

Abstract
Tripodal ligands (TMG3trphen-E) that feature heavy pnictogen elements (E = Sb(III), Bi(III)) and tetramethylguanidinyl (TMG) arms have been explored in stabilizing Cu(I) and Ag(I) sites and facilitating nitrene-transfer chemistry. Compounds [(TMG3trphen-E)M3(μ-X)3] (M = Cu(I), Ag(I); X = Cl, Br, I) have been generated upon extraction of M3(μ-X)3 units from MX sources, exhibiting support of the crown-shaped M3(μ-X)3 fragment by M–NTMG bonds and triply bridging E → M3 interactions. Orbital interactions between Cu(I) sites and NTMG residues are more dominant than Sb/Bi → Cu3 donor interactions between the Sb 5s or Bi 6s orbitals and admixed Cu 4s/3d orbitals, with larger interaction energies computed for Sb → Cu3. Nonhalogenated copper compounds [(TMG3trphen-E)2Cu2]2+2Y− (Y = PF6, B(C6F5)4) have been synthesized via dechlorination by TlPF6 or by application of halide-free Cu(I) sources with TMG3trphen-E ligands. Nitrene-transfer to olefins mediated by [(TMG3trphen-E)Cu3(μ-Cl)3] (E = Sb and Bi) affords aziridines in good yields, primarily for unencumbered styrenes and with the more robust Sb catalyst. Amination of C–H bonds is most effective with sec-benzylic substrates and requires a more electrophilic nitrene (NTces) to achieve practicable yields with halogenated or nonhalogenated copper precursors. Hammett plots indicate that the competitive amination of para-substituted ethylbenzenes enabled by [(TMG3trphen-Sb)Cu3(μ-Cl)3] involves stepwise C–H functionalization.
Graphical Abstract

INTRODUCTION
The development of C–N bond construction methodologies encompasses various pathways directed toward inserting nitrogen functionalities into carbonaceous feedstock for the synthesis of a diverse body of commodity and high-value chemicals (pharmaceuticals, agrochemicals, polymers, semiconductors, catalysts, solvents, and household chemicals).1 Among different approaches, the insertion of nitrenes (NR) or nitrenoids (NR(X)) into C–H bonds or their addition to C═C bonds affords valuable products of amination and aziridination, respectively, as well as derivatives (such as amidines or five-membered N-heterocycles) in the presence of additional substrates (usually unsaturated entities).2 This direct functionalization of C–H/C═C feedstock belongs to the general category of atom/group-transfer chemistry,3 pertaining to a wide variety of common atoms (e.g., H/D, N, O, S, and halogen) or groups (e.g., BR2, CR2/CR3, NR/NR2, and N3).4–12 Biological atom/group-transfer analogs have frequently provided inspiration and opportunities for further development via biomimetic approaches and enzyme engineering.13 The direct C–H/C═C functionalization obviates the need for constructing energetic C–X precursors (X = leaving or directing group) but raises challenges for achieving acceptable levels of reactivity and selectivity.14 Taking C–N bond construction from C–H/C═C feedstock as an example, both metal-free15 and metal-dependent catalytic systems2,16 have been advanced to overcome these challenges. Notwithstanding the expense and toxicity involved, platinum-group metal catalysts have been frequently invoked,17 but nonprecious, first-row transition metal reagents18 have found increasing use. The latter usually raise the possibility of single-electron steps that provide avenues to radical pathways, which may thus offer intriguing alternatives to closed-shell two-electron processes.
In recent work, in our laboratory, we have developed a family of divalent base metals (Mn, Fe, Co, and Ni) and monovalent coinage metal catalysts (Cu and Ag), supported by tripodal trisamido/imino-amine and bipodal bisamido/imino-amine ligands (Chart 1).19 The weak ligand field and the ability to decorate the equatorial amido/imino residues by a wide range of aryl, acyl, alkyl, and guanidinyl arms has given rise to a plethora of anionic, neutral, and cationic catalysts employed in nitrene transfer to C–H and C═C bonds of hydrocarbons. These tripodal and bipodal ligand scaffolds are analogs of the iconic N(CH2CH2NH2)3 (TREN) and bipodal N(CH2CH2NH2)2Me frameworks20–22 but rely on phenylene rather than ethylene linkers, resulting in metal reagents that tend to be more reactive in atom/group-transfer chemistry. Versions of the anionic tripodal metal compounds have also been nicely explored in bimetallic synthesis and catalysis.23 In general, anionic versions tend to be more appropriate for aziridination reactions,24 whereas the much more reactive cationic analogs, especially those decorated with tetramethylated guanidinyl arms (extensively studied in TREN chemistry),25,26 can be effectively employed both in C–H aminations/amidinations (Cu reagents)27 and C═C aziridinations, in tandem with five-membered N-heterocycle synthesis (Mn, Fe, or Co reagents).28
Chart 1.

Previously Explored Reagents Supported by Axial Namine Coordination
In all cases noted above, the axial coordination site is occupied by an Namine atom. With the present work, we are launching an effort toward replacing the apical Namine residue with more weakly electron-donating and potentially electron-accepting Sb(III) and Bi(III) residues.29 It is anticipated30 that electron-deficient apical elements can enhance the electro-philicity of any axially positioned metal-nitrene moiety and, therefore, increase reactivity for challenging C–H substrates. Of course, the pnictogen elements generate significantly more sizable ligand cavities than those examined with the tripodal [N3Namine] scaffolds noted above. Hence, in this publication, we aspire to explore the geometric and electronic characteristics of metal reagents that can be accessed with these axial Sb/Bi containing ligands and provide an initial account of their nitrene-transfer reactivity. We chose to first explore Cu(I) and to a lesser extent Ag(I) sites in tandem with equatorial tetramethylguanidinyl (TMG) residues, given our successful application of the analogous [(N3Namine)CuI]+ reagent in the past.27
RESULTS AND DISCUSSION
Synthesis of Ligands.
The tripodal ligands TMG3trphen-Sb (1) and TMG3trphen-Bi (2) used in this study are synthesized by a concise method (Scheme 1). First, the trimethylguanidinyl groups (TMG) are installed by coupling 2-bromoaniline and chlorotetramethylformamidinium chloride (prepared from tetramethylurea and oxalyl chloride) in the presence of triethylamine in acetonitrile, followed by low-temperature lithiation with sec-BuLi (or tert-BuLi) in diethyl ether (Sb) or tetrahydrofuran (Bi). The electrophiles SbCl3 and BiCl3 are then used in THF (0.33 equiv versus the lithiated product) to install the axial Sb(III) and Bi(III) elements, respectively. Both ligands can be crystallized from dichloromethane to provide pure products as white solids in moderate to good yields. The lower yields observed for 2 reflect the thermal instability of the Bi–C bond. 1H NMR spectra (in CD3CN) indicate that all aryl and methyl groups (NMe2; sharp single peak at δ = 2.57 (1), 2.58 (2)) are equivalent at 298 K, hence no significant rotational restrictions are observed at room temperature.
Scheme 1.

Synthesis of Ligands Employed in this Study
Synthesis of Metal Halide Compounds.
The reaction of ligand 1 with 3 equiv of anhydrous CuCl of high purity (beads, ≥99.99%) in THF provides a white solid, which can be dissolved in dichloromethane and obtained as colorless crystals of [(TMG3trphen-Sb)Cu3(μ-Cl)3]•4CH2Cl2 (3) from concentrated solutions (Scheme 2). The analogous reaction of ligand 2 with CuCl in THF affords the corresponding [(TMG3trphen-Bi)Cu3(μ-Cl)3]•4CH2Cl2 (4), which can be crystallized from dichloromethane as off-white crystals suitable for X-ray analysis. The corresponding bromo and iodo analogs of 3 can be obtained in a similar manner from the reaction of anhydrous CuX (X = Br, I) with ligand 1 in THF, followed by crystallization from dichloromethane to obtain colorless crystals of [(TMG3trphen-Sb)Cu3(μ-Br)3]•4CH2Cl2 (5) and [(TMG3trphen-Sb)Cu3(μ-I)3]•3CH2Cl2 (6), respectively.
Scheme 2.

Synthesis of Metal Halide Compounds
A similar trinuclear Ag3(μ-Cl)3 cluster can also be extracted by ligand 1 from anhydrous AgCl (beads, 99.998%) upon prolonged extraction times and crystallized from CH2Cl2 as off-white crystals of [(TMG3trphen-Sb)Ag3(μ-Cl)3]•3CH2Cl2 (7). The same reaction with ligand 2 was not productive even after several weeks of extraction at room temperature or upon heating.
Synthesis of Halide-Free Metal Compounds.
For comparative purposes, the coordination chemistry of the stibine (1) and bismuthine (2) ligand was examined with respect to Cu(I) sites in the absence of any halides. The reaction of either 1 or 2 with 1.5 equiv of [Cu(NCMe)4](PF6) in THF/MeCN (Scheme 3) affords colorless crystals of [(TMG3trphen-E)2Cu2](PF6)2•2 solv (E = Sb, solv = THF (8a); Bi, solv = Et2O (9)), upon crystallization from THF/MeCN (8a) or MeCN/Et2O (9). The Bi compound is only obtained in very low yields, most likely because of the thermal instability of the ligand, as noted above. Incidentally, the corresponding 1:1 ligand/Cu reaction affords intractable mixtures. On one occasion, a few crystals with stoichiometry [(TMG3trphen-Sb)2Cu3](PF6)3 (two ligands capping a Cu3 triangle) were identified by SCXRD analysis but were of poor quality. Compound 8a provides low-quality twinned crystals for single-crystal X-ray diffraction (see below), but the corresponding compound [(TMG3trphen-Sb)2Cu2](B(C6F5)4)2•2 MeCN (8b) with B(C6F5)4− as counteranion provides single crystals of superior XRD quality from MeCN. Compound 8b can be obtained from 8a upon addition of 2 equiv of KB(C6F5)4, or via the reaction of ligand 1 with 1.5 equiv of [Cu(NCMe)2][B(C6F5)4]31 in MeCN.
Scheme 3.

Synthesis of Halide-free Metal Compounds
We have also attempted to remove bridging chlorides from [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3) under various conditions. When TlPF6 (3 equiv) was employed in MeCN, the resulting needle-shaped crystalline material proved to be the same as compound 8a, albeit solvated by MeCN. On the other hand, the reaction of AgPF6 (3 equiv) with compound 3 in MeCN, provided a poorly soluble and difficult to crystallize white solid, whose NMR signature was identical to that obtained from a reaction of ligand 1 with AgPF6 in MeCN, thus indicating ligand transmetalation from Cu to Ag. We have been unable thus far to crystallize and fully characterize this product. We have also tried to use high-purity Ag(OTf) (3 equiv) for extracting chlorides from 3 in CH2Cl2, which led to the isolation of low yields of mononuclear [(TMG3H3trphen-Sb)Cu(OTf)3](OTf) (10), exhibiting protonation and detachment of the TMG arms, although the protonated ligand is still coordinated to Cu(I) via the Sb(III) element (Scheme 4). The supernatant affords an intractable mixture that also indicates partial ligand transformation. The provenance of protons, whether from the solvent or the ligand, is currently unknown, but notably, we have observed facile C–H activation of the NMe2 residues in the past.27 Instances in which metal triflate salts act as masked TfOH sources are also known in the literature.32
Scheme 4.

Extraction of Chlorides from 3 by Silver Triflate
Finally, the reaction of 3 equiv of KB(C6F5)4 with 3 in CH2Cl2 afforded an intriguing colorless product, [(TMG3trphen-Sb)Cu3(μ3-Cl)3Cu]2[B(C6F5)4]2•10CH2Cl2 (11), exhibiting partial chloride extraction (Scheme 5). While the crown-shaped Cu3(μ-Cl)3 fragment is largely retained, extraction of Cu(I) sites is also evident, contributing to the assembly of a new {[Cu3(μ3-Cl)3]2Cu2} core structure. The mechanism of formation is certainly complex, as we cannot generate the compound from the reaction of 3 with 2 equiv of [Cu(NCMe)2][B(C6F5)4].
Scheme 5.

Partial Extraction of Chloride from [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3)
Solid-State Structures.
Ligands TMG3trphen-Sb (1) and TMG3trphen-Bi (2) are essentially isostructural (Figure 1), featuring a rigorous C3-symmetric geometry that places all three guanidinyl groups toward the same side as the endo located Sb/Bi element. This preorganized cavity provides similar coordination space for both ligands given the longer Bi–C edges but more acute angles for the BiC3 pyramid.
Figure 1.

ORTEP Diagrams of TMG3trphen-Sb•3CH2Cl2 (1), TMG3trphen-Bi•3CH2Cl2 (2) drawn with 40% thermal ellipsoids. Selective interatomic distances [Å] and angles [°] for 1: Sb(1)–C(1) = 2.169(5), C(1)–Sb(1)–C(1) = 95.09(18). For 2: Bi(1)–C(1) = 2.2508(18), C(1)–Bi(1)–C(1) = 93.14(18).
Upon metalation with CuCl, both ligands extract a Cu3(μ-Cl)3 cluster in a manner similar to that previously noted in two rare cases, either with the assistance of ligand (o(iPr2P)-C6H4)3E (E = Sb, Bi)33 or with tris(2(2-pyridyl)ethyl)-phosphine.34 The resulting compounds (3, 4) feature an approximate C3-symmetric geometry and are distinguished by a crown-shaped Cu3(μ-Cl)3 unit embedded in the ligand framework. By contrast, the gas-phase structure of CuCl encompasses planar, D3h-symmetric Cu3Cl3 rings (Cu–Cu = 2.627 ± 0.012, Cu–Cl = 2.166 ± 0.008 Å, and Cu–Cl–Cu = 73.9 ± 0.6°, at 689 K).35
For compounds 3 and 4 (Figures 2 and 3), the Cu3(μ-Cl)3 cluster is supported by a Sb(III) (3) or Bi(III) (4) axial element, capping all three Cu sites (Sb–Cu3,cent = 2.187, Bi–Cu3,cent = 2.241 Å), as well as by three guanidinyl residues, each coordinated to a separate edge of the Cu3 triangle (ave. Cu–N = 2.023 ± 0.016 (3), 2.018 ± 0.003 (4) Å). The average Sb–Cu (2.657 ± 0.012 Å) and Bi–Cu (2.785 ± 0.013 Å) bond distances are shorter than those reported by Ke and Gabbaï (Sb–Cu = 2.802(2), Bi–Cu 2.934(2) Å) with (o(iPr2P)C6H4)3E (E = Sb and Bi) ligands,33 and very similar to the sum of covalent radii for Sb/Bi and Cu atoms (Sb–Cu = 2.71(6), Bi–Cu = 2.80(6) Å).36
Figure 2.
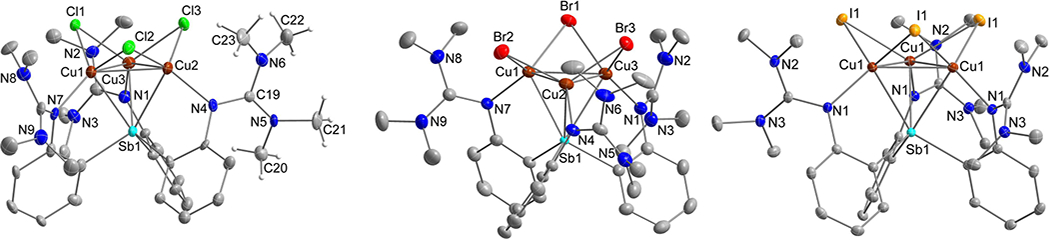
ORTEP diagrams of [(TMG3trphen-Sb)Cu3(μ-Cl)3]•4CH2Cl2 (3), [(TMG3trphen-Sb)Cu3(μ-Br)3]•4CH2Cl2 (5), [(TMG3trphen-Sb)Cu3(μ-I)3]•3CH2Cl2 (6) drawn with 40% thermal ellipsoids. Selective interatomic distances [Å] and angles [°] for 3: Sb(1)–Cu(1) = 2.652(2), Sb(1)–Cu(2) = 2.646(2), Sb(1)–Cu(3) = 2.673(2), Cu(1)–Cu(2) = 2.628(3), Cu(1)–Cu(3) = 2.594(3), Cu(2)–Cu(3) = 2.615(3), Cu(1)–Cl(1) = 2.320(4), Cu(1)–Cl(2) = 2.366(4), Cu(1)–N(7) = 2.027(11), Cu(2)–Cl(2) = 2.328(4), Cu(2)–Cl(3) = 2.337(4), Cu(2)–N(4) = 2.040(11), Cu(3)–Cl(1) = 2.342(4), Cu(3)–Cl(3) = 2.366(4), Cu(3)–N(1) = 2.001(11), Cu(2)–Cu(1)–Cu(3) = 60.11(7), Cu(1)–Cu(2)–Cu(3) = 59.29(7), Cu(1)–Cu(3)–Cu(2) = 60.60(7), Cu(1)–Cl(1)–Cu(3) = 67.60(11), Cu(1)–Cl(2)–Cu(2) = 68.09(12), Cu(2)–Cl(3)–Cu(3) = 67.57(11), Cu(1)–Sb(1)–Cu(2) = 59.48(6), Cu(1)–Sb(1)–Cu(3) = 58.29(6), Cu(2)–Sb(1)–Cu(3) = 58.91(6). For 5: Sb(1)–Cu(1) = 2.6430(5), Sb(1)–Cu(2) = 2.6510(5), Sb(1)–Cu(3) = 2.6676(5), Cu(1)–Cu(2) = 2.6203(7), Cu(1)–Cu(3) = 2.5958(7), Cu(2)–Cu(3) = 2.6012(7), Cu(1)–Br(1) = 2.4502(6), Cu(1)–Br(2) = 2.4785(6), Cu(1)–N(7) = 2.032(3), Cu(2)–Br(2) = 2.4488(6), Cu(2)–Br(3) = 2.4523(6), Cu(2)–N(4) = 2.032(3), Cu(3)–Br(1) = 2.4670(6), Cu(3)–Br(3) = 2.4719(6), Cu(3)–N(1) = 2.020(3), Cu(2)–Cu(1)–Cu(3) = 59.822(18), Cu(1)–Cu(2)–Cu(3) = 59.621(18), Cu(1)–Cu(3)–Cu(2) = 60.557(18), Cu(1)–Br(1)–Cu(3) = 63.728(18), Cu(1)–Br(2)–Cu(2) = 64.252(18), Cu(2)–Br(3)–Cu(3) = 63.773(18), Cu(1)–Sb(1)–Cu(2) = 59.335(15), Cu(1)–Sb(1)–Cu(3) = 58.522(15), Cu(2)–Sb(1)–Cu(3) = 58.558(15). For 6: Sb(1)–Cu(1) = 2.6619(5), Cu(1)–Cu(#1) = 2.6332(8), Cu(1)–I(1) = 2.6313(5), Cu(#1)–I(1) = 2.6410(5), Cu(1)–N(1) = 2.035(3), Cu(1)#1–Cu(1)–Cu(1)#2 = 60.0, Cu(1)–I(1)–Cu(1)#1 = 59.925(19), Cu(1)–Sb(1)–Cu(1)#1 = 59.289(17).
Figure 3.

ORTEP diagrams of [(TMG3trphen-Bi)Cu3(μ-Cl)3]•4CH2Cl2 (4) and [(TMG3trphen-Sb)Ag3(μ-Cl)3]•3CH2Cl2 (7) drawn with 40% thermal ellipsoids. Selective interatomic distances [Å] and angles [°] for 4: Bi(1)–Cu(1) = 2.7710(12), Bi(1)–Cu(2) = 2.7823(12), Bi(1)–Cu(3) = 2.8024(13), Cu(1)–Cu(2) = 2.8851(17), Cu(1)–Cu(3) = 2.8262(17), Cu(2)–Cu(3) = 2.8838(17), Cu(1)–Cl(1) = 2.259(3), Cu(1)–Cl(2) = 2.394(3), Cu(1)–N(7) = 2.021(7), Cu(2)–Cl(2) = 2.271(3), Cu(2)–Cl(3) = 2.339(3), Cu(2)–N(4) = 2.018(7), Cu(3)–Cl(1) = 2.360(3), Cu(3)–Cl(3) = 2.295(3), Cu(3)–N(1) = 2.015(8), Cu(2)–Cu(1)–Cu(3) = 60.64(4), Cu(1)–Cu(2)–Cu(3) = 58.67(4), Cu(1)–Cu(3)–Cu(2) = 60.69(4), Cu(1)–Cl(1)–Cu(3) = 75.42(8), Cu(1)–Cl(2)–Cu(2) = 76.36(8), Cu(2)–Cl(3)–Cu(3) = 76.95(8), Cu(1)–Bi(1)–Cu(2) = 62.60(4), Cu(1)–Bi(1)–Cu(3) = 60.94(3), Cu(2)–Bi(1)–Cu(3) = 62.18(4). For 7: Sb(1)–Ag(1) = 2.9174(6), Sb(1)–Ag(2) = 2.9505(7), Sb(1)–Ag(3) = 2.9331(7), Ag(1)–Ag(2) = 2.9256(7), Ag(1)–Ag(3) = 2.9227(7), Ag(2)–Ag(3) = 2.8822(7), Ag(1)–Cl(1) = 2.6443(18), Ag(1)–Cl(3) = 2.509(2), Ag(1)–N(1) = 2.266(6), Ag(2)–Cl(1) = 2.5078(17), Ag(2)–Cl(2) = 2.630(2), Ag(2)–N(4) = 2.274(6), Ag(3)–Cl(2) = 2.524(2), Ag(3)–Cl(3) = 2.6209(19), Ag(3)–N(7) = 2.271(5), Ag(2)–Ag(1)–Ag(3) = 59.054(17), Ag(1)–Ag(2)–Ag(3) = 60.423(18), Ag(1)–Ag(3)–Ag(2) = 60.523(18), Ag(1)–Cl(1)–Ag(2) = 69.14(4), Ag(2)–Cl(2)–Ag(3) = 67.97(5), Ag(1)–Cl(3)–Ag(3) = 69.43(5), Ag(1)–Sb(1)–Ag(2) = 59.808(16), Ag(1)–Sb(1)–Ag(3) = 59.941(17), Ag(2)–Sb(1)–Ag(3) = 58.664(17).
The presence of cuprophilic (d10–d10) interactions37 within the essentially equilateral Cu3 triangle of the Cu3(μ-Cl)3 unit is more nuanced, especially for the Bi supported compound 4, since the observed Cu–Cu distances (2.612 ± 0.014 (3), 2.865 ± 0.027 (4) Å) are either within range (3) or exceed (4) the sum of covalent radii (Cu–Cu = 2.64(6) Å)36 and may also be at the limit of the sum of the vdW radii (Cu–Cu = 2.80 Å), although the latter value may be an underestimation.38 Similar metallophilic Cu–Cu interactions were suggested and computationally justified, in the previously reported cases of [(o(iPr2P)C6H4)3ECu3(μ-Cl)3] (Cu–Cu = 2.660(2) (E = Sb), 2.853(2) (E = Bi) Å)33 and [(2(2-Py)-CH2CH2)3PCu3(μ-Cl)3] (Cu–Cu = 2.542 ± 0.049 Å).34 Single chloride bridges between pairs of Cu(I) sites (Cu–Cl = 2.343 ± 0.018 (3), 2.320 ± 0.049 (4) Å); Cu–Cl–Cu = 67.75 ± 0.24 (3), 76.24 ± 0.63 (4)°) complete the metal coordination sphere, with Cu–Cl distances that are very similar to those previously reported for the aforementioned Cu3(μ-Cl)3 units.33,34
The bromo- and iodo-bridged congeners [(TMG3trphen-Sb)Cu3(μ-X)3] (X = Br (5), I (6)) are essentially isostructural to the chloro analog 3. Compound 6 features a strict C3 axis penetrating the middle of the equilateral Cu3 triangle and the apical Sb element (Figure 2). Interestingly, metrical parameters associated with their [(L-Sb)Cu3] framework (Cu–Cu = 2.6058 ± 0.0105 (5), 2.6332(8) (6), Sb–Cu = 2.6539 ± 0.0102 (5), 2.6619(5) (6) Å) are similar to those of 3. The size of the halide is only reflected in the longer and slightly asymmetric halide bridges (Cu–Cl = 2.343 ± 0.018 (3), Cu–Br = 2.4615 ± 0.0116 (5), Cu–I = 2.6362 ± 0.0049 (6) Å) and the corresponding acute Cu–X–Cu angles (67.75 ± 0.24 (3), 63.918 ± 0.237 (5), 59.925(19) (6)°). The related Cu3X3 cluster embedded in tris(2(2-pyridyl)ethyl)phosphine (L-P) exhibits a [(L-P)Cu3] framework that is more tightly coordinated (Cu–Cu ranges narrowly from 2.528 to 2.542 Å) but is highly asymmetric in the cases of X = Cl and I, since one Cu–P bond is longer than the other two by a significant margin (0.40 (Cl), 0.37 (I) Å).34
The Ag3(μ-Cl)3 unit (7, Figure 3) is embedded in the cavity of ligand TMG3trphen-Sb in a similar fashion to that observed for Cu3(μ-Cl)3, as evidenced by the pseudo C3-symmetric attachment of the cluster to the tripodal Sb ligand via the three guanidinyl residues (ave. Ag–N = 2.270 ± 0.003 Å) and the axial Sb (ave. Sb–Ag = 2.934 ± 0.014 Å). Within the Ag3(μ-Cl)3 cluster, the average Ag–Ag (2.910 ± 0.020 Å) and Ag–Cl (2.573 ± 0.060 Å) bond distances are comparable to the sum of the corresponding covalent radii (Ag–Ag = 2.90(7), Ag–Cl = 2.47(6) Å)36 and shorter than the values reported for two independent molecules in the unit cell of [(o(iPr2P)-C6H4)3SbAg3(μ-Cl)3] (Ag–Ag = 3.0288(8), 3.1559(8); Ag–Cl = 2.5791(14), 2.6443(16) Å).33 One distinguishing feature of the Ag3(μ-Cl)3 cluster is the somewhat asymmetric chloride bridging (Δ(Ag–Cl) = 0.118 ± 0.013 Å) presumably due to distortion arising from the accommodation of a larger cluster in the ligand cavity.
The structures of the halide-free compounds [(TMG3trphen-E)2Cu2](X)2•2 solv (E = Sb (8a, 8b), Bi (9); solv = THF (8a), MeCN (8b), Et2O (9); X = PF6 (8a, 9), B(C6F5)4 (8b)) are all very similar (Figures 4 and S1), although significant differences in metrical parameters are observed. They all possess a Cu2E2 parallelogram with a short and a long Cu–E bond and exhibit a local inversion center applicable to the entire compound. Each stibine (1) or bismuthine (2) ligand is coordinated to both Cu sites via two separate TMG-substituted arms, whereas the third TMG residue remains noncoordinated, at a significant distance (>4 Å) from the closest Cu. The ligand moieties that bridge the shorter Cu–Sb bonds are oriented axially with respect to the Cu2E2 ring, whereas those spanning the longer Cu–Sb bonds are located equatorially. Since crystal data for 8a were of low quality (derived from a twinned specimen), we rely on the solid-state structure of 8b for further discussion of metrical parameters. Each symmetry-related Cu(I) site of 8b is coordinated by two Sb(III) elements (Cu–Sb = 2.5925(4), 2.8121(4) Å; Sb–Cu–Sb = 112.319(13)°) and two NTMG residues (Cu–N = 2.060(2), 2.022(2) Å; N–Cu–N = 122.35(9)°). Houser’s four-coordinate geometry index, τ4 (0.73),39 places the coordination of the Cu(I) site between trigonal pyramidal and seesaw. The Cu···Cu interatomic distance (3.0152(7) Å) is rather long to denote any significant cuprophilic interaction. The bismuth analog 9 is geometrically equivalent to 8a/8b yet the Cu2Bi2 parallelogram is characterized by metrical parameters (Cu–Bi = 2.9025(8), 3.3075(8) Å; Bi–Cu–Bi = 83.41(2)°) that include a bonafide covalent Cu–Bi bond and a longer Cu···Bi interaction that at best denotes a weak contact at the van der Waals sum of radii limit (3.47 Å).38,40 The Cu···Cu interatomic bond distance (4.644 Å) also precludes any cuprophilic contacts. Therefore, the Cu(I) sites of 9 are essentially three-coordinate with shorter Cu–N bonds (both 1.896(5) Å) and more obtuse N–Cu–N angle (155.0(2)°) than those observed with the stibine analogs (8a, 8b).
Figure 4.

ORTEP diagrams of [(TMG3trphen-Sb)2Cu2][B(C6F5)4]2•2MeCN (8b) and [(TMG3trphen-Bi)2Cu2](PF6)2•2Et2O (9) drawn with 40% thermal ellipsoids. Selective interatomic distances [Å] and angles [°] for 8b: Sb(1)–Cu(1) = 2.5925(4), Sb(1)–Cu(1)#1 = 2.8121(4), Cu(1)–Cu(1)#1 = 3.0152(7), Cu(1)–N(4) = 2.060(2), Cu(1)–N(7) = 2.022(2), Cu(1)–Sb(1)–Cu(1)#1 = 67.681(13), N(4)–Cu(1)–Sb(1) = 88.00(6), N(4)–Cu(1)–Sb(1)#1 = 126.81(6), N(4)–Cu(1)–N(7) = 122.35(9), N(7)–Cu(1)–Sb(1) = 129.86(6), N(7)–Cu(1)–Sb(1)#1 = 82.21(6). For 9: Bi(1)–Cu(1) = 2.9025(8), Bi(1)–Cu(1)#1 = 3.3075(8), Cu(1)–N(1) = 1.896(5), Cu(1)–N(4) = 1.896(5), Cu(1)–Bi(1)–Cu(1)#1 = 96.59(2), N(1)–Cu(1)–Bi(1) = 88.26(14), N(1)–Cu(1)–Bi(1)#1 = 120.96(16), N(1)–Cu(1)–N(4) = 155.0(2), N(4)–Cu(1)–Bi(1) = 112.73(15), N(4)–Cu(1)–Bi(1)#1 = 76.64(15).
A version of the coordination mode noted above was most recently unraveled in a comprehensive publication by Wright and co-workers41 for [{(2-Me-8-quinolyl)3Sb}2Cu2](PF6)2. In this case, the compound lacks an inversion center, and the Cu2Sb2 ring is essentially rhombic (Cu–Sb = 2.5451(19)–2.5884(19) Å) bridged by an ax/ax/eq/eq suite of ligand moieties rather than the alternating ax/eq/ax/eq motif observed in our work. Interestingly, the corresponding bismuth ligand (2-Me-8-quinolyl)3Bi did not provide compounds with Cu–Bi bonds.41
The structure of compound [(TMG3H3trphen-Sb)Cu(OTf)3](OTf) (10) (Figure S2) demonstrates an unusual decomplexation of the TMG residues of ligand 1 due to protonation (Cu–N interatomic distances vary from 3.561 to 3.902 Å), accompanied by retention of a strong Sb(III)–Cu(I) bond (2.4259(9) Å).42 The pseudotetrahedral ligand field around the Cu(I) site also includes three coordinated triflates (Cu–O = 2.090 ± 0.012 Å) with less obtuse O–Cu–O (99.30 ± 2.60°) than Sb–Cu–O (118.37 ± 2.83°) angles, reflecting the repulsive effect of the Cu–Sb bond and the bulk of the protonated ligand.
Finally, compound [(TMG3trphen-Sb)Cu3(μ3-Cl)3Cu]2[B(C6F5)4]2 (11) (Figure 5) relates two [(TMG3trphen-Sb)-Cu3Cl3] units via a central chloro-bridged Cu2 element by means of inversion. This unique structure demonstrates metrical parameters for the essentially equilateral CuI3 triangle and its coordination to the antimony ligand that are very similar to those demonstrated by 3, save for the shorter Cu–N bond distances (Cu–N = 1.957 ± 0.044 Å) due to the weaker Cu–Cl bonds in 10. The μ2-Cl bridges in 3 become triply bridged in 10 by incorporating an additional CuI site from the central Cu2 element. As a result, the Cu3Cl3 fragment in 10 exhibits longer Cu–Cl bond distances (2.413 ± 0.055 Å) and more acute Cu–Cl–Cu angles (65.27 ± 0.52°). Each Cu site of the Cu2 dimer is coordinated by three chlorides in a trigonal planar arrangement (Cu–Cl = 2.272 ± 0.080 Å, Cl–Cu–Cl = 118.7 ± 9.9°) and also features a cuprophilic contact (Cu–Cu = 2.772(7) Å).
Figure 5.

ORTEP diagram of [(TMG3trphen-Sb)Cu3(μ3-Cl)3Cu]2[B(C6F5)4]2•10CH2Cl2 (11) drawn with 40% thermal ellipsoids. Selective interatomic distances [Å] and angles [°]: Sb(1)–Cu(1) = 2.661(3), Sb(1)–Cu(2) = 2.649(3), Sb(1)–Cu(3) = 2.645(3), Cu(1)–Cu(2) = 2.648(4), Cu(1)–Cu(3) = 2.601(4), Cu(2)–Cu(3) = 2.564(4), Cu(4)–Cu(4)#1 = 2.772(7), Cu(1)–Cl(2) = 2.328(5), Cu(1)–Cl(3) = 2.465(6), Cu(1)–N(1) = 2.004(12), Cu(2)–Cl(1) = 2.386(5), Cu(2)–Cl(3) = 2.398(5), Cu(2)–N(4) = 1.968(14), Cu(3)–Cl(1) = 2.404(5), Cu(3)–Cl(2) = 2.497(6), Cu(3)–N(7) = 1.899(14), Cu(4)–Cl(1) = 2.375(7), Cu(4)–Cl(2) = 2.261(6), Cu(4)–Cl(3) = 2.180(6), Cu(2)–Cu(1)–Cu(3) = 58.48(11), Cu(1)–Cu(2)–Cu(3) = 59.83(10), Cu(1)–Cu(3)–Cu(2) = 61.69(9), Cu(2)–Cl(1)–Cu(3) = 64.72(14), Cu(2)–Cl(1)–Cu(4) = 103.8(2), Cu(3)–Cl(1)–Cu(4) = 74.62(19), Cu(1)–Cl(2)–Cu(3) = 65.12(15), Cu(1)–Cl(2)–Cu(4) = 101.9(2), Cu(3)–Cl(2)–Cu(4) = 74.8(2), Cu(1)–Cl(3)–Cu(2) = 65.97(14), Cu(1)–Cl(3)–Cu(4)#1 = 101.9(2), Cu(2)–Cl(3)–Cu(4)#1 = 97.9(2), Cu(4)#1–Cu(4)–Cl(1) = 82.2(2), Cu(4)#1–Cu(4)–Cl(2) = 98.8(2), Cu(4)#1–Cu(4)–Cl(3) = 105.0(2).
Summaries of crystallographic data for compounds 1–11 are collected in Tables S1–S4.
Solution Behavior.
1H NMR data for compounds [(TMG3trphen-Sb)Cu3(μ-X)3] (X = Cl (3), Br (5), I (6)) in CD2Cl2 at room temperature (298 K) exhibit broad peaks attributed to the four methyl groups of the TMG residues (fully resolved only for X = I), extending from δ 1.5 to 3.5 ppm for all compounds (Figure S3). The degree of resolution of the four distinct methyl peaks (all broad at 298 K) is enhanced with increasing size of the halide presumably due to restricted rotation of the TMG branches. Low-temperature 1H NMR data (243–298 K, Figure S4) indicate that all four methyl groups are progressively resolved with decreasing temperature, to afford four sharp peaks at −30 °C (for 3: δ (ppm, CD2Cl2, −30 °C) = 3.100 (H(23)), 2.758 (H(21/22)), 2.582 (H(21/22), 1.672 (H(20), see Figure 2 for atom labeling). Peak assignments were facilitated by computational (B3LYP/6–311+G(2d,p)) single point NMR property determination at the B3LYP/6–31G(d)-optimized geometry of 3, which indicates that the most upfield peak is due to protons residing in the shielded region of the phenylene rings, whereas the most downfield peak arises from protons pointing endo with respect to the ligand cavity. With respect to [(TMG3trphen-Bi)Cu3(μ-Cl)3] (4), a featureless broad band at room temperature progressively resolves to four distinct Me peaks at −30 °C in CD2Cl2. A similar broad band, assigned to TMG methyl groups, is also observed for [(TMG3trphen-Sb)Ag3(μ-Cl)3] (7). Apparently, for this more sizable ligand cavity, the rotational restrictions for the TMG arms are more relaxed than those applying to compounds 1–3 at room temperature. Notably, the 1H NMR spectra of the [(TMG3trphen-Sb)2Cu2]2+ compounds (8a, 8b) exhibit only a single peak for all Me groups of the three TMG arms (as well as equivalent aryl groups) in CD3CN at 298 K. On the other hand, the corresponding [(TMG3trphen-Bi)2Cu2]2+ (9) compound distinguishes the TMG arms (and the related aryl groups) in a 2:1 ratio, most likely reflecting the coordinated (two arms) and noncoordinated (one arm) TMG moieties observed in the solid-state structure of 9. Fast exchange between coordinated and noncoordinated TMG groups may account for the single TMG peak observed with the Sb analogs (8a, 8b), although dissociation to monomers with possible MeCN coordination cannot be excluded. The protonation of the TMG arms in compound 10 can easily be detected by 1H NMR. Finally, compound 11 demonstrates a 1H NMR spectrum not unlike that of 3 at 298 K, featuring broad overlapping peaks for the Me groups of TMG.
The metal coordination of the strongly superbasic TMG arms results in charge delocalization over the CN3 unit, as demonstrated by the essentially equivalent C–N bond distances (SCXRD data) and downfield shift of the central C peak (>160 ppm, 13C NMR data).
Computational Studies.
The structures of 3 and 4 were optimized and found to have an overall generally good agreement with the experimental crystallographic data. EDA calculations43 were conducted to decompose the interaction energy and give a more detailed view of the bonding for these complexes (Table 1). The results show that complexes 3 and 4 have similar bonding energetic patterns, as evidenced by the same percent contribution of attraction interactions and energy values. The leading attractive interaction is electrostatic interaction between two charge-neutral moieties, mainly arising from the Cu–N coordination bonds between two fragments. The steric effect of Sb (3) and Bi (4) to the Cu3Cl3 fragment accounts for the Pauli repulsion seen in both complexes, whereas the transfer of electrons synergistically increases their electrostatic attraction. Also, the Sb complex 3 has a slightly larger electrostatic (−287.8 kcal/mol) and orbital interaction energies (−141.7 kcal/mol) vs those for Bi complex 4 (−275.8 kcal/mol and −133.5 kcal/mol, respectively). The higher orbital interaction energy in the Sb complex 3 vs the Bi analogue 4, an attractive force, is offset by the correspondingly higher Pauli repulsion energy, resulting in the Bi complex 4 possessing a stronger overall interaction energy than that in the Sb analogue 3.
Table 1.
Energy Decomposition Analysis Results of Compounds 3 and 4a
| compound | ΔEelstat | ΔEPauli | ΔEOrbInt | ΔEdisp | ΔEint |
|---|---|---|---|---|---|
| 3 | −287.8 (63%) | 344.5 | −141.7 (31%) | −24.4 (6%) | − 109.4 |
| 4 | −275.8 (63%) | 323.5 | −133.5 (31%) | −25.4 (6%) | −111.2 |
Unit: kcal/mol, values in parentheses are the energy percentage to the total attractive interaction. Total interaction energy (ΔEint) is decomposed into electrostatic interaction (ΔEalstat), Pauli repulsion (ΔEPauli), orbital interaction (ΔEOrbInt), and dispersion energy (ΔEdisp).
As the Cu–N coordination bonds are broken during the fragmentation procedure for the EDA analysis, the total ΔEint does not directly reflect the interaction strengths between the Cu3Cl3 and ligand with Sb/Bi moieties. To overcome this limitation, we further divide ΔEOrbInt into pairwise interactions to reveal the detailed bonding pictures between fragments under the ETS-NOCV theoretical framework.43 The broken-down ΔEOrbInt is denoted as ΔEOrbInt,i (i = integer) in the energy strength order. NOCV results are consistent with Sb/Bi → Cu3 dative bonding in both complexes 3 and 4. The NOCV pairwise orbital deformation density maps are illustrated in Figure 6; the electron flows from the white region to the brown region comprising the bonding formation. The largest contributing NOCV pairwise orbitals (ΔEOrbInt,1) of 3 and 4 are the bonding interactions between Sb or Bi, respectively, and the Cu3Cl3 moieties, with interaction energies of −32.6 kcal/mol and −23.6 kcal/mol, respectively. The electrons that contribute to the bonding behavior originate from the filled Sb 5s or Bi 6s orbitals to the admixed Cu 4s0+x/3d10−x partially unoccupied/partially occupied orbitals, confirming the directional Sb/Bi→Cu3 dative bonding character (Figure 6a/c). The ΔEOrbInt,2–4 values for 3 and 4 clearly suggest Cu–N coordination bonds (Figure 6b/d), with comparable attractive bond strengths of −40.3 and −43.6 kcal/mol, respectively. The rest of the NOCV pairwise orbitals possess negligible interaction energies (<4 kcal/mol in total) suggest that the major bonding in these species is due to the Cu–N coordination, followed by Sb/Bi→Cu3 donor contributions. Any residual backdonation (Sb/Bi→Cu3) cannot be excluded at the present time, but this contribution is likely to be very small.
Figure 6.

NOCV deformation density maps of (a/c) ΔEOrbInt,1 and (b/d) ΔEOrbInt,2–4 of 3 and 4, respectively. Isovalue: 0.002 au 3 (for (a) ΔEOrbInt,1 = −32.6 kcal/mol; for (b) ΔEOrbInt,2–4 = −40.3 kcal/mol), 4 (for (c) ΔEOrbInt,1 = −23.6 kcal/mol; for (d) ΔEOrbInt,2–4 = −43.6 kcal/mol).
The independent gradient model based on the Hirshfeld partition (IGMH)44 analyzes noncovalent interactions between Sb/Bi and Cu3Cl3 moieties (Figure 7). The results correspond to the above NOCV results in that a stronger interfragment bonding character exists in 3 and 4. The plots confirm the attractive interaction between Sb/Bi atoms and Cu atoms, as illustrated by the blue region in Figure 7a–d. Comparing the IGMH results of 3 and 4, the blue color between Sb and Cu3 is deeper than that between Bi and Cu3, which suggests a stronger interaction in 3, in agreement with the larger interaction energy between Sb → Cu3 as indicated by the NOCV results (Figure 6a/c). These calculations are consistent with a decrease in electron-donating ability of the pnictine lone pair from Sb to Bi, concomitant with an increase in s-character. Also, the calculated average bond distance of 2.924 Å for Bi–Cu as compared to the average bond distance of 2.746 Å for Sb–Cu (2.68–2.84 Å) suggests that a slightly shorter bond contributes to the Sb → Cu3 interaction, given the rather similar van der Waals radii of Sb (2.06 Å) and Bi (2.07 Å).40
Figure 7.

Independent gradient model based on Hirshfeld partition (IGMH) analysis results of 3 and 4 in (a/c) top view and (b/d) side view, respectively. The value of the surface is set as 0.01 au.
Catalytic and Mechanistic Nitrene-Transfer Studies.
Aziridination of para-Substituted Styrenes.
An initial evaluation of nitrene addition to olefins was conducted with the assistance of imidoiodinane PhINTs (1 equiv, Ts = tosyl) and a panel of para-substituted styrenes (8 equiv) in the presence of different catalytic systems (5 mol % with respect to Cu) at 30 °C (drybox temperature) over 12 h (Table 2). The choice of the solvent (MeCN) was suggested by an initial screening of various chlorinated and nonchlorinated solvents (MeCN, benzene, CH2Cl2, 1,2-dichloroethane, and chlorobenzene) in catalytic reactions with styrene/PhINTs mediated by [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3). Acetonitrile has an edge over all other solvents, even if the catalyst is more soluble in CH2Cl2. Molecular sieves (5 Å) are also needed for obtaining good yields, thus denoting sensitivity to any adventitious water.
Table 2.
Yields of Aziridination Products of para-Substituted Styrenes Catalyzed by [(TMG3trphen-E)Cu3(μ-Cl)3] (E = Sb (3), Bi (4))a
| Entry No. | Substrate | Products | Yield (%) [(L-Sb)Cu3Cl3](3) | Yield (%) 3/KPFe (3 equiv.) | Yield (%) [(L-Bi)Cu3Cl3] (4) | |
|---|---|---|---|---|---|---|
|
| ||||||
| 1. | R = H | 92 | 80 | 85 | ||
| 2. | R = Me | 73 | 77 | 80 | ||
| 3. | R = tBu | 77 | 63 | 82 | ||
| 4. | R = OMe | 28 | polymer | polymer | ||
| 5. |

|
|
R = OtBu | 53 | polymer | polymer |
| 6. | R = F | 77 | 74 | 83 | ||
| 7. | R = Cl | 88 | 79 | 89 | ||
| 8. | R = CF3 | 87 | 96 | 82 | ||
| 9. | R=NO2 | 83 | 87 | 73 | ||
Conditions: catalyst, 0.0125 mmol with respect to Cu (5 mol %); PhINTs, 0.25 mmol; olefin, 2.0 mmol; KPF6, 0.0125 mmol; MS 5 Å, 20 mg; solvent (MeCN) 0.250 g; 30 °C; 12 h.
Gratifyingly, both catalysts 3 and 4 provide good to excellent yields of the corresponding aziridines as evaluated by 1H NMR spectra of the purified products (with respect to an internal standard). Both 3 (87%) and 4 (62%) can be recovered at the end of the reaction, albeit to a much lower extent for 4, potentially reflecting the fragility of the Bi–C bond.45
To evaluate the effect of partial removal of chlorides as suggested by the synthesis of compound 11 (see above), KPF6 (3 equiv) was added to the catalytic mixture containing [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3). Yields of aziridines (Table 2) are not unlike those obtained in the absence of KPF6, save for the most electron-withdrawing styrenes (CF3, NO2), for which unusually high yields are observed. Interestingly, SbAg3(μ-Cl)3 reagent 7 is totally unproductive.
Aziridination of Other Alkenes.
The more robust catalyst 3 was further employed in the aziridination of a wider panel of aromatic and aliphatic olefins under the conditions noted above (Table 3). In the presence of ortho styrene substitution (entry 1), significantly lower yields are obtained, due to steric and electronic reasons (orthogonal orientation of the aromatic/olefinic planes).46 Benzylic amination was not observed in this case. Nitrene transfer to α-substituted styrenes (Me, Ph; entries 2, 3) is also sterically hampered, especially for the α-Ph congener. The α-methylstyrene affords substantial allylic amination in almost 1:1 ratio with respect to aziridination (entry 2). Surprisingly, the major product for α-phenylstyrene is a hydroxyamine rather than the more common enamine (entry 3), even under conditions that are purported to exclude water. In this case, aziridine is known to undergo facile ring opening (and rearrangement).47 The corresponding cis- and trans-β-substituted styrenes (Me, Ph; entries 4–7) are less hampered by sterics and afford good yields of the corresponding cis- and trans-aziridines. A minor amount of allylic amination is only observed for the Me-substituted analog. On the other hand, the retention of stereochemistry for aziridination of the mechanistically diagnostic cis-β-R-styrenes is rather modest (cis:trans = 1:1.3, R = Me; 1.8:1, R = Ph). Amination of allylic/benzylic C–H sites is very competitive with styrenyl aziridinations (entries 8, 9), provided that acyclic trans substrates are involved (entry 8) that usually restrict aziridination yields. Allylic aminations are also dominant with cycloalkenes (entries 10, 11), although the more electron-rich cyclooctene (entry 12) favors aziridination by a significant margin since H atom abstraction from the allylic C–H bonds is associated with large energy barriers, due to modest σC–H/πC=C orbital overlap.48 Other aliphatic alkenes (entries 13–15) exhibit significantly lower yields than those encountered with aromatic congeners.
Table 3.
Yields of Aziridination/Amination Products of Various Olefins Catalyzed by [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3)a
| Entry No. | Substrate | Products | Yields (%) |
|---|---|---|---|
|
| |||
| 1. |

|

|
22 |
| 2. |

|
|
24, 23 |
| 3. |

|
|
tr, 2, 17 |
| 4. |

|
|
28, 36, 3 |
| 5. |

|
|
49, 2, 4 |
| 6. |

|
|
30, 17 |
| 7. |

|

|
45 |
| 8. |

|
|
25, 37 |
| 9. |

|
|
48, nd |
| 10. |

|
|
4, 14 |
| 11. | 2, 42 | ||
| 12. | 51, nd | ||
| 13. |

|
|
8, 5 |
| 14. |

|
|
19, 12 |
| 15. |
|
|
28 |
Conditions: 3, 0.0125 mmol with respect to Cu (5 mol %); PhINTs, 0.25 mmol; olefin, 2.0 mmol; MS 5 Å, 20 mg; solvent (MeCN) 0.250 g; 30 °C; 12 h.
Amination of Benzylic Substrates.
The more challenging amination of C–H-bond containing substrates was subsequently examined. The benchmark substrate ethylbenzene (1 equiv) was initially explored with PhINTs (2 equiv) in the presence of catalytic amounts of [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3) (5 mol % with respect to Cu) in several solvent matrices. Under all circumstances, benzylic amination yields did not exceed 10%. The more electrophilic nitrene source PhINTces (Tces = 2,2,2-trichloroethoxysulfonyl) provided significantly better yields and was further applied in the amination of ethylbenzene. An initial screening of chlorinated and nonchlorinated solvents indicated that PhCl and PhCF3 are the most productive solvents, especially if a small amount of HFIP (10%) is added. Presynthesized PhINTces49 is significantly more soluble than PhINTs but is unstable in halogenated solvents. The selection of PhCF3/HFIP (10:1 v/v) for further experimentation was indicated by the superior stability it offered to PhINTces at 30 °C over a period of 24 h in the absence of light, according to 1H NMR experiments.
The amination of a panel of benzylic substrates (Table 4) was pursued most effectively with a 2:1 ratio of PhINTces/substrate in PhCF3/HFIP (10:1 v/v) at 30 °C, over 24 h. Several catalysts (5 mol % with respect to Cu) were explored, namely, [(TMG3trphen-Sb)Cu3(μ-X)3] (X = Cl (3), Br (5), I (6)), [(TMG3trphen-Bi)Cu3(μ-Cl)3] (4), and [(TMG3trphen-Sb)2Cu2](PF6)2 (8a). As a general characteristic, all catalysts afford moderate to good amination yields specifically for secbenzylic sites. Prim-benzylic C–H bonds are unaffected (entry 11), as are C–H bonds adjacent to O/N atoms and tert/sec/prim C–H bonds of unactivated alkanes and cycloalkanes (yields ≤10%, not shown). Notably, amination of ethylbenzene does not occur in the presence of catalytic amounts (5 mol %) of TMG3trphen-Sb (1) or CuCl alone.
Table 4.
Yields of Benzylic Amination of Various Substrates Mediated by [(TMG3trphen-Sb)Cu3(μ-X)3] (X = Cl (3) Br (5), I (6)), [(TMG3trphen-Bi)Cu3(μ-Cl)3] (4), and [(TMG3trphen-Sb)2Cu2](PF6)2 (8a)a
| Entry No. | Substrate | Product |
Yield (%) [(L-Sb)Cu3X3] |
Yield (%) [(L-Bi)Cu3Cl3] (4) |
Yield (%) [(L-Sb)2Cu2]2+ (8a) |
||
|---|---|---|---|---|---|---|---|
| X = Cl (3) | X = Br (5) | X = I (6) | |||||
|
| |||||||
| 1. |
|

|
50 | 35 | 49 | 49 | 46 |
| 2. |

|

|
54 | 41 | 42 | 41 | 32 |
| 3. |

|

|
60 | 29 | 56 | 50 | 41 |
| 4. |

|

|
48 | 44 | 42 | 50 | 49 |
| 5. |

|

|
49 | 67 | 63 | 43 | 54 |
| 6. |

|

|
61 | 39 | 37 | 57 | 45 |
| 7. |

|

|
52 | 46 | 31 | 51 | 41 |
| 8. |

|
|
13 | 18 | 10 | 20 | 13 |
| 9. |

|

|
31 | 12 | 17 | 26 | 9 |
| 10. |

|

|
12 | 31 | 34 | 20 | 26 |
| 11. |
|

|
5 | 7 | 2 | 6 | 8 |
| 12. |

|

|
23 | 34 | 36 | 31 | 20 |
| 13. |

|

|
26 | 50 | 36 | 29 | 26 |
| 14. |

|

|
30 | 22 | 25 | 24 | 23 |
| 15. |

|

|
7 | 9 | 10 | 13 | 25 |
| 16. |
|

|
26 | 2 | 5 | 25 | 25 |
Conditions: catalyst, 0.0125 mmol with respect to Cu (5 mol %); PhINTces, 0.50 mmol; benzylic substrate, 0.25 mmol; MS 5 Å, 20 mg; solvent (PhCF3/HFIP 10:1 v/v), 0.300 g (0.500 g for 8a); 30 °C; 24 h.
A cursory look at the yields afforded for the amination of all benzylic sites shown in Table 4 by catalysts 3 and 4 indicates that no significant variations exist in the operation of these axially supported Sb and Bi reagents. This is likely consistent with nitrene docking in the Cu3 plane (cis with respect to the E–Cu3,cent axis) rather than on the encumbered side of the halide crown. Inspection of the solid-state structures of 3 and 4 provides evidence of ample coordination space externally to the Cu3 ring (in-plane). Additionally, the difference between Sb(III) and Bi(III) in terms of electron donicity to the Cu3 cluster may be insufficient to significantly alter the metal-nitrene electrophilicity.
The amination yields for the functionalization of the benzylic substrates by catalysts [(TMG3trphen-Sb)Cu3(μ-X)3] (X = Cl (3), Br (5), I (6)) vary more appreciably (Table 4). For the panel of the para-substituted ethylbenzenes (entries 1–9), each catalyst provides good yields not only with substrates bearing ED substituents (entries 1–3) but also with para-X (X = F, Cl, Br, I) substituted congeners (entries 4–7), a trend that usually denotes significant benzyl radical stabilization via resonance effects. Only strongly EW substituents (entries 8, 9) are associated with low yields, as anticipated for electrophilic metal nitrene-driven reactions. The yields for the amination of sec-benzylic sites (entry 10) are modest most likely due to steric encumbrance, and those for prim-benzylic sites (entry 11) are below ≤10% given the higher aliphatic C–H bond energy of toluene (90 kcal/mol50). Steric hampering is also reflected in the moderate yields for the benzylic amination of 1,2,3,4-tetrahydronaphthalene and derivatives (entries 12, 13). Competitions between benzylic and tert-C–H sites (entries 14, 15) singularly result in benzylic aminations with yields increasing with increasing distance between the two sites. Finally, acetate not only deactivates adjacent C–H sites (entry 16), as anticipated, but curiously seems to interfere with the operation of the Br- and I-possessing catalysts 5 and 6. Lateral comparisons between catalysts 3, 5, and 6 do not reveal any particular trends possibly because the effect of the halide is likely to be multivariate in regulating (i) the electrophilicity of the putative metal nitrene; (ii) the approach of the substrate to the active catalytic site; and (iii) the stability offered to any substrate-centered carboradical (or carbocation) upon H atom abstraction. Finally, the halide-free catalyst [(TMG3trphen-Sb)2Cu2](PF6)2 (8a) affords similar yields and trends to those encountered with the aforementioned reagents but also shows signs of more tolerance to steric encumbrance and/or electron-withdrawing functionalities (entries 14–16).
Direct comparisons to the halide-rich reagents 3–6 cannot be made at the present time given the structural uniqueness of catalyst 8a. On average, the previously explored catalyst [(TMG3trphen)Cu](PF6),27 featuring Namine apical coordination, is more active than the present reagents, but this may be due to its mononuclear character. Incidentally, an interesting mechanistic distinction between Cu(I) catalysts that carry halide ligands and others that are halide-free, has been recently reported by Peréz and co-workers.51
Hammett Plots.
To start placing these reagents in a mechanistic context, Hammett plots were constructed for the competitive amination (PhINTces) of ethylbenzene versus seven para-X-ethylbenzenes (X = MeO, Me, F, Br, I, CF3, and NO2) as mediated by catalyst 3 (5 mol % with respect to Cu) in PhCF3/HFIP (10:1 v/v) at 30 °C (5 h). Reasonably, good linear free-energy correlations of log (kX/kH) versus the polar substituent parameter σp (ρp = −1.70, R2 = 0.95) and, even better, versus the resonance-sensitive σ + (ρ+ = −1.22, R2 = 0.98) can be obtained (Figure 8 and Table S5). The negative ρp and ρ+ coefficients indicate that significant positive charge develops en route to the transition state, presumably at the benzylic carbon, by means of an incipient hydrogen-atom abstraction by an electrophilic oxidant. These sizable ρ values are more consistent with stepwise benzylic C–H functionalization ([Ru2(hp)4Cl], ρ+ = −0.90; [Ru2(esp)2SbF6], ρ+ = −1.49)52 by comparison to the more modest values found in Rh amination chemistry (−0.47,17g −0.55,17b −0.66,53 −0.73,17a −0.9017f); the latter is characterized by a concerted asynchronous C–H insertion pathway.
Figure 8.

Linear free energy correlation of log(kX/kH) as a function of σp (left) (ρp = −1.70, R2 = 0.95) and σ+ (right) (ρ+ = −1.22, R2 = 0.98) for the competitive amination of para-substituted ethylbenzenes versus ethylbenzene catalyzed by [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3).
CONCLUSIONS
The following are the major findings and insights of the present study:
The synthesized tripodal ligands TMG3trphen-E (E = Sb (1) and Bi (2)) generate extended cavities that permit capturing of M3(μ-X)3 fragments (M = Cu, Ag; X = Cl, Br, I) extracted from anhydrous MX precursors. Single-crystal crystallographic analysis indicates that the crown-shaped M3(μ-X)3 units are supported by axial Sb(III) and Bi(III) elements, triply bridging the M3 triangle, and equatorial N-donor residues provided by the superbasic TMG ligand arms. 1H NMR data suggest that the rotation of the CN3 branches of TMG is partially restricted at ambient temperatures to various degrees, commensurate with the bulk of the halide, with resolution of all four methyl groups emerging at low temperatures.
Although abstraction of the halides in select cases can best be achieved by means of TlPF6, the most convenient path for obtaining halide-free copper(I) compounds with ligands 1 and 2 involves the use of [Cu(NCMe)4](PF6) or [Cu(NCMe)2][B(C6F5)4]. The resulting complexes [(TMG3trphen-E)2Cu2]2+ (E = Sb and Bi) are dimeric in the solid state and possess an asymmetric Cu2(μ-E)2 core that is retained in solution for E = Bi and tentatively for E = Sb, according to 1H NMR data. Other efforts to dechlorinate [(TMG3trphen-Sb)Cu3(μ-Cl)3] by means of AgPF6 and AgOTf lead to ligand transmetalation and protonation, respectively. Partial dechlorination and cluster rearrangement is achieved with KB(C6F5)4.
Energy decomposition analysis (EDA) studies performed on [(TMG3trphen-E)Cu3(μ-Cl)3] (E = Sb (3) and Bi (4)) indicate that among the attractive interactions, the electrostatic component contributions (63%) far exceed those associated with orbital interactions (31%) and dispersion energy (6%) for both compounds. The natural orbitals for chemical valence (NOCV) analysis, further supported by the independent gradient model based on Hirshfeld partition (IGMH) method, suggest that the dominant orbital interaction is that between the Cu(I) sites and NTMG residues, followed by the Sb/Bi → Cu3 donor interaction between the Sb 5s or Bi 6s orbitals and admixed Cu 4s0+x/3d10−x orbitals. The attractive Sb → Cu3 interaction is more pronounced than that of Bi, given the increased s-character of the 6s lone pair. Little, if any, π-backdonation can be extracted from the analysis, suggesting that 1 and 2 are largely L-type ligands.54
An initial evaluation of [(TMG3trphen-E)Cu3(μ-Cl)3] (E = Sb (3), Bi (4)) as nitrene-transfer agents in the aziridination of various p-substituted styrenes by PhINTs afforded very good to excellent product yields, although catalyst 4 showed signs of fragility. Further evaluation of 3 as the catalyst for the aziridination of other aromatic and aliphatic alkenes provided good to modest yields and selectivities, respectively, in response to steric (aromatic alkenes) and electronic (aliphatic alkenes) challenges.
The amination of C–H bonds is best accomplished by the more electrophilic PhINTces in PhCF3/HFIP (10:1 v/v). An extensive study of catalysts [(TMG3trphen-E)Cu3(μ-X)3] (E = Sb, X = Cl (3) Br (5), I (6)); E = Bi, X = Cl (4) and [(TMG3trphen-Sb)2Cu2](PF6)2 (8a) demonstrates good yields, almost exclusively for the amination of electron-rich and unencumbered secbenzylic-C–H bonds (mostly para-substituted ethyl-benzenes). Although no discernible patterns of reactivity emerge that can be correlated with catalyst characteristics, reagents 3 and 8a are more consistent in their behavior. Their activity is still inferior to the previously explored [(TMG3trphen)Cu](PF6) catalyst27 presumably because the latter catalyst is strictly mononuclear. Hammett plots for the competitive p-X-ethylbenzene/ethylbenzene amination (PhINTces) mediated by 3, provide evidence that a stepwise C–H activation takes place by an electrophilic oxidant, featuring an intermediate benzylic carboradical.
Future studies will address more precisely the interaction between these reagents and the N-donor sources, to explore the location of nitrene docking and further establish the mode of operation of the resulting active oxidant responsible for C–H aminations. In addition, catalyst modifications that are purported to incorporate halide substituents in the ligand framework will be pursued, given the known role of halide substituents in Z-type ligands.29,55
EXPERIMENTAL SECTION
Safety Warning.
Antimony and thallium compounds can be highly toxic and should be handled and disposed carefully.
Synthesis and Characterization of Ligands and Metal Compounds.
TMG3trphen-Sb•3CH2Cl2 (1).
2-(2′-bromophenyl)-1,1,3,3-tetramethylguanidine (1.10 g, 4.08 mmol) was dissolved in 20 mL of Et2O in a Schlenk flask. The solution was cooled in a dry ice/acetone bath to −78 °C, after which a cyclohexane solution of sec-butyllithium (1.4 M, 6.4 mL, 8.99 mmol) was added, effecting a color change to bright orange. After stirring for 1 h while being kept cold, SbCl3 (0.307 g, 1.34 mmol) dissolved in THF (10 mL) was added at −78 °C via cannula. The reaction mixture was kept at −78 °C for 2 h and then was allowed to warm to room temperature and stirred overnight. After the addition of 1.0 mL of methanol, the volatiles were removed in vacuo, producing a yellow-orange residue. The crude was dissolved in EtOAc (20 mL) and washed with water (10 mL). The aqueous phase was extracted with additional EtOAc (20 mL). The combined organic phases were dried over sodium sulfate and concentrated by rotary evaporation to give an oily mass. The oily mass was dissolved in 10 mL of CH2Cl2 and chilled to 0 °C for 1 h, separating colorless crystals. The white solid was collected by filtration, washed with cooled CH2Cl2 (5 mL), and dried in vacuum (yield = 0.580 g, 62%). 1H NMR (CD3CN, 1.94 ppm): δ 7.08 (t, 3H, aryl), 6.97(d, 3H, aryl), 6.60 (t, 3H, aryl), 6.41(d, 3H, aryl), 2.57 (s, 36H, CH3).13C NMR (CD3CN, 118.26, 1.32 ppm): δ 159.0, 157.5, 137.6, 135.3, 129.1, 120.6, 120.2, 39.9. IR (KBr, cm−1): ν 3031, 2932, 2889, 1539,1452, 1419, 1378, 1136, 1018, 791, 723, 535. UV–vis (CH3CN): λmax (ε (M−1 cm−1)) 220(76263). Elem. Anal. calcd for C36H54Cl6N9Sb (1): C, 45.64; H, 5.75, N, 13.31. Found: C, 45.71; H, 5.76, N, 13.29.
TMG3trphen-Bi•3CH2Cl2 (2).
2-(2′-bromophenyl)-1,1,3,3-tetramethylguanidine (1.16 g, 4.31 mmol) was dissolved in 20 mL of THF in a Schlenk flask. The solution was cooled in a dry ice/acetone bath to −78 °C, after which a cyclohexane solution of sec-butyllithium (1.4 M, 6.8 mL, 9.48 mmol) was added, effecting a color change to bright orange. After stirring for 1 h while being kept cold, BiCl3 (0.448 g, 1.42 mmol), dissolved in THF (10 mL), was added at −78 °C via cannula. The reaction mixture was kept at −78 °C for 2 h and then was allowed to warm to room temperature. The reaction mixture was refluxed for 16 h, and subsequently, the solvent was removed to give an oily residue that was redissolved in toluene (20 mL). The reaction mixture was heated again and stirred at 90–95 °C for 3 h. The temperature of the reaction was lowered to ambient conditions, and the solvent was evacuated to dryness to give an oily mass. The residual oily mass was suspended in CH2Cl2 (10 mL) and was stirred vigorously at room temperature for 30 min, separating white crystals that were collected by filtration (yield = 0.550 g, 49%). 1H NMR (CD3CN, 1.94 ppm): δ 7.49 (d, 3H, aryl), 7.09 (t, 3H, aryl), 6.65 (t, 3H, aryl), 6.51(d, 3H, aryl), 2.58 (s, 36H, CH3) 13C NMR (CD3CN, 118.26, 1.32 ppm): δ 159.2, 157.6, 139.5, 128.5, 122.7, 120.7, 39.9. IR (KBr, cm−1): ν 3031, 2918, 2791, 1545, 1443, 1419, 1372, 1133, 1012, 781, 747, 724, 534. UV–vis (CH3CN): λmax (ε (M−1 cm−1)) 214 (59181). Elem. Anal. calcd for C36H54BiCl6N9 (2): C, 41.79; H, 5.26, N, 12.18. Found: C, 41.73; H, 5.23, N, 12.12.
[(TMG3trphen-Sb)Cu3(μ-Cl)3]•4CH2Cl2 (3).
To a suspension of CuCl (138 mg, 1.39 mmol) in THF (12 mL) was added a solution of TMG3trphen-Sb (1) (321 mg, 0.463 mmol) in THF (18 mL) at room temperature. The solution was stirred for 24 h. The volatiles were removed under vacuum, and the residue was redissolved in CH2Cl2 (10 mL) and stirred for 6 h. The solution was filtered and concentrated to give an off-white-colored powder (yield = 0.380 g, 83%). The compound was recrystallized from CH2Cl2 to obtain X-ray-quality crystals. 1H NMR (CD2Cl2, 5.32 ppm, 243 K): δ 7.17–7.14 (t, 3H, aryl), 7.02–7.01 (d, 3H, aryl), 6.69–6.66 (t, 3H, aryl), 6.46–6.44 (d, 3H, aryl), 3.10(s, 9H, CH3), 2.76 (s, 9H, CH3), 2.58 (s, 9H, CH3), 1.67 (s, 9H, CH3). 13C NMR (CD2Cl2, 54.0 ppm): δ 166.2, 156.3, 136.5, 131.7, 126.0, 122.0, 121.7, 40.4, 30.6. IR (KBr, cm−1): ν 3476, 3044, 2929, 2879, 2791, 1519, 1453, 1405, 1392, 1330, 1266, 1155, 1027, 853, 750, 725, 536. UV–vis (CH2Cl2): λmax (ε (M−1 cm−1)) 235 (225820). Elem. Anal. calcd for C33H48Cl3Cu3N9Sb (3 – 4CH2Cl2): C, 40.05; H, 4.89, N, 12.74. Found: C, 39.99; H, 4.84, N, 12.80.
[(TMG3trphen-Bi)Cu3(μ-Cl)3]•4CH2Cl2 (4).
To a suspension of CuCl (57.2 mg, 0.58 mmol) in THF (12 mL) was added a solution of TMG3trphen-Bi (2) (150 mg, 0.192 mmol) in THF (18 mL) at room temperature. The solution was stirred for 24 h. The volatiles were removed under vacuum, and the residue was redissolved in 1,2-dichloroethane (10 mL) and stirred for 6 h. The solution was filtered and concentrated to give off-white colored powder (yield = 0.205g, 99%). The compound was recrystallized from CH2Cl2 to obtain X-ray-quality crystals. 1H NMR (CD2Cl2, 5.32 ppm, 243 K): δ 7.50–7.48 (d, 3H, aryl), 7.21–7.18 (t, 3H, aryl), 6.80–6.77 (t, 3H, aryl), 6.58–6.56 (d, 3H, aryl), 3.19(s, 9H, CH3), 2.82 (s, 9H, CH3), 2.61 (s, 9H, CH3), 1.69 (s, 9H, CH3).13C NMR (CD2Cl2, 54.0 ppm): δ 162.5, 150.2, 143.4, 127.7, 121.9, 118.5, 115.7, 40.7, 39.9. IR: 3040, 2924, 2878,2114, 1518, 1447, 1415, 1406, 1388, 1330, 1260, 1234, 1199, 1152, 1109, 1064, 1041, 1025, 923, 850, 805, 751, 720, 536. UV–vis (CH2Cl2): λmax (ε (M−1 cm−1)) 235 (26520). Elem. Anal. calcd for C33H48BiCl3Cu3N9 (4 – 4CH2Cl2): C, 36.81; H, 4.49, N, 11.71. Found: C, 36.77; H, 4.45, N, 11.78.
[(TMG3trphen-Sb)Cu3(μ-Br)3]•4CH2Cl2 (5).
To a suspension of CuBr (205 mg, 1.43 mmol) in THF (12 mL) was added a solution of TMG3trphen-Sb (1) (330 mg, 0.477 mmol) in THF (18 mL) at room temperature. The solution was stirred for 24 h. The volatiles were removed under vacuum, and the residue was redissolved in CH2Cl2 (10 mL) and stirred for 6 h. The solution was filtered and concentrated to give off-white colored powder (yield = 0.402 g, 85%). The compound was recrystallized in CH2Cl2 to obtain X-ray-quality crystals. 1H NMR (CD2Cl2, 5.32 ppm, 243 K): δ 7.25–7.21 (t, 3H, aryl), 7.08–7.07 (d, 3H, aryl), 6.75–6.71 (t, 3H, aryl), 6.51–6.49 (d, 3H, aryl), 3.20(s, 9H, CH3), 2.85 (s, 9H, CH3), 2.65 (s, 9H, CH3), 1.74 (s, 9H, CH3). 13C NMR (CD2Cl2, 54.0 ppm): δ 165.9, 156.0, 136.0, 130.9, 121.5, 120.7, 40.8, 15.0. IR (KBr, cm−1): ν 3043, 2924, 2881,2792, 1517,1453,1416, 1404, 1392, 1330, 1282, 1265, 1202, 1155, 1116, 1063, 1047, 1028, 926, 854, 807, 753, 726, 705, 537, 452. UV–vis (CH2Cl2): λmax (ε (M−1 cm−1)) 230 (225450). Elem. Anal. calcd for C33H48Br3Cu3N9Sb (5 – 4CH2Cl2): C, 35.30; H, 4.31, N, 11.23. Found: C, 35.34; H, 4.34, N, 11.19.
[(TMG3trphen-Sb)Cu3(μ-I)3]•3CH2Cl2 (6).
To a suspension of CuI (372 mg, 1.95 mmol) in THF (12 mL) was added a solution of TMG3trphen-Sb (1) (450 mg, 0.650 mmol) in THF (18 mL) at room temperature. The solution was stirred for 24 h. The volatiles were removed under vacuum and the residue was redissolved in CH2Cl2 (10 mL) and stirred for 6 h. The solution was filtered and concentrated to give off-white colored powder (yield = 0.530 g, 82%). The compound was recrystallized from CH2Cl2 to obtain X-ray-quality crystals. 1H NMR (CD2Cl2, 5.32 ppm, 243 K): δ 7.22–7.18 (t, 3H, aryl), 7.05–7.03 (d, 3H, aryl), 6.73–6.69 (t, 3H, aryl), 6.45–6.43 (d, 3H, aryl), 3.19(s, 9H, CH3), 2.84 (s, 9H, CH3), 2.61 (s, 9H, CH3), 1.70 (s, 9H, CH3). 13C NMR (CD2Cl2, 54.0 ppm): δ 166.6, 156.3, 136.1, 130.7, 121.5, 120.3, 40.7, 14.8. IR (KBr, cm−1): ν 3042, 2923,2880, 2792, 1557,1544, 1512, 1454, 1415, 1404, 1392, 1329, 128. UV–vis (CH2Cl2): λmax (ε (M−1 cm−1)) 340(18032). Elem. Anal. calcd for C33H48Cu3I3N9Sb (6 – 3CH2Cl2): C, 31.36; H, 3.83, N, 9.97. Found: C, 31.38; H, 3.85, N, 9.93.
[(TMG3trphen-Sb)Ag3(μ-Cl)3]•3CH2Cl2 (7).
To a solution of TMG3trphen-Sb (1) (186 mg, 0.268 mmol) in THF (20 mL), AgCl (115 mg, 0.805 mmol) was added at room temperature. The suspension was stirred for 2 weeks, protected from light. The volatiles were removed under vacuum, and the residue was redissolved in CH2Cl2 (20 mL) and stirred for 12 h. The solution was filtered and concentrated to give off-white colored powder (yield = 0.215 g, 71%). The compound was recrystallized from CH2Cl2 to obtain X-ray-quality crystals. 1H NMR (CD2Cl2, 5.32 ppm, 243 K): δ 7.25–7.22 (t, 3H, aryl), 7.13–7.11 (d, 3H, aryl), 6.77–6.73(t, 3H, aryl), 6.52–6.50 (d, 3H, aryl), 2.66 (s, 36H, CH3). 13C NMR (CD2Cl2, 54.0 ppm): 165.1, 156.6, 137.2, 130.9, 122.1, 120.8, 39.8, 29.6. IR (KBr, cm−1): ν 3020, 2930, 2870, 1519, 1454, 1417, 1406, 191, 1263, 1153, 1026, 852, 758, 725, 536. UV–vis (CH2Cl2): λmax (ε (M−1 cm−1)) 295(12534). Elem. Anal. calcd for C33H48 Ag3Cl3N9Sb (7 – 3CH2Cl2): C, 35.31; H, 4.31, N, 11.23. Found: C, 35.36; H, 4.33, N, 11.19.
[(TMG3trphen-Sb)2Cu2](PF6)2•2THF (8a).
A solution of [Cu(CH3CN)4](PF6) (108 mg, 0.288 mmol) in acetonitrile/THF (5 mL each) was added to a prestirred solution of TMG3trphen-Sb (1) (200 mg, 0.288 mmol) in acetonitrile (20 mL) at room temperature. The suspension was stirred for 24 h. The reaction mixture was filtered, reduced to 5 mL, and stored at −35 °C for 2 weeks to obtain X-ray quality crystals (yield = 0.145 g, 51%). 1H NMR (CD3CN, 1.94 ppm): δ 7.45–7.44 (d, 6H, aryl), 7.17–7.13 (t, 6H, aryl), 6.80–6.76 (t, 6H, aryl), 6.42–6.39 (d, 6H, aryl), 2.67 (s, 72H, CH3). 13C NMR (CD3CN, 118.26, 1.32 ppm): 163.9, 157.0, 137.2, 131.6, 130.0, 122.3, 121.6, 120.9, 40.1. IR: 3672, 2935, 2883, 1515, 1454, 1416, 1394, 1324, 1281, 1265, 1231, 1201, 1142, 1111, 1062, 1046, 1021, 875, 834, 783, 762, 750, 726, 710, 556, 536, 467, 448. UV–vis (CH3CN): λmax (ε (M−1 cm−1)) 275(70831). Elem. Anal. calcd for C66H96Cu2F12N18P2Sb2 (8a – 2THF): C, 43.99; H, 5.37, N, 13.99. Found: C, 44.02; H, 5.38, N, 13.92.
[(TMG3trphen-Sb)2Cu2][B(C6F5)4]2•2MeCN (8b).
An acetonitrile solution (10 mL) of [Cu(CH3CN)2][B(C6F5)31 (64.4 mg, 0.078 mmol) was added to a prestirred solution of TMG3trphen-Sb (1) (36.05 mg, 0.052 mmol) in acetonitrile (10 mL) at room temperature. The suspension was stirred for 24 h. The reaction mixture was filtered, reduced to 5 mL under vacuum, and stored at −35 °C for 1 week to obtain X-ray quality colorless crystals (yield = 0.054 g, 70%). 1H NMR (CD3CN, 1.94 ppm): δ 7.50–7.46 (d, 6H, aryl), 7.19–7.15(t, 6H, aryl), 6.82–6.78 (t, 6H, aryl), 6.44–6.42 (d, 6H, aryl), 2.69 (s, 72H, CH3). 13C NMR (CD3CN, 118.26, 1.32 ppm): δ 165.07, 157.0, 137.1, 129.5, 121.9, 121.7, 120.3, 52.1, 39.7. IR (cm−1): 2932, 1643, 1513, 1460, 1420, 1396, 1324, 1274, 1156, 1085, 1021, 978, 850, 807, 774, 755, 727, 683, 661, 611, 574, 537. UV–vis (CH3CN): λmax (ε (M−1 cm−1)) 215(147250). Elem. Anal. calcd for C118H102B2Cu2F40N20Sb2 (8b): C, 48.01; H, 3.48, N, 9.49. Found: C, 48.07; H, 3.50, N, 9.45.
[(TMG3trphen-Bi)2Cu2](PF6)2•2Et2O (9).
A solution of [Cu(CH3CN)4](PF6) (143.4 mg, 0.385 mmol) in acetonitrile (5 mL) was added to a prestirred solution of TMG3trphen-Bi (2) (200 mg, 0.256 mmol) in acetonitrile (20 mL) at room temperature. The suspension was stirred for 24 h. The reaction mixture was filtered, reduced to 5 mL under vacuum, and carefully layered with diethyl ether to obtain X-ray quality crystals at −35 °C over a period of 2 weeks (yield = 0.035 g, 6.4%). 1H NMR (CD3CN, 1.94 ppm): δ 7.85–7.83 (d, 2H, aryl), 7.26–7.22 (t, 6H, aryl), 7.19–7.15 (t, 2H, aryl), 6.95–6.91 (t, 4H, aryl), 6.88–6.84 (d, 2H, aryl), 6.83–6.80 (d, 6H, aryl), 6.56–6.54 (d, 2H, aryl), 2.75 (s, 42H, CH3), 2.67(s, 30H, CH3). 13C NMR (CD3CN, 118.26, 1.32 ppm): 164.9, 157.0, 139.3, 130.0, 129.3, 124.0, 123.1, 122.1, 40.3. IR: 3677, 3367, 2937, 1630, 1526, 1485, 1472, 1451, 1424, 1398, 1322, 1263, 1233, 1205, 1158, 1066, 1038, 831, 750, 696, 606, 556, 442. UV–vis (CH3CN): λmax (ε (M− 1 cm− 1 )) 265(58035). Elem. Anal. calcd for C66H96Bi2Cu2F12N18P2 (9 – 2Et2O): C, 40.11; H, 4.90, N, 12.76. Found: C, 40.08; H, 4.88, N, 12.80.
[(TMG3H3trphen-Sb)Cu(OTf)3](OTf)•1.21CH2Cl2 (10).
A methylene chloride solution (15 mL) of [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3) (64.1 mg, 0.065 mmol) was added to solid Ag(OTf) (49.93 mg, 0.195 mmol), and the reaction was stirred for 12 h in the absence of light. The resulting mixture was filtered to remove AgCl and reduced to 5.0 mL under vacuum. The clear solution was kept at −35 °C over a period of 1 week, affording large colorless crystals of the titled compound (yield = 0.036 g, 14%; based on Cu). 1H NMR (CD2Cl2, 5.32 ppm): δ 7.36–7.32 (t, 3H, aryl), 7.25–7.23 (d, 3H, aryl), 6.93–6.89 (t, 3H, aryl), 6.61–6.59 (d, 3H, aryl), 2.85 (bs, 36H, CH3), 1.26 (s, 3H, = NH). 13C NMR (CD2Cl2, 54.0 ppm): 166.5, 155.6, 136.5, 132.5, 128.0, 122.6, 122.0, 40.7, 30.2. IR: 3225, 3053, 2930, 1630, 1547, 1520, 1456, 1419, 1398, 1328, 1262, 1222, 1154, 1028, 855, 753, 636, 517, 447. UV–vis (CH2Cl2): λmax (ε (M−1 cm−1)) 235(10664). Elem. Anal. calcd for C37H51CuF12N9O12S4Sb (10 – 1.21CH2Cl2): C, 32.79; H, 3.79, N, 9.30. Found: C, 32.75; H, 3.78, N, 9.35.
[(TMG3trphen-Sb)Cu3(μ3-Cl)3Cu]2[B(C6F5)4]2•10CH2Cl2 (11).
A methylene chloride solution (20 mL) of [(TMG3trphen-Sb)Cu3(μ-Cl)3] (3) (47.0 mg, 0.047 mmol) was added to K[B(C6F5)4] (102.3 mg, 0.142 mmol), and the reaction mixture was stirred for 24 h. The resulting mixture was filtered to remove KCl and reduced to 5.0 mL under vacuum. The solution was kept at −35 °C over a period of 1 week, to provide off-white crystals of the titled compound (yield = 0.046 g, 75%; based on Cu). 1H NMR (CDCl3, 7.26 ppm): δ 7.27–7.24 (t, 6H, aryl), 7.19–7.17 (d, 6H, aryl), 6.85–6.82 (t, 6H, aryl), 6.49–6.47 (d, 6H, aryl), 3.15 (bs, 12H, CH3), 2.81 (bs, 40H, CH3), 1.90 (bs, 20H, CH3). 13C NMR (CDCl3, 77.2 ppm): 166.0, 155.4, 137.8, 135.4, 132.2, 122.4, 121.5, 53.9, 40.0. IR: 2928, 1643, 1513, 1457, 1420,1407, 1396, 1330, 1270, 1233, 1158, 1085, 1031, 977, 854, 809,774, 755, 725, 683,661, 611, 574, 537, 450. UV–vis (CH3CN): λmax (ε (M−1 cm−1)) 230(14172). Elem. Anal. calcd for C114H96B2Cl6Cu8F40N18Sb2 (11 – 10CH2Cl2): C, 39.53; H, 2.79, N, 7.28. Found: C, 39.49; H, 2.78, N, 7.31.
Catalytic and Mechanistic Studies.
General Catalytic Olefin Aziridination Procedure.
In a typical experiment, a 20 mL screw-cap vial containing a small magnetic bar was charged in sequence with the catalyst (0.0125 mmol with respect to Cu), N(p-tolylsulfonyl)-imido]phenyliodinane (93.3 mg, 0.25 mmol), molecular sieves (5 Å) (20 mg), olefin (2.0 mmol), and solvent (0.200 g) (acetonitrile, unless otherwise stated). The reaction mixture was stirred vigorously at 30 °C for 24 h (unless otherwise stated). After completion of the reaction, the products were isolated by column chromatography (silica gel) and quantified by 1H NMR (in CDCl3) versus an internal standard (4′-methoxyacetophenone). All aziridination and/or allylic/benzylic amination products are known compounds and have been identified with the assistance of 1H by comparison to spectroscopic features reported for authentic samples in the literature.
General Catalytic Benzylic Amination Procedure.
In a standard method for copper-catalyzed amination, a tiny magnetic stirrer was placed in a flame-dried 20 mL screw cap vial. To this vial, catalyst (0.0125 mmol with respect to copper), 2,2,2-trichloroethyl(phenyl-λ3-iodanylidene)sulfamate (PhINTces) (214.4 mg, 0.500 mmol), and molecular sieves (5 Å) (20 mg) were added sequentially. A solution of the substrate (0.250 mmol) in a mixture of trifluorotoluene and HFIP (10:1 v/v, 300 mg) was added to the vial that contained the solids. The reaction was stirred at 30 °C for 24 h. After completion of the reaction, the products were isolated by column chromatography (silica gel) using a mixture of hexane and ethyl acetate and were quantified by 1H NMR (in CDCl3) versus an internal standard (4′-methoxyacetophenone).
Hammett Plots.
The general procedure for the amination of benzylic substrates was conducted with the assistance of the catalyst [(TMG3trphen-Sb)Cu3(μ-Cl)3]•4CH2Cl2 (3) (0.0125 mmol with respect to Cu). The reaction mixture was composed of 1.0 mmol of ethylbenzene, 1.0 mmol of p-X-ethylbenzene (X = Me, MeO, F, Cl, Br, CF3, and NO2), and PhINTces (2 mmol) in the PhCF3:HFIP mixture (10:1 v/v). The reaction vial was sealed tightly and wrapped with aluminum foil to protect the reaction mixture from light. The reactions were stirred for 6 h. At the end of the reaction, the mixture was flash chromatographed on silica gel (CH2Cl2) in order to recover the benzylic aminated products and evaluate their ratio by quantitative 1H NMR analysis in CDCl3.
Supplementary Material
ACKNOWLEDGMENTS
Dr. Steven Kelley is acknowledged for collecting single-crystal X-ray diffraction data at the University of Missouri-Columbia.
Funding
The authors acknowledge generous funding awarded by NIH/NIGMS (R15GM117508 and R15GM139071) for this work (to P.S.) and partial support of this research through grant DE-FG02–03ER15387 (to T.R.C.) from the U.S. Department of Energy, Basic Sciences, Catalysis Science program.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.3c00493.
General considerations; synthesis and characterization; X-ray crystallography; catalytic and mechanistic studies; computational methods; crystallographic data; competitive amination reactions; ORTEP; and 1H and 13C NMR (PDF)
Atomic coordinates of compound 4 (XYZ)
Atomic coordinates of compound 3 (XYZ)
Accession Codes
CCDC 2309022–2309033 contain the supplementary crystallographic data for this paper. This data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223 336033.
Contributor Information
Meenakshi Sharma, Department of Chemistry, Missouri University of Science and Technology, Rolla, Missouri 65409, United States.
Reece M. Fritz, Department of Chemistry, Missouri University of Science and Technology, Rolla, Missouri 65409, United States
Joseph O. Adebanjo, Department of Chemistry, University of North Texas, Denton, Texas 76203, United States
Zhou Lu, Department of Chemistry, University of North Texas, Denton, Texas 76203, United States.
Thomas R. Cundari, Department of Chemistry, University of North Texas, Denton, Texas 76203, United States
Mohammad A. Omary, Department of Chemistry, University of North Texas, Denton, Texas 76203, United States
Amitava Choudhury, Department of Chemistry, Missouri University of Science and Technology, Rolla, Missouri 65409, United States.
Pericles Stavropoulos, Department of Chemistry, Missouri University of Science and Technology, Rolla, Missouri 65409, United States.
REFERENCES
- (1).Roose P; Eller K; Henkes E; Rossbacher R; Höke H Amines, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Wenheim, Germany, 2015. [Google Scholar]
- (2).(a) Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]; (b) Roizen JL; Harvey ME; Du Bois J Metal-Catalyzed Nitrogen-Atom Transfer Methods for the Oxidation of Aliphatic C–H Bonds. Acc. Chem. Res. 2012, 45, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ju M; Schomaker JM Nitrene Transfer Catalysts for Enantioselective C–N Bond Formation. Nat. Rev. Chem. 2021, 5, 580–594. [DOI] [PubMed] [Google Scholar]; (d) Chandrachud PP; Jenkins DM Transition Metal Aziridination Catalysts. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Wiley Online Library, 2017; pp 1–11. [Google Scholar]; (e) Cardoso AL; Pinho e Melo TMVD Aziridines in Formal [3 + 2] Cycloadditions: Synthesis of Five-Membered Heterocycles. Eur. J. Org. Chem. 2012, 6479–6501. [Google Scholar]
- (3).Taube H Observations on Atom-Transfer Reactions. In Mechanistic Aspects of Inorganic Reactions, Rorabacher DB; Endicott JF, Eds.; ACS Symposium Series; 1982; Chapter 7, Vol. 198, pp 151–179. [Google Scholar]
- (4).(a) Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roth JP; Lovell S; Mayer JM Intrinsic Barriers for Electron and Hydrogen Atom Transfer Reactions of Biomimetic Iron Complexes. J. Am. Chem. Soc. 2000, 122, 5486–5498. [Google Scholar]; (c) Waidmann CR; Zhou X; Tsai EA; Kaminsky W; Hrovat DA; Borden WT; Mayer JM Slow Hydrogen Atom Transfer Reactions of Oxo- and Hydroxo-Vanadium Compounds: The Importance of Intrinsic Barriers. J. Am. Chem. Soc. 2009, 131, 4729–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Atienza CCH; Bowman AC; Lobkovsky E; Chirik PJ Photolysis and Thermolysis of Bis(imino)pyridine Cobalt Azides: C–H Activation from Putative Cobalt Nitrido Complexes. J. Am. Chem. Soc. 2010, 132, 16343–16345. [DOI] [PubMed] [Google Scholar]; (b) Berry JF; Bill E; Bothe E; DeBeer George S; Mienert B; Neese F; Wieghardt K An Octahedral Coordination Complex of Iron(VI). Science 2006, 312, 1937–1941. [DOI] [PubMed] [Google Scholar]; (c) DuBois J; Tomooka CS; Hong J; Carreira EM Nitridomanganese(V) Complexes: Design, Preparation, and Use as Nitrogen Atom-Transfer Reagents. Acc. Chem. Res. 1997, 30, 364–372. [Google Scholar]; (d) Scepaniak JJ; Vogel CS; Khusniyarov MM; Heinemann FW; Meyer K; Smith JM Synthesis, Structure, and Reactivity of an Iron(V) Nitride. Science 2011, 331, 1049–1052. [DOI] [PubMed] [Google Scholar]; (e) Betley TA; Peters JC A Tetrahedrally Coordinated L3Fe–Nx Platform that Accommodates Terminal Nitride (FeIV⋮N) and Dinitrogen (FeI–N2–FeI) Ligands. J. Am. Chem. Soc. 2004, 126, 6252–6254. [DOI] [PubMed] [Google Scholar]
- (6).(a) Holm RH Metal-centered oxygen atom transfer reactions. Chem. Rev. 1987, 87, 1401–1449. [Google Scholar]; (b) Murray LJ; Lippard SJ Substrate Trafficking and Dioxygen Activation in Bacterial Multi-component Monooxygenases. Acc. Chem. Res. 2007, 40, 466–474. [DOI] [PubMed] [Google Scholar]; (c) Cooper HLR; Mishra G; Huang X; Pendler-Cudlip M; Austin RN; Shanklin J; Groves JT Parallel and Competitive Pathways for Substrate Desaturation, Hydroxylation, and Radical Rearrangement by the Non-heme Diiron Hydroxylase AlkB. J. Am. Chem. Soc. 2012, 134, 20365–20375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Que L Jr.; Tolman WB Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]; (e) McDonald AR; Que L Jr High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar]; (f) Chen MS; White MC Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]; (g) Nam W; Lee Y-M; Fukuzumi S Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47, 1146–1154. [DOI] [PubMed] [Google Scholar]; (h) Abu-Omar MM In Physical Inorganic Chemistry, Reactions, Processes, and Applications, Bakac A, Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2010; pp 75–108. [Google Scholar]; (i) Peterson RL; Himes RA; Kotani H; Suenobu T; Tian L; Siegler M; Solomon EI; Fukuzumi S; Karlin KD Cupric Superoxo-Mediated Intermolecular C–H Activation Chemistry. J. Am. Chem. Soc. 2011, 133, 1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kundu S; Thompson JVK; Ryabov AD; Collins TJ On the Reactivity of Mononuclear Iron(V)oxo Complexes. J. Am. Chem. Soc. 2011, 133, 18546–18549. [DOI] [PubMed] [Google Scholar]; (k) Taguchi T; Gupta R; Lassalle-Kaiser B; Boyce DW; Yachandra VK; Tolman WB; Yano J; Hendrich MP; Borovik AS Preparation and Properties of a Monomeric High-Spin MnV–Oxo Complex. J. Am. Chem. Soc. 2012, 134, 1996–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Donahue JP Thermodynamic Scales for Sulfur Atom Transfer and Oxo-for-Sulfido Exchange Reactions. Chem. Rev. 2006, 106, 4747–4783. [DOI] [PubMed] [Google Scholar]; (b) Yang L; Tehranchi J; Tolman WB Reactions of Ph3Sb = S with Copper(I) Complexes Supported by N-Donor Ligands: Formation of Stable Adducts and S-Transfer Reactivity. Inorg. Chem. 2011, 50, 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang J-J; Kryatova OP; Rybak-Akimova EV; Holm RH Comparative Kinetics and Mechanism of Oxygen and Sulfur Atom Transfer Reactions Mediated by Bis(dithiolene) Complexes of Molybdenum and Tungsten. Inorg. Chem. 2004, 43, 8092–8101. [DOI] [PubMed] [Google Scholar]
- (8).(a) Matyjaszewski K Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar]; (b) Pintauer T; Matyjaszewski K Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes. Chem. Soc. Rev. 2008, 37, 1087–1097. [DOI] [PubMed] [Google Scholar]; (c) Ouchi M; Terashima T; Sawamoto M Transition Metal-Catalyzed Living Radical Polymerization: Toward Perfection in Catalysis and Precision Polymer Synthesis. Chem. Rev. 2009, 109, 4963–5050. [DOI] [PubMed] [Google Scholar]
- (9).(a) Hartwig JF Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [DOI] [PubMed] [Google Scholar]; (b) Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF C–H Activation for the Construction of C–B Bonds. Chem. Rev. 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]; (c) Obligacion JV; Semproni SP; Chirik PJ Cobalt-Catalyzed C–H Borylation. J. Am. Chem. Soc. 2014, 136, 4133–4136. [DOI] [PubMed] [Google Scholar]
- (10).(a) Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–935. [DOI] [PubMed] [Google Scholar]; (b) Davies HML; Manning JR Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Davies HML; Morton D Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. [DOI] [PubMed] [Google Scholar]; (d) Deng Y; Qiu H; Srinivas HD; Doyle MP Chiral Dirhodium(II) Catalysts for Selective Metal Carbene Reactions. Curr. Org. Chem. 2016, 20, 61–81. [Google Scholar]; (e) Ford A; Miel H; Ring A; Slattery CN; Maguire AR; McKervey MA Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [DOI] [PubMed] [Google Scholar]; (f) Ebner C; Carreira EM Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev. 2017, 117, 11651–11679. [DOI] [PubMed] [Google Scholar]; (i) Wang B; Qiu D; Zhang Y; Wang J Recent advances in C(sp3)–H bond functionalization via metal–carbene insertions. Beilstein J. Org. Chem. 2016, 12, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Zalatan DN; Du Bois J Metal-Catalyzed Oxidations of C–H to C–N Bonds. Top. Curr. Chem. 2010, 292, 347–378. [DOI] [PubMed] [Google Scholar]; (b) Roizen JL; Harvey ME; Du Bois J Metal-Catalyzed Nitrogen-Atom Transfer Methods for the Oxidation of Aliphatic C–H Bonds. Acc. Chem. Res. 2012, 45, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gephart RT III; Warren TH Copper-Catalyzed sp3 C–H Amination. Organometallics 2012, 31, 7728–7752. [Google Scholar]; (d) Collet F; Lescot C; Dauban P Catalytic C–H amination: the stereoselectivity issue. Chem. Soc. Rev. 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]; (e) Dequirez G; Pons V; Dauban P Nitrene Chemistry in Organic Synthesis: Still in Its Infancy? Angew. Chem., Int. Ed. 2012, 51, 7384–7395. [DOI] [PubMed] [Google Scholar]; (f) Hazelard D; Nocquet P-A; Compain P Catalytic C–H amination at its limits: challenges and solutions. Org. Chem. Front. 2017, 4, 2500–2521. [Google Scholar]; (g) Uchida T; Katsuki T Asymmetric Nitrene Transfer Reactions: Sulfimidation, Aziridination and C–H Amination Using Azide Compounds as Nitrene Precursors. Chem. Rec. 2014, 14, 117–129. [DOI] [PubMed] [Google Scholar]; (h) Pellissier H Recent Developments in Asymmetric Aziridination. Adv. Synth. Catal. 2014, 356, 1899–1935. [Google Scholar]; (i) Díaz-Requejo MM; Pérez PJ Coinage Metal Catalyzed C–H Bond Functionalization of Hydrocarbons. Chem. Rev. 2008, 108, 3379–3394. [DOI] [PubMed] [Google Scholar]; (j) Müller P; Fruit C Enantioselective Catalytic Aziridinations and Asymmetric Nitrene Insertions into CH Bonds. Chem. Rev. 2003, 103, 2905–2919. [DOI] [PubMed] [Google Scholar]
- (12).(a) Huang X; Bergsten TV; Groves JT Manganese-Catalyzed Late-Stage Aliphatic C–H Azidation. J. Am. Chem. Soc. 2015, 137, 5300–5303. [DOI] [PubMed] [Google Scholar]; (b) Sharma A; Hartwig JF Metal-catalysed azidation of tertiary C–H bonds suitable for late–stage functionalization. Nature 2015, 517, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karimov RR; Sharma A; Hartwig JF Late Stage Azidation of Complex Molecules. ACS Cent. Sci. 2016, 2, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Coin G; Latour J-M Nitrene transfers mediated by natural and artificial iron enzymes. J. Inorg. Biochem. 2021, 225, 111613. [DOI] [PubMed] [Google Scholar]; (b) Bollinger JM; Broderick JB Frontiers in enzymatic C–H-bond activation. Curr. Opin. Chem. Biol. 2009, 13, 51–57. [DOI] [PubMed] [Google Scholar]; (c) Lewis JC; Coelho PS; Arnold FH Enzymatic functionalization of carbon–hydrogen bonds. Chem. Soc. Rev. 2011, 40, 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang Y; Arnold FH Navigating the Unnatural Reaction Space: Directed Evolution of Heme Proteins for Selective Carbene and Nitrene Transfer. Acc. Chem. Res. 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Brandenberg OF; Fasan R; Arnold FH Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol. 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hartwig JF; Larsen MA Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Combee LA; Raya B; Wang D; Hilinski MK Organocatalytic nitrenoid transfer: metal-free selective intermolecular C(sp3)–H amination catalyzed by an iminium salt. Chem. Sci. 2018, 9, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qin Q; Yu S Visible-Light-Promoted Remote C(sp3)–H Amidation and Chlorination. Org. Lett. 2015, 17, 1894–1897. [DOI] [PubMed] [Google Scholar]; (c) Pandey G; Laha R Visible-Light-Catalyzed Direct Benzylic C(sp3)–H Amination Reaction by Cross-Dehydrogenative Coupling. Angew. Chem., Int. Ed. 2015, 54, 14875–14879. [DOI] [PubMed] [Google Scholar]; (d) Zhu C; Liang Y; Hong X; Sun H; Sun W-Y; Houk KN; Shi Z Iodoarene-Catalyzed Stereospecific Intramolecular sp3 C–H Amination: Reaction Development and Mechanistic Insights. J. Am. Chem. Soc. 2015, 137, 7564–7567. [DOI] [PubMed] [Google Scholar]; (e) Souto JA; Martínez C; Velilla I; Muñiz K Defined Hypervalent Iodine(III) Reagents Incorporating Transferrable Nitrogen Groups: Nucleophilic Amination through Electrophilic Activation. Angew. Chem. Int. Ed. 2013, 52, 1324–1328. [DOI] [PubMed] [Google Scholar]; (f) Souto JA; Zian D; Muñiz K Iodine(III)-Mediated Intermolecular Allylic Amination under Metal-Free Conditions. J. Am. Chem. Soc. 2012, 134, 7242–7245. [DOI] [PubMed] [Google Scholar]; (g) Kim HJ; Kim J; Cho SH; Chang S Intermolecular Oxidative C–N Bond Formation under Metal-Free Conditions: Control of Chemoselectivity between Aryl sp2 and Benzylic sp3 C–H Bond Imidation. J. Am. Chem. Soc. 2011, 133, 16382–16385. [DOI] [PubMed] [Google Scholar]; (h) Kantak AA; Potavathri S; Barham RA; Romano KM; DeBoef B Metal-Free Intermolecular Oxidative C–N Bond Formation via Tandem C–H and N–H Bond Functionalization. J. Am. Chem. Soc. 2011, 133, 19960–19965. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Antonchick AP; Samanta R; Kulikov K; Lategahn J Organocatalytic, Oxidative, Intramolecular C–H Bond Amination and Metal-free Cross-Amination of Unactivated Arenes at Ambient Temperature. Angew. Chem., Int. Ed. 2011, 50, 8605–8608. [DOI] [PubMed] [Google Scholar]; (j) Ochiai M; Miyamoto K; Kaneaki T; Hayashi S; Nakanishi W Highly Regioselective Amination of Unactivated Alkanes by Hypervalent Sulfonylimino-λ3-Bromane. Science 2011, 332, 448–451. [DOI] [PubMed] [Google Scholar]
- (16).(a) Chu JCK; Rovis T Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem., Int. Ed. 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Selective examples of typical Rh-centered C–N Bond Formation: Fiori KW; Du Bois J Catalytic Intermolecular Amination of C–H Bonds: Method Development and Mechanistic Insights. J. Am. Chem. Soc. 2007, 129, 562–568. Fiori KW; Espino CG; Brodsky BH; Du Bois J A mechanistic analysis of the Rh-catalyzed intramolecular C–H amination reaction. Tetrahedron 2009, 65, 3042–3051.Zalatan DN; Du Bois J Understanding the Differential Performance of Rh2(esp)2 as a Catalyst for C–H Amination. J. Am. Chem. Soc. 2009, 131, 7558–7559. Liang C; Collet F; Robert-Peillard F; Müller P; Dodd RH; Dauban P Toward a Synthetically Useful Stereoselective C–H Amination of Hydrocarbons. J. Am. Chem. Soc. 2008, 130, 343–350. Liang C; Robert-Peillard F; Fruit C; Müller P; Dodd RH; Dauban P Efficient Diastereoselective Intermolecular Rhodium-Catalyzed C-H Amination. Angew. Chem., Int. Ed. 2006, 45, 4641–4644.Nägeli I; Baud C; Bernardinelli G; Jacquier Y; Moran M; Müller P Rhodium(II)-Catalyzed CH Insertions with {[(4-Nitrophenyl)-sulfonyl]imino}phenyl-λ3-iodane. Helv. Chim. Acta 1997, 80, 1087–1105.Huard K; Lebel H N-Tosyloxycarbamates as Reagents in Rhodium-Catalyzed C–H Amination Reactions. Chem.—Eur. J. 2008, 14, 6222–6230. Lebel H; Huard K; Lectard S N-Tosyloxycarbamates as a Source of Metal Nitrenes: Rhodium-Catalyzed C–H Insertion and Aziridination Reactions. J. Am. Chem. Soc. 2005, 127, 14198–14199. Reddy RP; Davies HML Dirhodium Tetracarboxylates Derived from Adamantylglycine as Chiral Catalysts for Enantioselective C–H Aminations. Org. Lett. 2006, 8, 5013–5016. Nguyen Q; Sun K; Driver TG Rh2(II)-Catalyzed Intramolecular Aliphatic C–H Bond Amination Reactions Using Aryl Azides as the N-Atom Source. J. Am. Chem. Soc. 2012, 134, 7262–7265. Jat JL; Paudyal MP; Gao H; Xu Q-L; Yousufuddin M; Devarajan D; Ess DH; Kürti L; Falck JR Direct Stereospecific Synthesis of Unprotected N-H and N-Me Aziridines from Olefins. Science 2014, 343, 61–65. Ma Z; Zhou Z; Kürti L Direct and Stereospecific Synthesis of N-H and N-Alkyl Aziridines from Unactivated Olefins Using Hydroxylamine-O-Sulfonic Acids. Angew. Chem., Int. Ed. 2017, 56, 9886–9890.
- (18).Selective examples of typical Cu-centered C–N Bond Formation: Aguila MJB; Badiei YM; Warren TH Mechanistic Insights into C–H Amination via Dicopper Nitrenes. J. Am. Chem. Soc. 2013, 135, 9399–9406. Badiei YM; Dinescu A; Dai X; Palomino RM; Heinemann FW; Cundari TR; Warren TH Copper–Nitrene Complexes in Catalytic C–H Amination. Angew. Chem., Int. Ed. 2008, 47, 9961–9964.Barman DN; Liu P; Houk KN; Nicholas KM On the Mechanism of Ligand-Assisted, Copper-Catalyzed Benzylic Amination by Chloramine-T. Organometallics 2010, 29, 3404–3412.Barman D; Nicholas KM Ligand-assisted, copper-catalyzed enantioselective benzylic amination. Tetrahedron Lett. 2010, 51, 1815–1818.Fructos MR; Trofimenko S; Díaz-Requejo MM; Pérez PJ Facile Amine Formation by Intermolecular Catalytic Amidation of Carbon–Hydrogen Bonds. J. Am. Chem. Soc. 2006, 128, 11784–11791. Gómez-Emeterio BP; Urbano J; Díaz-Requejo MM; Pérez PJ Easy Alkane Catalytic Functionalization. Organometallics 2008, 27, 4126–4130.Díaz-Requejo MM; Pérez PJ; Brookhart M; Templeton JL Substituent Effects on the Reaction Rates of Copper-Catalyzed Cyclopropanation and Aziridination of para-Substituted Styrenes. Organometallics 1997, 16, 4399–4402.Lebel H; Parmentier M Copper-catalyzed enantioselective aziridination of styrenes. Pure Appl. Chem. 2010, 82, 1827–1833.Lebel H; Lectard S; Parmentier M Copper-Catalyzed Alkene Aziridination with N-Tosyloxycarbamates. Org. Lett. 2007, 9, 4797–4800. Powell DA; Fan H Copper-Catalyzed Amination of Primary Benzylic C–H Bonds with Primary and Secondary Sulfonamides. J. Org. Chem. 2010, 75, 2726–2729. Pelletier G; Powell DA Copper-Catalyzed Amidation of Allylic and Benzylic C-H Bonds. Org. Lett. 2006, 8, 6031–6034. Comba P; Haaf C; Lienke A; Muruganantham A; Wadepohl H Synthesis, Structure, and Highly Efficient Copper-Catalyzed Aziridination with a Tetraaza-Bispidine Ligand. Chem.—Eur. J. 2009, 15, 10880–10887. Vedernikov AN; Caulton KG Facile alkane functionalization in copper-[2.1.1]-(2,6)-pyridinophane-PhINTs systems. Chem. Commun. 2004, 162–163.Evans DA; Faul MM; Bilodeau MT Development of the Copper-Catalyzed Olefin Aziridination Reaction. J. Am. Chem. Soc. 1994, 116, 2742–2753.Li Z; Quan RW; Jacobsen EN Mechanism of the (Diimine)copper-Catalyzed Asymmetric Aziridination of Alkenes. Nitrene Transfer via Ligand-Accelerated Catalysis. J. Am. Chem. Soc. 1995, 117, 5889–5890.
- (19).(a) Paraskevopoulou P; Lin A; Wang Q; Pinnapareddy D; Acharrya R; Dinda R; Çelenligil-Çetin R; Floros G; Sanakis Y; Choudhury A; Rath NP; Stavropoulos P Synthesis and Characterization of a Series of Structurally and Electronically Diverse Fe(II) Complexes Featuring a Family of Triphenylamido-amine Ligands. Inorg. Chem. 2010, 49, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Çelenligil-Çetin R; Paraskevopoulou P; Dinda R; Staples RJ; Sinn E; Rath NP; Stavropoulos P Synthesis, Characterization, and Reactivity of Iron Trisamidoamine Complexes that Undergo both Metal- and Ligand-Centered Oxidative Transformations. Inorg. Chem. 2008, 47, 1165–1172. [DOI] [PubMed] [Google Scholar]; (c) Çelenligil-Çetin R; Paraskevopoulou P; Lalioti N; Sanakis Y; Staples RJ; Rath NP; Stavropoulos P Metalloradical Complexes of Manganese and Chromium Featuring an Oxidatively Rearranged Ligand. Inorg. Chem. 2008, 47, 10998–11009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Çelenligil-Çetin R; Paraskevopoulou P; Dinda R; Lalioti N; Sanakis Y; Rawashdeh AM; Staples RJ; Sinn E; Stavropoulos P Oxidative Ligand Rearrangement due to Incipient Aminyl Radicals in the Oxidation of Iron(II) Species with Dioxygen. Eur. J. Inorg. Chem. 2008, 2008, 673–677. [Google Scholar]
- (20).(a) Schrock RR Transition Metal Complexes that Contain a Triamidoamine Ligand. Acc. Chem. Res. 1997, 30, 9–16. [Google Scholar]; (b) Verkade JG Atranes: New Examples with Unexpected Properties. Acc. Chem. Res. 1993, 26, 483–489. [Google Scholar]
- (21).(a) Borovik AS Bio-Inspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc. Chem. Res. 2005, 38, 54–61. [DOI] [PubMed] [Google Scholar]; (b) Gupta R; Borovik AS Monomeric MnIII/II and FeIII/II Complexes with Terminal Hydroxo and Oxo Ligands: Probing Reactivity via O–H Bond Dissociation Energies. J. Am. Chem. Soc. 2003, 125, 13234–13242. [DOI] [PubMed] [Google Scholar]; (c) MacBeth CE; Golombek AP; Young VG Jr.; Yang C; Kuczera K; Hendrich MP; Borovik AS O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)–Oxo Complex Derived From O2. Science 2000, 289, 938–941. [DOI] [PubMed] [Google Scholar]
- (22).(a) Freundlich JS; Schrock RR; Davis WM Synthetic and Mechanistic Investigations of Trimethylsilyl-Substituted Triamidoamine Complexes of Tantalum That Contain Metal-Ligand Multiple Bonds. J. Am. Chem. Soc. 1996, 118, 3643–3655. [Google Scholar]; (b) Yandulov DV; Schrock RR Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]; (c) Yandulov DV; Schrock RR Reduction of Dinitrogen to Ammonia at a Well-Protected Reaction Site in a Molybdenum Triamidoamine Complex. J. Am. Chem. Soc. 2002, 124, 6252–6253. [DOI] [PubMed] [Google Scholar]; (d) Greco GE; Schrock RR Synthesis of Triamidoamine Ligands of the Type (ArylNHCH2CH2)3N and Molybdenum and Tungsten Complexes That Contain an [(ArylNCH2CH2)3N]3− Ligand . Inorg. Chem. 2001, 40, 3850–3860. [DOI] [PubMed] [Google Scholar]; (e) Greco GE; Schrock RR Synthesis, Structure, and Electrochemical Studies of Molybdenum and Tungsten Dinitrogen, Diazenido, and Hydrazido Complexes that Contain Aryl-Substituted Triamidoamine Ligands. Inorg. Chem. 2001, 40, 3861–3878. [DOI] [PubMed] [Google Scholar]; (f) Cummins CC; Schrock RR Synthesis of an Iron(IV) Cyanide Complex that Contains the Triamido Amine Ligand [(t-BuMe2SiNCH2CH2)3N]3−. Inorg. Chem. 1994, 33, 395–396. [Google Scholar]
- (23).(a) Villanueva O; Weldy NM; Blakey SB; MacBeth CE Cobalt catalyzed sp3 C–H amination utilizing aryl azides. Chem. Sci. 2015, 6, 6672–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Corcos AR; Villanueva O; Walroth RC; Sharma SK; Basca J; Lancaster KM; MacBeth CE; Berry JF Oxygen Activation by Co(II) and a Redox Non-Innocent Ligand: Spectroscopic Characterization of a Radical–Co(II)–Superoxide Complex with Divergent Catalytic Reactivity. J. Am. Chem. Soc. 2016, 138, 1796–1799. [DOI] [PubMed] [Google Scholar]; (c) Krogman JP; Thomas CM Metal-metal multiple bonding in C3-symmetric bimetallic complexes of the first row transition metals. Chem. Commun. 2014, 50, 5115–5127. [DOI] [PubMed] [Google Scholar]; (d) Cammarota RC; Vollmer MV; Xie J; Ye J; Linehan JC; Burgess SA; Appel AM; Gagliardi L; Lu CC A Bimetallic Nickel–Gallium Complex Catalyzes CO2 Hydrogenation via the Intermediacy of an Anionic d10 Nickel Hydride. J. Am. Chem. Soc. 2017, 139, 14244–14250. [DOI] [PubMed] [Google Scholar]
- (24).(a) Kalra A; Bagchi V; Paraskevopoulou P; Das P; Ai L; Sanakis Y; Raptopoulos G; Mohapatra S; Choudhury A; Sun Z; Cundari TR; Stavropoulos P Is the Electrophilicity of the Metal Nitrene the Sole Predictor of Metal-Mediated Nitrene Transfer to Olefins? Secondary Contributing Factors as Revealed by a Library of High-Spin Co(II) Reagents. Organometallics 2021, 40, 1974–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bagchi V; Kalra A; Das P; Paraskevopoulou P; Gorla S; Ai L; Wang Q; Mohapatra S; Choudhury A; Sun Z; Cundari TR; Stavropoulos P Comparative Nitrene-Transfer Chemistry to Olefinic Substrates Mediated by a Library of Anionic Mn(II) Triphenylamido-Amine Reagents and M(II) Congeners (M = Fe, Co, Ni) Favoring Aromatic over Aliphatic Alkenes. ACS Catal. 2018, 8, 9183–9206. [Google Scholar]
- (25).(a) Wittman H; Raab V; Schorm A; Plackmeyer J; Sundermeyer J Complexes of Manganese, Iron, Zinc, and Molybdenum with a Superbasic Tris(guanidine) Derivative of Tris(2-ethylamino)amine (Tren) as a Tripod Ligand. Eur. J. Inorg. Chem. 2001, 1937–1948. [Google Scholar]; (b) Stanek J; Rösener T; Metz A; Mannsperger J; Hoffmann A; Herres-Pawlis S Guanidine Metal Complexes for Bioinorganic Chemistry and Polymerisation Catalysis. Top. Heterocycl. Chem. 2015, 51, 95–164. [Google Scholar]; (c) Bienemann O; Hoffman A; Herres-Pawlis S (Guanidine)copper complexes: structural variety and applications in bioinorganic chemistry and catalysis. Rev. Inorg. Chem. 2011, 31, 83–108. [Google Scholar]; (d) Cui X-Y; Tan C-H; Leow D Metal-catalysed reactions enabled by guanidine-type ligands. Org. Biomol. Chem. 2019, 17, 4689–4699. [DOI] [PubMed] [Google Scholar]
- (26).(a) Malik DD; Chandra A; Seo MS; Lee Y-M; Farquhar ER; Mebs S; Dau H; Ray K; Nam W Formation of cobalt-oxygen intermediates by dioxygen activation at a mononuclear nonheme cobalt(II) center. Dalton Trans. 2021, 50, 11889–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Comba P; Löhr A-M; Pfaff F; Ray K Redox Potentials of High-Valent Iron-, Cobalt-, and Nickel-Oxido Complexes: Evidence for Exchange Enhanced Reactivity. Isr. J. Chem. 2020, 60, 957–962. [Google Scholar]; (c) Liu JJ; Siegler MA; Karlin KD; Moënne-Loccoz P Direct Resonance Raman Characterization of a Peroxynitrito Copper Complex Generated from O2 and NO and Mechanistic Insights into Metal-Mediated Peroxynitrite Decomposition. Angew. Chem., Int. Ed. 2019, 58, 10936–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Speelman AL; White CJ; Zhang B; Alp EE; Zhao J; Hu M; Krebs C; Penner-Hahn J; Lehnert N Non-heme High-Spin {FeNO}6–8 Complexes: One Ligand Platform Can Do it All. J. Am. Chem. Soc. 2018, 140, 11341–11359. [DOI] [PubMed] [Google Scholar]; (e) Saracini C; Liakos DG; Rivera JEZ; Neese F; Meyer GJ; Karlin KD Excitation Wavelength Dependent O2 Release from Copper(II)–Superoxide Compounds: Laser Flash-Photolysis Experiments and Theoretical Studies. J. Am. Chem. Soc. 2014, 136, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Peterson RL; Ginsbach JW; Cowley RE; Qayyum MF; Himes RA; Siegler MA; Moore CD; Hedman B; Hodgson KO; Fukuzumi S; Solomon EI; Karlin KD Stepwise Protonation and Electron-Transfer Reduction of a Primary Copper–Dioxygen Adduct. J. Am. Chem. Soc. 2013, 135, 16454–16467. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Pfaff FF; Kundu S; Risch M; Pandian S; Heims F; Pryjomska-Ray I; Haack P; Metzinger R; Bill E; Dau H; Comba P; Ray K An Oxocobalt(IV) Complex Stabilized by Lewis Acid Interactions with Scandium(III) Ions. Angew. Chem., Int. Ed. 2011, 50, 1711–1715. [DOI] [PubMed] [Google Scholar]; (h) England J; Guo Y; Van Heuvelen KM; Cranswick MA; Rohde GT; Bominaar EL; Münck E; Que L Jr. A More Reactive Trigonal-Bipyramidal High-Spin Oxoiron(IV) Complex with a cis-Labile Site. J. Am. Chem. Soc. 2011, 133, 11880–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bagchi V; Paraskevopoulou P; Das P; Chi L; Wang Q; Choudhury A; Mathieson JS; Cronin L; Pardue DB; Cundari TR; Mitrikas G; Sanakis Y; Stavropoulos P A versatile Tripodal Cu(I) Reagent for C–N Bond Construction via Nitrene-Transfer Chemistry: Catalytic Perspectives and Mechanistic Insights on C–H Aminations/Amidinations and Olefin Aziridinations. J. Am. Chem. Soc. 2014, 136, 11362–11381. [DOI] [PubMed] [Google Scholar]
- (28).Sahoo SK; Harfmann B; Ai L; Wang Q; Mohapatra S; Choudhury A; Stavropoulos P Cationic Divalent Metal Sites (M = Mn, Fe, Co) Operating as Both Nitrene-Transfer Agents and Lewis Acids toward Mediating the Synthesis of Three- and Five-Membered N-Heterocycles. Inorg. Chem. 2023, 62, 10743–10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).You D; Gabbaï FP Tunable σ-Accepting, Z-Type Ligands for Organometallic Catalysis. Trends Chem. 2019, 1, 485–496. [Google Scholar]
- (30).Bernasconi L; Louwerse MJ; Baerends EJ The Role of Equatorial and Axial Ligands in Promoting the Activity of Non-Heme Oxidoiron(IV) Catalysts in Alkane Hydroxylation. Eur. J. Inorg. Chem. 2007, 2007, 3023–3033. [Google Scholar]
- (31).Liang H-C; Kim E; Incarvito CD; Rheingold AL; Karlin KD A Bis-Acetonitrile Two-Coordinate Copper(I) Complex: Synthesis and Characterization of Highly Soluble B(C6F5)4− Salts of [Cu(MeCN)2]+ and [Cu(MeCN)4]+. Inorg. Chem. 2002, 41, 2209–2212. [DOI] [PubMed] [Google Scholar]
- (32).(a) Sletten ET; Tu Y-J; Schlegel HB; Nguyen HM Are Brønsted Acids the True Promoter of Metal-Triflate-Catalyzed Glycosylations? A Mechanistic Probe into 1,2-cis-Aminoglycoside Formation by Nickel Triflate. ACS Catal. 2019, 9, 2110–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen J; Goforth SK; McKeown BA; Gunnoe TB Brønsted acid-catalysed intramolecular hydroamination of unactivated alkenes: metal triflates as an in situ source of triflic acid. Dalton Trans. 2017, 46, 2884–2891. [DOI] [PubMed] [Google Scholar]; (c) Tschan MJ-L; Thomas CM; Strub H; Carpentier J-F Copper(II) Triflate as a Source of Triflic Acid: Effective, Green Catalysis of Hydroalkoxylation Reactions. Adv. Synth. Catal. 2009, 351, 2496–2504. [Google Scholar]; (d) Rosenfeld DC; Shekhar S; Takemiya A; Utsunomiya M; Hartwig JF Hydroamination and Hydroalkoxylation Catalyzed by Triflic Acid. Parallels to Reactions Initiated with Metal Triflates. Org. Lett. 2006, 8, 4179–4182. [DOI] [PubMed] [Google Scholar]; (e) Dumeunier R; Markó IE On the role of triflic acid in the metal triflate-catalysed acylation of alcohols. Tetrahedron Lett. 2004, 45, 825–829. [Google Scholar]
- (33).Ke I-S; Gabbaï FP Cu3(μ2-Cl)3 and Ag3(μ2-Cl)3 Complexes Supported by Tetradentate Trisphosphino-stibine and -bismuthine Ligands: Structural Evidence for Triply Bridging Heavy Pnictines. Aust. J. Chem. 2013, 66, 1281–1287. [Google Scholar]
- (34).Baranov AY; Pritchina EA; Berezin AS; Samsonenko DG; Fedin VP; Belogorlova NA; Gritsan NP; Artem′ev AV Beyond Classical Coordination Chemistry: The First Case of a Triply Bridging Phosphine Ligand. Angew. Chem., Int. Ed. 2021, 60, 12577–12584. [DOI] [PubMed] [Google Scholar]
- (35).Hargittai M; Schwerdtfeger P; Réffy B; Brown R The Molecular Structure of Different Species of Cuprous Chloride from Gas-Phase Electron Diffraction and Quantum Chemical Calculations. Chem.—Eur. J. 2003, 9, 327–333. [DOI] [PubMed] [Google Scholar]
- (36).Cordero B; Gómez V; Platero-Prats AE; Revés M; Echeverría J; Cremades E; Barragán F; Alvarez S Covalent radii revisited. Dalton Trans. 2008, 2832–2838. [DOI] [PubMed] [Google Scholar]
- (37).(a) Zhang L; Li X-X; Lang Z-L; Liu Y; Liu J; Yuan L; Lu W-Y; Xia Y-S; Dong L-Z; Yuan D-Q; Lan Y-Q Enhanced Cuprophilic Interactions in Crystalline Catalysts Facilitate the Highly Selective Electroreduction of CO2 to CH4. J. Am. Chem. Soc. 2021, 143, 3808–3816. [DOI] [PubMed] [Google Scholar]; (b) Harisomayajula NVS; Makovetskyi S; Tsai Y-C Cuprophilic Interactions in and between Molecular Entities. Chem. - Eur. J. 2019, 25, 8936–8954. [DOI] [PubMed] [Google Scholar]; (c) Singh K; Long JR; Stavropoulos P Ligand-Unsupported Metal–Metal (M = Cu, Ag) Interactions between Closed-Shell d10 Trinuclear Systems. J. Am. Chem. Soc. 1997, 119, 2942–2943. [Google Scholar]
- (38).Alvarez S A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [DOI] [PubMed] [Google Scholar]
- (39).Yang L; Powell DR; Houser RP Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- (40).Mantina M; Chamberlin AC; Valero R; Cramer CJ; Truhlar DG Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).García-Romero Á; Waters JE; Jethwa RB; Bond AD; Colebatch AL; García-Rodríguez R; Wright DS Inorg. Chem. 2023, 62, 4625–4636. [DOI] [PubMed] [Google Scholar]
- (42).Taylor WV; Cammack CX; Shubert SA; Rose MJ Thermoluminescent Antimony-Supported Copper-Iodo Cuboids: Approaching NIR Emission via High Crystallographic Symmetry. Inorg. Chem. 2019, 58, 16330–16345. [DOI] [PubMed] [Google Scholar]
- (43).(a) Mitoraj MP; Michalak A; Ziegler T A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [DOI] [PubMed] [Google Scholar]; (b) Ziegler T; Rauk A Carbon Monoxide, Carbon Monosulfide, Molecular Nitrogen, Phosphorus Trifluoride, and Methyl Isocyanide as σ Donors and π Acceptors. A Theoretical Study by the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar]; (c) Ziegler T; Rauk A A Theoretical Study of the Ethylene-Metal Bond in Complexes between Cu+, Ag+, Au+, Pt0 or Pt2+ and Ethylene, Based on the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar]
- (44).Lu T; Chen Q Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [DOI] [PubMed] [Google Scholar]
- (45).Greenacre VK; Levason W; Reid G Developments in the chemistry of stibine and bismuthine complexes. Coord. Chem. Rev. 2021, 432, 213698. [Google Scholar]
- (46).Al-Ajlouni A; Espenson JH Epoxidation of Styrenes by Hydrogen Peroxide As Catalyzed by Methylrhenium Trioxide. J. Am. Chem. Soc. 1995, 117, 9243–9250. [Google Scholar]
- (47).Müller P; Baud C; Jacquier Y; Moran M; Nägeli I Rhodium(II)-Catalyzed Aziridinations and CH Insertions with [N(p-Nitrobenzenesulfonyl)Imino]Phenyliodinane. J. Phys. Org. Chem. 1996, 9, 341–347. [Google Scholar]
- (48).Neuenschwander U; Hermans I The Conformations of Cyclooctene: Consequences for Epoxidation Chemistry. J. Org. Chem. 2011, 76, 10236–10240. [DOI] [PubMed] [Google Scholar]
- (49).Van Leest NP; Grooten L; van der Vlugt JI; de Bruin B Uncatalyzed Oxidative C–H Amination of 9,10-Dihydro-9-Heteroanthracenes: A Mechanistic Study. Chem.—Eur. J. 2019, 25, 5987–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Nam P-C; Nguyen MT; Chandra AK The C–H and α(C–X) Bond Dissociation Enthalpies of Toluene, C6H5-CH2X (X = F, Cl), and Their Substituted Derivatives: A DFT Study. J. Phys. Chem. A 2005, 109, 10342–10347. [DOI] [PubMed] [Google Scholar]
- (51).Rodríguez MR; Rodríguez AM; López-Resano S; Pericàs MA; Díaz-Requejo MM; Maseras F; Pérez PJ Non-innocent Role of the Halide Ligand in the Copper-Catalyzed Olefin Aziridination Reaction. ACS Catal. 2023, 13, 706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Harvey ME; Musaev D; Du Bois J A Diruthenium Catalyst for Selective, Intramolecular Allylic C–H Amination: Reaction Development and Mechanistic Insight Gained through Experiment and Theory. J. Am. Chem. Soc. 2011, 133, 17207–17216. [DOI] [PubMed] [Google Scholar]
- (53).Park SH; Kwak J; Shin K; Ryu J; Park Y; Chang S Mechanistic Studies of the Rhodium-Catalyzed Direct C–H Amination Reaction Using Azides as the Nitrogen Source. J. Am. Chem. Soc. 2014, 136, 2492–2502. [DOI] [PubMed] [Google Scholar]
- (54).(a) Wächtler E; Gericke R; Block T; Pöttgen R; Wagler J Trivalent Antimony as L-, X-, and Z-Type Ligand: The Full Set of Possible Coordination Modes in Pt–Sb Bonds. Inorg. Chem. 2020, 59, 15541–15552. [DOI] [PubMed] [Google Scholar]; (b) Benjamin SL; Krämer T; Levason W; Light ME; Macgregor SA; Reid G [Pd4(μ3-SbMe3)4(SbMe3)4]: A Pd(0) Tetrahedron with μ3-Bridging Trimethylantimony Ligands. J. Am. Chem. Soc. 2016, 138, 6964–6967. [DOI] [PubMed] [Google Scholar]
- (55).(a) Lin T-P; Ke I-S; Gabbaï FP σ-Accepting Properties of a Chlorobismuthine Ligand. Angew. Chem., Int. Ed. 2012, 51, 4985–4988. [DOI] [PubMed] [Google Scholar]; (b) Jones JS; Wade CR; Gabbaï FP Redox and Anion Exchange Chemistry of a Stibine–Nickel Complex: Writing the L, X, Z Ligand Alphabet with a Single Element. Angew. Chem., Int. Ed. 2014, 53, 8876–8879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.