions, yielding 5(2) equiv of NH4Cl following workup.
Figure 9.

Catalytic dinitrogen silylation reaction scheme to form NH4Cl and the series of catalysts used in this study.
We next evaluated N2 reduction catalysis by other dicobalt precatalysts (Figure 9). Our recent investigations have yielded a host of [(3PDI2)Co2(μ-X)(PMe3)2]+ ([Co2X]+, X = Cl, Me, N3, CN, H) species. We anticipated that under highly reducing conditions, these complexes would funnel into the same reduced dicobalt unit, as has been observed previously on a series of cyclophane-supported trimetallic complexes.48 However, a systematic study of the catalytic activity of the [Co2X]+ complexes revealed a marked dependence on the identity of the μ-X unit.
A screen of the solvent identity, reaction temperature, and catalyst loading revealed optimized conditions with 0.2 mol % of catalyst in THF at room temperature (Table 1; see Supporting Information). The use of [Co2Cl]+ as a pre-catalyst under these conditions provided results that were comparable to those of [Co2(PMe3)2]0 [TON = 7(5)], as expected based on the synthetic pathway used to form [Co2(PMe3)2]0. The complex [Co2Me]+ performed similarly, with a TON of 4(2). The performance improved, however, for [Co2N3]+ and [Co2CN]+. A TON of 31(13) was achieved with [Co2N3]+, and the TON was observed to increase further with [Co2CN]+, which provided a TON of 42(5), corresponding to a 51% yield of NH3.
Table 1.
Catalytic Dinitrogen Silylation by Dinuclear Complexes
| Entry | Precatalyst | Solvent | T/°C | TON | Yield |
|---|---|---|---|---|---|
| 1 | [Co 2 CN] + | THF | −78 | 17(4) | 20% |
| 2 | [Co 2 CN] + | THF | −40 | 26(9) | 31% |
| 3 | [Co 2 CN] + | THF | 0 | 28(6) | 34% |
| 4 | [Co 2 CN] + | THF | 25 | 42(5) | 50% |
| 5 | [Co 2 CN] + | toluene | 25 | 10(2) | 12% |
| 6 | [Co 2 CN] + | Et2O | 25 | 30(11) | 36% |
| 7 | [Fe 2 N 2 ] 0 | THF | 25 | 5(2) | 6% |
| 8 | [Co 2 (PMe 3 ) 2 ] 0 | THF | 25 | 12(3) | 14% |
| 9 | [Co 2 Cl] + | THF | 25 | 7(5) | 8% |
| 10 | [Co 2 CH 3 ] + | THF | 25 | 4(2) | 5% |
| 11 | [Co 2 H] + | THF | 25 | 15(5) | 18% |
| 12 | [Co 2 N 3 ] + | THF | 25 | 31(13) | 37% |
| 13 | no catalyst | THF | 25 | 0 | 0% |
The improved performance with [Co2CN]+ is notable in that it is the only example from this series that is known to form a higher nuclearity complex. Phosphine dissociation from [Co2CN]+ was shown to allow for dimerization of the monophosphine complexes [(3PDI2)Co2(CN)(PMe3)]+ to form a tetracobalt dication in which the cyanide ions bridged between dicobalt units. The tetracobalt dication exhibits low solubility, but exposure to a reducing environment would be expected to increase its availability for further reactivity.
CONCLUSION
We have explored the synthesis and reactivity of a versatile and highly reduced dicobalt complex supported by a geometrically and electronically flexible macrocyclic ligand. The dicobalt core in [Co2(PMe3)2]0 is stabilized by coordination of two PMe3 ligands; however, they are labile and readily prone to partial or full substitution with a wide variety of substrates. While the physical oxidation states of the metal centers in [Co2(PMe3)2]0 are near Co(II) (like its more oxidized synthon, [Co2Cl]+), the metal-based oxidation states belie the reducing power of the complex as a whole, which leverages the electron equivalents stored in the redox-active 3PDI2 framework for small molecule activation chemistry. Upon exposure to the small gaseous molecules CO2 and H2, [Co2(PMe3)2]0 coordinates CO2 and oxidatively adds H2 between the metal center, with concomitant oxidation of the ligand. Similarly, the addition of diphenylacetylene to [Co2(PMe3)2]0 led to reduction of the alkynyl group, and in this case, the geometric constraints of the macrocycle appear to foster an unusual dimetallacyclobutene binding mode for the alkynyl group. This latter aspect highlights that in addition to the reducing power of this complex, the 3PDI2 ligand is able to adopt multiple configurations, often leading to unique or fluxional small molecule binding modes. This is further demonstrated with [Co2H]+, which was formed from the addition of a weak acid to [Co2(PMe3)2]0 and appeared to display multiple bridging hydride geometries in solution.
Finally, the ability of [Co2(PMe3)2]0 to react with and form stable complexes with a wide variety of substrates demonstrates its potential for further small molecule activation chemistry. This was initially explored through catalytic N2 silylation chemistry, for which [Co2(PMe3)2]0 was found to be a competent catalyst, even though the precatalyst was not observed to bind N2. A screen of related dicobalt complexes found improved N2 fixation performance for a dicobalt μ-CN complex. Future work with these dicobalt systems will endeavor to leverage the geometric and electronic flexibility of the 3PDI2 scaffold and its analogs for supporting multinuclear catalytic transformations.
EXPERIMENTAL
General Considerations.
All reactions containing transition metals were performed under an inert atmosphere of N2, using standard Schlenk line or glovebox techniques. Glassware, stir bars, filter aid (Celite), and 3 Å molecular sieves were dried in an oven at 150 °C for at least 12 h prior to use. All solvents (THF, fluorobenzene, n-pentane, diethyl ether, benzene, and toluene) were dried by passage through a column of activated alumina, deoxygenated by passage through a copper Q5 column where applicable, sparged with N2, and stored over activated 3 Å molecular sieves under an inert atmosphere. Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc., and dried over Na/benzophenone (THF-d8, C6D6), then isolated by either vacuum transfer (THF-d8) or distillation (C6D6) and stored under an inert atmosphere over 3 Å sieves. [(3PDI2)Co2(μ-Cl)(PMe3)2][OTf] ([Co2Cl]+)13 and (3PDI2)Sr(OTf)2 ([Sr]2+)49 were synthesized following previously published procedures. KC8 was synthesized according to a literature procedure50 and stored under nitrogen at − 35 °C in a glovebox before use. PMe3 (98%) was either purchased from Strem Chemicals or synthesized according to a literature procedure.51Lutidinium triflate was synthesized according to a literature procedure52, dried over P2O5, and recrystallized from dry THF/pentane layering at − 35 °C. Diphenylacetylene was recrystallized from dry pentane at − 35 °C. Ultra-high purity grade H2 and CO2 were passed through columns of activated Drierite before use. [nBu4N][PF6] (98%) was purchased from MilliporeSigma and recrystallized twice from hot ethanol, followed by drying at 60 °C under 30 mbar for 10 h before being stored in a glovebox under dry nitrogen prior to use.53 Ferrocene (98%) was purchased from Acros Organics, purified by sublimation three times,54 and stored in a glovebox under dry nitrogen prior to use. All other chemicals were used as received.
1H, 13C{1H}, 31P{1H}, 1H-13C HSQC, 1H-13C HMBC, and 1H-1H COSY NMR spectra were recorded on UNI 400, UNI 500, Bruker AV3Bio 500, and Cryo 500 spectrometers. All chemical shifts (δ) are reported in units of ppm, with references to the residual protio-solvent resonance for proton and carbon chemical shifts. External neat H3PO4 was used for referencing 31P NMR spectra. For the complexes with asymmetry in the 3PDI2 signals, the apostrophes in the assignments given below refer to the portion of the ligand closest to the coordinated PMe3. Elemental analysis was performed by the CENTC Elemental Analysis Facility, Department of Chemistry, University of Rochester. Many of the complexes did not meet the bulk purity standard of 0.4% for elemental analysis despite repeated attempts. The data are included for completeness. The air sensitivity, low yields, and difficulty in isolating usable quantities of material may all contribute to the deviations from theoretical values. The purity of diamagnetic complexes was determined by NMR with use of an internal standard. IR spectra (KBr pellet) were collected on a JASCO FT/IR-480 Plus spectrometer FTIR. Solution phase effective magnetic moment data were determined using Evans’ method.55
UV–vis–NIR absorption spectra were collected from 200 to 1000 nm using an Agilent Cary 60 UV–vis spectrophotometer at room temperature. The samples were prepared under an N2 atmosphere in a glovebox. For each species, at least four spectra at different concentrations were collected. The absorption intensities of various peak maxima were plotted vs. concentration to ensure that absorption data were being collected in the linear response range of the spectrometer. Linear regression fits to the plotted data resulted in R2 values ≥ 0.994.
Crystallographic X-ray intensity data were collected on a Bruker D8QUEST CMOS ([Co2H]+, [Co2(PMe3)2]0), a Bruker APEXII CCD ([Co2H2]0) or Rigaku XtaLAB Synergy-S (Dectris Pilatus3 R 200K) ([Co2PhCCPh]0) using Mo-Kα radiation (λ=0.71073 Å) at a temperature of 100 K. X-ray intensity data for [Co2CO2]0 were collected on a Rigaku XtaLAB Synergy-S diffractometer (HyPix-6000HE) using Cu-Kα radiation (λ=1.54184 Å) at a temperature of 100 K. Rotation frames were integrated using SAINT56 or CrysAlisPro,57 producing a listing of unaveraged F2 and σ(F2) values. The intensity data were corrected for Lorentz and polarization effects and for absorption using SADABS58 or SCALE3 ABSPACK.59 The structures were solved by dual space methods by using either SHELXT60 ([Co2H]+, [Co2(PMe3)2]0, [Co2PhCCPh]0, [Co2CO2]0, [Co2(dmpm)2]0, [Co2(Ph2Me)]0) or direct methods with SHELXS-9761 ([Co2H2]0) and refined by full-matrix least-squares, based on F2 using SHELXL-2018.62 The crystal of [Co2H2]0 grew as a non-merohedral twin; the program CELL_NOW63 was used to index the diffraction images and to determine the twinning mechanism (180° about the 001 real direction). Non-hydrogen atoms on all structures were refined anisotropically and hydrogen atoms were refined using a riding model. All hydride hydrogen atoms were located in the difference map and refined, except for the hydride hydrogens in [Co2H2]0.
Synthesis of [(3PDI2)2Co2(PMe3)2] ([Co2(PMe3)2]).
(3PDI2)Sr(OTf)2 (772.5 mg, 0.86 mmol) and CoCl2 (222.2 mg, 1.71 mmol) were suspended in 15 mL of THF and stirred for 4 h to yield a light brown slurry. PMe3 (180 μL, 1.87 mmol) was added at room temperature, turning the slurry bright red. The mixture was frozen, then removed from the cold well of a glovebox. KC8 (235.4 mg, 1.74 mmol) was added to the thawing mixture with vigorous stirring, quickly giving a dark green solution. The solution was allowed to warm to room temperature and stirred for 15 min. The solution was refrozen, removed from the cold well, and a second aliquot of KC8 (232.2 mg, 1.72 mmol) was added the thawing solution. Upon warming, the solution first turned blue, then a greenish-brown and was allowed to stir for 30 min at room temperature. The volatile materials were removed in vacuo and the resulting dark green solid was then triturated with pentane (3 × 1 mL). The solid was washed with diethyl ether (20 mL) and the remaining green solid was extracted into benzene, filtered through Celite, and the volatile materials were removed in vacuo to yield a green powder. The green solid was again washed with diethyl ether and collected by filtration; the filtrand was dried and collected to yield 222.2 mg (33% yield) of green powder that was used without further purification. Crystals suitable for X-ray diffraction were grown from concentrated solutions of [Co2(PMe3)2]0 in diethyl ether at −35 °C. 1H NMR (500 MHz, C6D6, 300 K): δ = 8.58 (s, 4H, py m-H), 4.18–3.92 (m, 4H, CH2CH2CH2), 3.75–3.67 (m, 4H, CH2CH2CH2), 2.62–2.52 (m, 4H, CH2CH2CH2), 2.00 (d, 5JPH = 2.4 Hz, 12H, N=C-CH3), 1.70 (s, 18H, C(CH3)3), 0.26 (d, 2JPH = 6.2 Hz, 18H, P(CH3)3) ppm. 13C{1H} NMR (126 MHz, C6D6, 300 K): δ = 144.6 (s, o-ArC), 140.6 (d, 3JPC = 5.3 Hz, Cimine), 136.8 (s, p-ArC), 109.7 (s, m-ArC), 58.5 (d, 3JPC = 6.4 Hz, CH2CH2CH2), 35.6 (s, C(CH3)3), 33.2 (s, CH2CH2CH2), 32.0 (s, C(CH3)3), 18.9 (d, 1JPC = 15.9 Hz, P(CH3)3), 16.2 (s, N=C-CH3) ppm. 31P{1H} NMR (162 MHz, C6D6, 300 K): δ = −9.78 (s, P(CH3)3) ppm. Anal. Calcd. for C38H64Co2N6P2 (784.79 g/mol): C, 58.16; H, 8.22; N, 10.61%. Found: C, 57.84; H, 8.22; N, 10.52%.
Synthesis of [(3PDI2)2Co2(H)(PMe3)2][OTf] ([Co2H]+).
[Co2(PMe3)2]0 (28.8 mg, 0.037 mmol) was dissolved in 2 mL of toluene, and a suspension of lutidinium triflate (9.5 mg. 0.037 mmol) in 2 mL of toluene was added dropwise to the cobalt solution at room temperature with stirring over ~1 min. The lutidinium triflate vial was rinsed successively with 1 mL of toluene and 1 mL of THF, with the rinses added to the green cobalt solution. The reaction turned bright green with a small amount of precipitate, and the mixture was stirred for 5 min at room temperature before the volatile materials were removed in vacuo. The solid was washed with diethyl ether (2 × 1 mL), and the remaining solid was extracted into fluorobenzene (2 mL) and filtered through Celite. A drop of PMe3 was added into the fluorobenzene solution before pentane (10 mL) was allowed to diffuse into the fluorobenzene solution at room temperature for 2 d. This process yielded dark green, needle-like crystals (26.8 mg, 78%). Compound A (major): 1H NMR (500 MHz, THF-d8, 300 K): δ = 7.46 (s, 4H, py m-H), 4.02 (br s, 4H, CH2), 2.33 (br s, 2H, CH2), 2.05 (d, 5JPH = 4.1 Hz, 12H, N=C-CH3), 1.35 (d, 2JPH = 7.1 Hz, 18H, P(CH3)3), 1.28 (s, 18H, C(CH3)3), −20.72 (s, 1H, Co-H), −21.77 (s, 1H, Co-H) ppm. 1H resonances for the two remaining CH2 signals could not be located. 13C NMR (126 MHz, THF-d8, 300 K): δ = 150.7 (br s, N=C-CH3), 145.2 (d, 3JPC = 2.1 Hz, o-ArC), 140.1 (s, p-ArC), 122.8 (q, 1JCF = 323.6 Hz, OTf),116.5 (br s, m-ArC), 36.0 (s, C(CH3)3), 31.1 (s, C(CH3)3), 19.5 (br s, P(CH3)3), 14.9 (br s, N=C-CH3).
13C resonances for the backbone methylene signals could not be located. 31P NMR (202 MHz, THF-d8): δ = 0.85 ppm.Compound B (minor): 1H NMR (500 MHz, THF-d8, 300 K): δ = 8.08 (s, 4H, py m-H), 4.11 (m, 4H, CH2CH2CH2), 3.54 (m, 4H, CH2CH2CH2, overlapped with THF signal), 2.53 (t, J = 6.2 Hz, 4H, CH2CH2CH2), 2.18 (d, 5JPH = 5.7 Hz, 12H, N=C-CH3), 1.51 (s, 18H, C(CH3)3), 0.65 (d, 2JPH = 6.8 Hz, 18H, P(CH3)3), −26.76 (s, 1H, Co-H) ppm. 13C NMR (126 MHz, THF-d8, 300 K): δ = 155.4 (s, N=C-CH3), 146.8 (s, o-ArC), 140.5 (s, p-ArC), 122.8 (q, 1JCF = 323.6 Hz, OTf), 118.5 (s, m-ArC), 57.3 (s, CH2CH2CH2), 36.6 (s, C(CH3)3), 32.4 (s, CH2CH2CH2), 31.0 (s, C(CH3)3), 17.1 (d, 1JPC = 16.3 Hz, P(CH3)3), 15.3 (s, N=C-CH3). 31P NMR (202 MHz, THF-d8): δ = 13.9 ppm. Anal. Calcd. for C39H65Co2F3N6O3P2S1 · 1 C6H5F (1030.96 g/mol): C, 52.43; H, 6.84; N, 8.15%. Found: C, 51.43; H, 6.85; N, 8.34%.
Synthesis of [(3PDI2)2Co2(H2)] [Co2H2].
Method A: [Co2(PMe3)2]0 (12.4 mg, 0.016 mmol) was dissolved in 0.5 mL of C6D6 in a J. Young NMR tube, and the headspace was evacuated. The tube was backfilled with 1 atm of H2, leading to a greenish-brown solution with some green precipitate. The spectroscopic features were consistent with previously isolated [Co2H2]0. Removal of the volatile materials in vacuo led to a dark green solid that was washed with diethyl ether (2 × 1 mL). Extraction of the material into toluene (2 mL), filtration through Celite, and diffusion of pentane (6 mL) into the solution at room temperature for 5 d led to 1.2 mg of crystalline [Co2H2]0 (8% yield). Method B: (3PDI2)Sr(OTf)2 (102.8 mg, 0.11 mmol) and CoCl2 (28.0 mg, 0.21 mmol) were suspended in 5 mL of THF and stirred overnight to yield a light brown slurry. KC8 (61.9 mg, 0.46 mmol) was added as a solid to the mixture at room temperature, leading to a rapid color change of the solution to dark blue. The solution was stirred for 10 minutes at room temperature, then the volatile materials were removed in vacuo. The greenish-blue solid was triturated with pentane (3 × 1 mL) and washed with pentane and ether (2 × 1 mL each). A small amount of material was extracted into toluene (3 mL) and filtered through Celite. Most of the remaining material was extracted into fluorobenzene (2.5 mL) and filtered through Celite. Pentane (10 mL each) was allowed to diffuse into the toluene and fluorobenzene solutions at room temperature for 1 d, leading to a small amount of crystals in both solutions that were isolated, washed with pentane, and dried under vacuum. Yield: 1.7 mg, 2%. Please note that the yield and purity of the reaction by Method B is not improved if performed under an atmosphere of H2. 1H NMR (500 MHz, C6D6): δ = 8.02 (s, 4H, py m-H), 5.21–4.97 (m, 4H, CH2CH2CH2), 3.61 (d, J = 12.2 Hz, 4H, CH2CH2CH2), 1.95 (s, 4H, CH2CH2CH2), 1.55 (s, 18H, C(CH3)3), 1.02 (s, 12H, N=C-CH3), −27.30 (s, 2H, Co-H) ppm. 13C NMR (126 MHz, C6D6, 300 K): δ = 152.2 (s, o-ArC), 151.3 (s, N=C-CH3), 139.4 (s, p-ArC), 114.9 (s, m-ArC), 58.1 (s, CH2CH2CH2), 36.7 (s, C(CH3)3), 30.5 (s, C(CH3)3), 20.0 (s, CH2CH2CH2), 15.9 (s, N=C-CH3) ppm. Anal. Calcd. for C32H48Co2N6 (634.64 g/mol): C, 60.56; H, 7.62; N, 13.24%. Found: C, 55.21; H, 6.44; N, 11.62 %.
Synthesis of [(3PDI2)2Co2(PhCCPh)(PMe3)] [Co2PhCCPh].
[Co2(PMe3)2]0 (9.7 mg, 0.012 mmol) was dissolved in 2 mL of toluene, and a solution of diphenylacetylene (2.0 mg, 0.011 mmol) in 1 mL toluene was added dropwise to the cobalt solution at room temperature over 1 min with stirring. The bright green solution was stirred for 5 min at room temperature and the volatile materials were removed in vacuo. Most of the green solid was extracted into 2 mL of pentane, then the green solution was filtered through Celite, concentrated down to ca. 1 mL, and left at –35 °C overnight to yield large green plates. The plates were washed with minimal HMDSO and dried under vacuum. Yield: 1.2 mg (11%). 1H NMR (500 MHz, C6D6, 300 K): δ = 8.29 (s, 2H, py m-H), 7.84 (d, J = 6.6 Hz, 2H, Ph o-H), 7.46 (t, J = 7.6 Hz, 2H, Ph m-H), 7.35–7.24 (m, 1H, Ph p-H), 7.07 ( br s, 2H, Ph o-H), 7.03–6.96 (m, 2H, Ph m-H), 6.91–6.84 (m, 1H, Ph p-H), 6.80 (s, 2H, py’ m-H), 5.15–4.91 (m, 2H, CH2CH2C’H2), 4.64 (d, J = 3.6 Hz, 2H, CH2CH2C’H2), 3.18 (dd, J = 18.2, 13.0 Hz, 4H, CH2CH2C’H2 and CH2CH2C’H2 overlapped), 2.76 – 2.40 (m, 2H, CH2CH2C’H2), 1.80 (d, 5JPH = 7.2 Hz, 6H, N=C-C’H3 ), 1.71 (dt, J = 14.1, 3.0 Hz, 2H, CH2CH2C’H2), 1.51 (s, 9H, C(CH3)3), 1.28 (s, 6H, N=C-CH3), 1.02 (s, 9H, C(C’H3)3), 0.02 (d, 2JPH = 7.7 Hz, 9H, P(CH3)3) ppm. 13C NMR (126 MHz, C6D6, 300 K): δ = 156.6 (d, J = 10.8 Hz, Ph ipso-C), 152.2 (s, py o-C), 149.7 (d, J = 11.0 Hz, Calkyne), 146.4 (s, py p-C), 146.3 (d, 3JPC = 3.7 Hz, py’ o-C), 144.7 (d, 5JPC = 3.4 Hz, py’ p-C), 143.6 (d, J = 5.5 Hz, Ph ipso-C), 143.12 (d, J = 7.7 Hz, N=C’-CH3),140.1 (s, N=C-CH3), 137.5 (s, Calkyne), 130.3 (s, Ph m-C), 128.8 (s, Ph m-C), 128.0 (s, Ph o-C), 126.8 (s, Ph o-C), 122.14 (s, Ph p-C), 122.04 (s, Ph p-C), 112.0 (d, 4JPC = 2.2 Hz, py’ m-C), 104.9 (s, py m-C), 55.9 (s, CH2CH2C’H2), 55.1 (s, CH2CH2C’H2), 35.9 (s, C(CH3)3), 34.5 (s, C’(CH3)3), 31.8 (s, C(CH3)3), 31.4 (s, C(C’H3)3), 30.1 (s, CH2CH2C’H2), 15.7 (s, N=C-CH3), 13.3 (d, 4JPC = 4.1 Hz, N=C-C’H3), 12.7 (d, 1JPC = 15.6 Hz, P(CH3)3) ppm. 31P{1H} NMR (162 MHz, C6D6, 300 K): δ = −11.5 (s, P(CH3)3) ppm. Unambiguous determination of which alkyne resonances were on the side nearest to the phosphine could not be determined with the available data. Anal. Calcd. For C49H65Co2N6P1 (886.94 g/mol): C, 66.36; H, 7.39; N, 9.48%. Found: C, 68.04; H, 7.54; N, 8.67%.
Synthesis of [(3PDI2)2Co2(CO2)(PMe3)] [Co2CO2].
[Co2(PMe3)2]0 (6.1 mg, 0.008 mmol) was dissolved in 0.5 mL of C6D6 in a J. Young NMR tube, and the headspace was evacuated. The tube was backfilled with 1 atm of CO2, leading to a green-blue solution with some precipitate. Removal of the volatile materials in vacuo led to a green-blue solid that was washed with diethyl ether (2 × 1 mL). The remaining solid was extracted into fluorobenzene (2 mL) and filtered through Celite, then pentane (6 mL) was allowed to diffuse into the solution at room temperature for 2 d, leading to 4.3 mg (73%) of crystalline [Co2CO2]0. 1H NMR (500 MHz, THF-d8, 300 K): δ = 7.45 (s, 2H, py m-H), 6.96 (s, 2H, py’ m-H), 5.19 (td, J = 13.0, 3.0 Hz, 2H, CH2CH2C’H2), 4.68–4.54 (m, 2H, CH2CH2C’H2), 4.09 (ddd, J = 12.0, 4.7, 2.4 Hz, 2H, CH2CH2C’H2), 3.59 (m, 2H, CH2CH2C’H2, overlapped THF residual), 3.15 (dt, J = 13.8, 4.4 Hz, 2H, CH2CH2C’H2), 2.41 (d, 5JPH = 6.7 Hz, 6H, N=C-C’H3), 2.23 (d, J = 14.1 Hz, 2H, CH2CH2C’H2), 1.76 (s, 6H, N=C-CH3), 1.21 (s, 9H, C(CH3)3), 1.02 (s, 9H, C(C’H3)3), 0.58 (d, J = 9.1 Hz, 9H, P(CH3)3) ppm. 13C NMR (126 MHz, THF-d8, 300 K): δ = 196.2 (d, 3JPC = 6.8 Hz, CO2), 150.7 (s, o-ArC), 146.0 (d, 3JPC = 7.7 Hz, N=C’-CH3), 145.4 (d, 3JPC = 4.2 Hz, o-ArC’), 144.6 (d, 5JPC = 2.4 Hz, p-ArC’), 144.1 (s, p-ArC), 142.3 (s, N=C-CH3), 114.2 (s, m-ArC’), 108.3 (s, m-ArC), 57.9 (s, CH2CH2C’H2), 52.3 (s, CH2CH2C’H2), 36.4 (s, C(CH3)3), 35.2 (s, C’(CH3)3), 32.5 (d, 4JPC = 2.7 Hz, CH2CH2C’H2), 31.7 (s, C(C’H3)3), 31.2 (s, C(CH3)3), 15.7 (s, N=C-CH3), 13.4 (d, 1JPC = 18.6 Hz, P(CH3)3), 13.4 (d, 4JPC = 4.1 Hz, N=C-C’H3) ppm. 31P{1H} NMR (162 MHz, C6D6, 300 K): δ = −5.25 (s, P(CH3)3) ppm. Anal. Calcd. For C36H55Co2N6O2P (752.72 g/mol): C, 57.45; H, 7.36; N, 11.16%. Found: C, 55.50; H, 7.21; N, 9.61%.
Synthesis of [(3PDI2)Co2(μ-N3)(PMe3)2][OTf] [Co2N3]+.
[Co2Cl]+ (193.2 mg, 0.20 mmol, 1.0 equiv) was added to a 20 mL scintillation vial and dissolved in THF ( ca. 10 mL). NaN3 (13.0 mg, 0.20 mmol, 1.0 equiv) was added as a solid to the dark green solution at ambient temperature and stirred for 3 days. The solvent was removed in vacuo and the dark green solid was mostly extracted with fluorobenzene (ca. 5 mL) and filtered through Celite. The remaining solid was extracted with THF (ca. 2 mL) and filtered through Celite. Vapor diffusion of pentane into both the fluorobenzene and THF solutions overnight yielded dark green crystals of [Co2N3]+ (143.8 mg, 74%). 1H NMR (500 MHz, THF-d8, 300 K): δ = 7.57 (s, 4H, py m-H), 4.45 (t, J = 12.1 Hz, 4H, CH2CH2CH2), 4.20–4.17 (m, 4H, CH2CH2CH2), 2.65–2.58 (m, 2H, CH2CH2CH2), 2.53–2.49 (m, 2H, CH2CH2CH2), 1.80 (s, 12H, N=C-CH3), 1.26 (s, 18H, C(CH3)3), and 0.87 (d, 2JPH = 10.7 Hz, 18H, P(CH3)3) ppm.13C{1H} NMR (126 MHz, THF-d8, 300 K): δ = 153.9 (s, Cimine), 145.0 (s, o-ArC), 139.6 (s, p-ArC), 118.3 (s, m-ArC) ppm, 58.7 (s, CH2CH2CH2), 35.4 (s, C(CH3)3), 29.4 (s, C(CH3)3), 28.1 (s, CH2CH2CH2), 14.2 (s, N=C-CH3), 13.6 (br s, P(CH3)3). 31P{1H} NMR (162 MHz, THF-d8, 300 K): δ = −30.52 (s, P(CH3)3) ppm. Anal. Calcd. For C39H64Co2F3N9O3P2S1 (975.86 g/mol): C, 48.00; H, 6.61; N,12.92 %. Found C, 47.42; H, 6.43; N, 12.47 %. IR (KBr): νN3 = 2040 cm−1
Catalytic N2 Reduction.
To a stirred suspension of KC8 (266.3 mg, 1.97 mmol) in THF (20 mL) at room temperature was added a THF solution (5 mL) of a dicobalt catalyst (0.004 mmol). Neat TMSCl (0.25 mL, 1.97 mmol) was then added. For the trial using [Fe2N2]0 as the catalyst, KC8 and TMSCl were mixed immediately prior to the addition of the catalyst. After stirring for 24 h, the system was exposed to air before being filtered through Celite. A diethyl ether solution of HCl (2.0 M, 10 mL) was added to the filtrate, then the volatile materials were removed under vacuum. 1,3,5-Trimethoxybenzene (TMB, 10 mg, 0.059 mmol) was added to the flask containing the resulting powder, and the mixture was dissolved in DMSO-d6. The yields of NH4Cl were then determined by 1H NMR spectroscopy, using TMB as an internal standard. 1H NMR (400 MHz, DMSO-d6, 300 K): δ = 7.38 (t, 4H, NH4Cl) ppm.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Science Foundation (CHE-1945265) and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128794) for financial support.
Footnotes
Notes
The authors declare no competing financial interest.
ACCESSION CODES
CCDC 2328016–2328018, 2347312, 2347317, 2347318, 2347320, 2347923, and 2347924 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; Fax: + 44 1223 336033.
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website.
NMR spectra, IR spectra, and crystallographic details (PDF)
REFERENCES
- (1).Powers IG; Uyeda C Metal-Metal Bonds in Catalysis. ACS Catal. 2017, 7 (2), 936–958. DOI: 10.1021/acscatal.6b02692 [DOI] [Google Scholar]
- (2).Buchwalter P; Rosé J; Braunstein P Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. DOI: 10.1021/cr500208k [DOI] [PubMed] [Google Scholar]
- (3).Wang Q; Brooks SH; Liu T; Tomson NC Tuning metal–metal interactions for cooperative small molecule activation. Chem. Commun. 2021, 57 (23), 2839–2853. DOI: 10.1039/D0CC07721F [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Uyeda C; Farley CM Dinickel Active Sites Supported by Redox-Active Ligands. Acc. Chem. Res. 2021, 54 (19), 3710–3719. DOI: 10.1021/acs.accounts.1c00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gavrilova AL; Bosnich B Principles of Mononucleating and Binucleating Ligand Design. Chem. Rev. 2004, 104 (2), 349–384. DOI: 10.1021/cr020604g [DOI] [PubMed] [Google Scholar]
- (6).Dammann W; Buban T; Schiller C; Burger P Dinuclear tethered pyridine, diimine complexes. Dalton Trans. 2018, 47 (35), 12105–12117. DOI: 10.1039/C8DT02347F [DOI] [PubMed] [Google Scholar]
- (7).Chen Q; Suo H; Zhang W; Zhang R; Solan GA; Liang T; Sun W-H 1,5-Naphthyl-linked bis(imino)pyridines as binucleating scaffolds for dicobalt ethylene oligo-/polymerization catalysts: exploring temperature and steric effects. Dalton Trans. 2019, 48 (23), 8264–8278. DOI: 10.1039/C9DT01235D [DOI] [PubMed] [Google Scholar]
- (8).Chang CJ; Loh Z-H; Deng Y; Nocera DG The Pacman Effect: A Supramolecular Strategy for Controlling the Excited-State Dynamics of Pillared Cofacial Bisporphyrins. Inorg. Chem. 2003, 42 (25), 8262–8269. DOI: 10.1021/ic034750w [DOI] [PubMed] [Google Scholar]
- (9).Hollingsworth RL; Hollingsworth TS; Groysman S Synthesis and Reactivity of Bimetallic Systems Tethered with a 4,5-Diaminoxanthene Linker; Zhou M, Ed.; ACS Symposium Series, Vol. 1317; American Chemical Society, 2019. DOI: 10.1021/bk-2019-1317.ch001 [DOI] [Google Scholar]
- (10).Hollingsworth RL; Beattie JW; Grass A; Martin PD; Groysman S; Lord RL Reactions of dicobalt octacarbonyl with dinucleating and mononucleating bis(imino)pyridine ligands. Dalton Trans. 2018, 47 (43), 15353–15363. DOI: 10.1039/C8DT03405B [DOI] [PubMed] [Google Scholar]
- (11).Zhang S; Wang Q; Thierer LM; Weberg AB; Gau MR; Carroll PJ; Tomson NC Tuning Metal-Metal Interactions through Reversible Ligand Folding in a Series of Dinuclear Iron Complexes. Inorg. Chem. 2019, 58 (18), 12234–12244. DOI: 10.1021/acs.inorgchem.9b01673 [DOI] [PubMed] [Google Scholar]
- (12).Spentzos AZ; Gau MR; Carroll PJ; Tomson NC Unusual cyanide and methyl binding modes at a dicobalt macrocycle following acetonitrile C–C bond activation. Chem. Commun. 2020, 56 (67), 9675–9678. DOI: 10.1039/D0CC03521A [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang Q; Zhang S; Cui P; Weberg AB; Thierer LM; Manor BC; Gau MR; Carroll PJ; Tomson NC Interdependent Metal-Metal Bonding and Ligand Redox-Activity in a Series of Dinuclear Macrocyclic Complexes of Iron, Cobalt, and Nickel. Inorg. Chem. 2020, 59 (7), 4200–4214. DOI: 10.1021/acs.inorgchem.9b02339 [DOI] [PubMed] [Google Scholar]
- (14).Liu T; Gau MR; Tomson NC Mimicking the Constrained Geometry of a Nitrogen-Fixation Intermediate. J. Am. Chem. Soc. 2020, 142 (18), 8142–8146. DOI: 10.1021/jacs.0c01861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Green JC; Green MLH; Parkin G The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds. Chem. Commun. 2012, 48 (94), 11481–11503. DOI: 10.1039/C2CC35304K [DOI] [PubMed] [Google Scholar]
- (16).Ashley AE; Balázs G; Cowley AR; Green JC; O’Hare D syn-Permethylpentalene Iron and Cobalt Carbonyl Complexes: Proximity Bimetallics Lacking Metal–Metal Bonding. Organometallics 2007, 26 (23), 5517–5521. DOI: 10.1021/om700702v [DOI] [Google Scholar]
- (17).Römelt C; Weyhermüller T; Wieghardt K Structural characteristics of redox-active pyridine-1,6-diimine complexes: Electronic structures and ligand oxidation levels. Coord. Chem. Rev. 2019, 380, 287–317. DOI: 10.1016/j.ccr.2018.09.018 [DOI] [Google Scholar]
- (18).Cui P; Wang Q; McCollom SP; Manor BC; Carroll PJ; Tomson NC Ring-Size-Modulated Reactivity of Putative Dicobalt-Bridging Nitrides: C–H Activation versus Phosphinimide Formation. Angew. Chem. Int. Ed. 2017, 56 (50), 15979–15983. DOI: 10.1002/anie.201708966 [DOI] [PubMed] [Google Scholar]
- (19).When there is a mixture of [Co2H]+ and [Co2Cl]+, we have grown both disordered crystals and crystals containing one equivalent each of [Co2H]+ and [Co2Cl]+, but with only one phosphine coordinated to the [Co2H]+ molecule. [Google Scholar]
- (20).Kaesz HD; Saillant RB Hydride complexes of the transition metals. Chem. Rev. 1972, 72 (3), 231–281. DOI: 10.1021/cr60277a003 [DOI] [Google Scholar]
- (21).Spentzos AZ; Tomson NC Mapping the Reactivity of Dicobalt Bridging Nitrides in Constrained Geometries. Inorg. Chem. 2021, 60 (10), 6889–6899. DOI: 10.1021/acs.inorgchem.0c03774 [DOI] [PubMed] [Google Scholar]
- (22).Brooks SH; Richards CA; Carroll PJ; Gau MR; Tomson NC Anion Capture at the Open Core of a Geometrically Flexible Dicopper(II,II) Macrocycle Complex. Inorganics 2023, 11 (9), 348. DOI: 10.3390/inorganics11090348 [DOI] [Google Scholar]
- (23).Zhang S; Cui P; Liu T; Wang Q; Longo TJ; Thierer LM; Manor BC; Gau MR; Carroll PJ; Papaefthymiou GC; Tomson NC N–H Bond Formation at a Diiron Bridging Nitride. Angew. Chem. Int. Ed. 2020, 59 (35), 15215–15219. DOI: 10.1002/anie.202006391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Thierer LM; Brooks SH; Weberg AB; Cui P; Zhang S; Gau MR; Manor BC; Carroll PJ; Tomson NC Macrocycle-Induced Modulation of Internuclear Interactions in Homobimetallic Complexes. Inorg. Chem. 2022, 61 (16), 6263–6280. DOI: 10.1021/acs.inorgchem.2c00522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dugan TR; Goldberg JM; Brennessel WW; Holland PL Low-Coordinate Cobalt Fluoride Complexes: Synthesis, Reactions, and Production from C–F Activation Reactions. Organometallics 2012, 31 (4), 1349–1360. DOI: 10.1021/om200991k [DOI] [Google Scholar]
- (26).Suzuki T; Masuda H; Fryzuk MD Variable coordination geometries via an amine-tethered-enamidophosphinimine ligand on cobalt. Dalton Trans. 2017, 46 (20), 6612–6622. DOI: 10.1039/C7DT00930E [DOI] [PubMed] [Google Scholar]
- (27).Dugan TR; Holland PL New routes to low-coordinate iron hydride complexes: The binuclear oxidative addition of H2. J. Organomet. Chem. 2009, 694 (17), 2825–2830. DOI: 10.1016/J.JORGANCHEM.2009.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pfirrmann S; Yao S; Ziemer B; Stösser R; Driess M; Limberg C β-Diketiminato Nickel(I) Complexes with Very Weak Ligation Allowing for H2 and N2 Activation. Organometallics 2009, 28 (24), 6855–6860. DOI: 10.1021/om9007983 [DOI] [Google Scholar]
- (29).Bach I; Goddard R; Kopiske C; Seevogel K; Pörschke K-R Synthesis, Structure, and Reactivity of (tBu2PC2H4PtBu2)Ni(CH3)2 and {(tBu2PC2H4PtBu2)Ni}2(μ-H)2. Organometallics 1999, 18 (1), 10–20. DOI: 10.1021/om980705y [DOI] [Google Scholar]
- (30).Klein H-F; Mager M; Schmidt A; Hüber M; Haase W; Flörke U; Haupt H-J; Boca R Synthesis, Structure, and Magnetic Properties of Low-Valent Triangulo Cobalt–Hydride Clusters [XCo3(μ-CO)3(PMe3)6]. Inorg. Chem. 1997, 36 (20), 4303–4306. DOI: 10.1021/ic970431r [DOI] [PubMed] [Google Scholar]
- (31).Fryzuk MD; Ng JB; Rettig SJ; Huffman JC; Jonas K Nature of the catalytically inactive cobalt hydride formed upon hydrogenation of aromatic substrates. Structure and characterization of the binuclear cobalt hydride [{Pri2P(CH2)3PPri2}Co]2(H)(μ-H)3. Inorg. Chem. 1991, 30 (10), 2437–2441. DOI: 10.1021/ic00010a039 [DOI] [Google Scholar]
- (32).Hoffman DM; Hoffmann R; Fisel CR Perpendicular and parallel acetylene complexes. J. Am. Chem. Soc. 1982, 104 (14), 3858–3875. DOI: 10.1021/ja00378a014 [DOI] [Google Scholar]
- (33).Examples are known of tri- or tetra-cobalt clusters that adopt this orientation between the alkyne and two of the metal atoms in the cluster, with the remaining metal centers interacting with the face of the Co2C2 metallacycle: Chetcuti MJ; Fanwick PE; Gordon JC, Reactions of a cobalt-molybdenum and related cobalt-tungsten tetrahedral clusters with phenylacetylene: formation of tetrametallic alkyne clusters or cluster fragmentation? Inorg. Chem. 1991, 30 (25), 4710–4717. DOI: 10.1021/ic00025a008 [DOI] [Google Scholar]
- (34).Pepermans H; Hoogzand C; Geerlings P A reinvestigation of the isomerization of alkyne dicobalt hexacarbonyl complexes by the perpendicular-to-parallel twist of the alkyne: Influence of the induced carbonyl reorientations. J. Organomet. Chem. 1986, 306 (3), 395–405. DOI: 10.1016/S0022-328X(00)99001-6 [DOI] [Google Scholar]
- (35).Liu T; Murphy RP; Carroll PJ; Gau MR; Tomson NC C–C σ-Bond Oxidative Addition and Hydrofunctionalization by a Macrocycle-Supported Diiron Complex. J. Am. Chem. Soc. 2022, 144 (31), 14037–14041. DOI: 10.1021/jacs.2c06266 [DOI] [PubMed] [Google Scholar]
- (36).Abramenkov A; Almenningen A; Cyvin B; Cyvin S; Jonvik T; Khaikin L; Rømming C; Vilkov L Internal rotation in tolane: molecular structure in gas and crystal phases. Acta Chem. Scand. A 1988, 42, 674–684. [Google Scholar]
- (37).Sly WG The Molecular Configuration of Dicobalt Hexacarbonyl Diphenylacetylene. J. Am. Chem. Soc. 1959, 81 (1), 18–20. DOI: 10.1021/ja01510a005 [DOI] [Google Scholar]
- (38).Enachi A; Baabe D; Zaretzke M-K; Schweyen P; Freytag M; Raeder J; Walter MD [(NHC)CoR2]: pre-catalysts for homogeneous olefin and alkyne hydrogenation. Chem. Commun. 2018, 54 (98), 13798–13801. DOI: 10.1039/C8CC08891H [DOI] [PubMed] [Google Scholar]
- (39).Pilato RS; Housmekerides CE; Jernakoff P; Rubin D; Geoffroy GL; Rheingold AL Net [2 + 2] cycloaddition reactions of the oxo complexes Cp2M=O (M = Mo, W) with electrophilic organic and organometallic substrates. Formation of bimetallic μ2-η3-CO2 complexes. Organometallics 1990, 9 (8), 2333–2341. DOI: 10.1021/om00158a034 [DOI] [Google Scholar]
- (40).Aresta M; Dibenedetto A; Quaranta E CO2 Coordination to Metal Centres: Modes of Bonding and Reactivity. In Reaction Mechanisms in Carbon Dioxide Conversion, Aresta M, Dibenedetto A, Quaranta E. Eds.; Springer Berlin; Heidelberg, 2016; pp 35–69. [Google Scholar]
- (41).Audett JD; Collins TJ; Santarsiero BD; Spies GH Substrate Organometallic Chemistry of Osmium Tetraoxide: Formation of a Novel Type of Carbon Dioxide Coordination. J. Am. Chem. Soc. 1982, 104 (25), 7352–7353. DOI: 10.1021/ja00389a093 [DOI] [Google Scholar]
- (42).Fujita E; Szalda DJ; Creutz C; Sutin N Carbon dioxide activation: thermodynamics of carbon dioxide binding and the involvement of two cobalt centers in the reduction of carbon dioxide by a cobalt(I) macrocycle. J. Am. Chem. Soc. 1988, 110 (14), 4870–4871. DOI: 10.1021/ja00222a079 [DOI] [Google Scholar]
- (43).Szalda DJ; Chou MH; Fujita E; Creutz C Properties and reactivity of metallocarboxylates. Crystal and molecular structure of the –CO22–-bridged “polymer” {[CoIII(en)2(CO2)](ClO4)•H2O}n. Inorg. Chem. 1992, 31 (22), 4712–4714. DOI: 10.1021/ic00048a050 [DOI] [Google Scholar]
- (44).Polyansky DE; Grills DC; Ertem MZ; Ngo KT; Fujita E Role of Bimetallic Interactions in the Enhancement of Catalytic CO2 Reduction by a Macrocyclic Cobalt Catalyst. ACS Catal. 2022, 12 (3), 1706–1717. DOI: 10.1021/acscatal.1c05043 [DOI] [Google Scholar]
- (45).Siedschlag RB; Bernales V; Vogiatzis KD; Planas N; Clouston LJ; Bill E; Gagliardi L; Lu CC Catalytic Silylation of Dinitrogen with a Dicobalt Complex. J. Am. Chem. Soc. 2015, 137 (14), 4638–4641. DOI: 10.1021/jacs.5b01445 [DOI] [PubMed] [Google Scholar]
- (46).Imayoshi R; Tanaka H; Matsuo Y; Yuki M; Nakajima K; Yoshizawa K; Nishibayashi Y Cobalt-Catalyzed Transformation of Molecular Dinitrogen into Silylamine under Ambient Reaction Conditions. Chem. Eur. J. 2015, 21 (24), 8905–8909. DOI: 10.1002/chem.201501088 [DOI] [PubMed] [Google Scholar]
- (47).Tanabe Y; Nishibayashi Y Recent advances in catalytic silylation of dinitrogen using transition metal complexes. Coord. Chem. Rev. 2019, 389, 73–93. DOI: 10.1016/j.ccr.2019.03.004 [DOI] [Google Scholar]
- (48).Ferreira RB; Cook BJ; Knight BJ; Catalano VJ; García-Serres R; Murray LJ Catalytic Silylation of Dinitrogen by a Family of Triiron Complexes. ACS Catal. 2018, 8 (8), 7208–7212. DOI: 10.1021/acscatal.8b02021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Thierer LM; Wang Q; Brooks SH; Cui P; Qi J; Gau MR; Manor BC; Carroll PJ; Tomson NC Pyridyldiimine macrocyclic ligands: Influences of template ion, linker length and imine substitution on ligand synthesis, structure and redox properties. Polyhedron 2021, 198, 115044. DOI: 10.1016/j.poly.2021.115044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Schwindt MA; Lejon T; Hegedus LS Improved Synthesis of (Aminocarbene)chromium(0) Complexes with Use of C8K-Generated Cr(CO)52–. Multivariant Optimization of an Organometallic Reaction. Organometallics 1990, 9 (10), 2814–2819. DOI: 10.1021/om00160a033 [DOI] [Google Scholar]
- (51).Luetkens ML; Sattelberger AP; Murray HH; Basil JD; Fackler JP; Jones RA; Heaton DE Trimethylphosphine. In Inorganic Syntheses, Angelici RJ. Ed.; Vol. 28; John Wiley & Sons, Ltd, 2007; pp 305–310. [Google Scholar]
- (52).Curley JJ; Bergman RG; Tilley TD Preparation and physical properties of early-late heterobimetallic compounds featuring Ir–M bonds (M = Ti, Zr, Hf). Dalton Trans. 2012, 41 (1), 192–200. DOI: 10.1039/C1DT11753J [DOI] [PubMed] [Google Scholar]
- (53).Zoski C Handbook of Electrochemistry; Elsevier Science, 2007. [Google Scholar]
- (54).Kaplan L; Kester WL; Katz JJ Some Properties of Iron Biscyclopentadienyl. J. Am. Chem. Soc. 1952, 74 (21), 5531–5532. DOI: 10.1021/ja01141a518 [DOI] [Google Scholar]
- (55).Evans DF The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, (0), 2003–2005. DOI: 10.1039/jr9590002003 [DOI] [Google Scholar]
- (56).SAINT v8.38A: Bruker-AXS; Madison, Wisconsin, USA. 2014. [Google Scholar]
- (57).CrysAlisPro 1.171.43.94a: Rigaku Oxford Diffraction, Rigaku Corporation, Oxford, UK. 2023. [Google Scholar]
- (58).Krause L; Herbst-Irmer R; Sheldrick GM; Stalke D Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48 (1), 3–10. DOI: 10.1107/S1600576714022985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).SCALE3 ABSPACK v1.0.7: an Oxford Diffraction program; Oxford Diffraction Ltd: Abingdon, UK. 2005. [Google Scholar]
- (60).Sheldrick GM SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71 (1), 3–8. DOI: 10.1107/S2053273314026370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sheldrick GM A short history of SHELX. International Union of Crystallography: 2008; Vol. 64, pp 112–122. [DOI] [PubMed] [Google Scholar]
- (62).Sheldrick GM Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71 (1), 3–8. DOI: 10.1107/S2053229614024218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).CELL_NOW v2008/4: Sheldrick GM. 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.