

ABSTRACT
A novel series of compounds was designed and synthesized by combining the distal piperazine nitrogen of the antifungal ketoconazole (KTZ) with primary arylsulfonamides. The aim of this study is to present the basis for a new generation of Malassezia antifungal agents able to inhibit the enzyme lanosterol‐14α‐demethylase (CYP51; EC 1.14.13.70) as well as a newly emergent therapeutic target: carbonic anhydrases (CAs; EC 4.2.1.1). The final compounds showed effective interactions with the intended targets in vitro, as well as KTZ comparable minimum inhibitory concentrations on yeast strains of the Malassezia genus: Malassezia furfur ATCC 14521; Malassezia globosa ATCC MYA 4612; and Malassezia pachydermatis DSM 6172. Overall, the data obtained account for the reported compounds as promising antifungal candidates with high safety profiles for the management of fungal infections.
Keywords: 14α‐demethylase, antifungals, carbonic anhydrases, fungi, Malassezia
A series of ketoconazole‐based carbonic anhydrase (CA) inhibitors were synthesized and characterized, and their inhibitory activities were assessed both against human and fungal CA. The novel compounds showed antifungal activity and minimal inhibitory concentrations comparable to the reference standard ketoconazole, with higher safety profiles when tested on human keratinocytes at maximal dosages.

1. Introduction
Malassezia species (spp.) are high lipophilic yeasts symbiotically present as part of the natural microbiota on the skin of mammals particularly rich in sebum (i.e., in humans, the scalp, face, and chest among others) [1]. Any physical or physiological cause that disrupts the microbioma equilibrium may lead to Malassezia spp. overgrowth, which in turn triggers acute infections such as pityriasis versicolor, seborrheic dermatitis, dandruff, and folliculitis, among others [2, 3]. Such diseases may spread over the host surface as well as deeper within tissues with significant systemic effects when adequate pharmacological treatments are absent [2, 3]. Malassezia spp., such as Malassezia furfur, Malassezia globosa, Malassezia restricta, and Malassezia sympodialis, are the most common yeasts on human skin [3]. In animals, Malassezia pachydermatis is the most represented yeast in the cutaneous mycobiome, both as a commensal and as a pathogen, and its overgrowth is related to the onset of severe dermatitis and otitis externa in companion animals, mainly dogs and cats, and therefore assumes substantial clinical significance [4, 5]. The standard of care treatment of Malassezia‐promoted infections largely depends on the specific clinical conditions, but more in general, any pharmacological approach accounts for the use of antifungal agents targeting the etiological yeast [1, 6, 7]. Representative drugs used for human and veterinary purposes are azole‐containing derivatives shown in Figure 1, which are quite effective in inhibiting the cytochrome P450 14α‐demethylase enzyme (CYP51; EC 1.14.13.70). As a result, such compounds affect the conversion of lanosterol into ergosterol and compromise the specific lipidomic composition in yeast biomembranes.
Figure 1.

Chemical structures of drugs used for the management of Malassezia spp.‐promoted infections. * denotes chiral centers.
Compounds such as ketoconazole (KTZ) and itraconazole (ITZ) contain multiple stereogenic centers and are therefore associated with extra costs of production and quality assurance. For instance, in KTZ and ITZ, the triazolomethylene and aryloxymethylene moieties anchored to the dioxolane ring are always fixed in cis relation to each another, thus yielding the clinical formulation cis‐(2S,4 R)‐(‐) and cis‐(2 R,4S)‐(+) enantiomers in a 1/1 ratio for the former [8] and cis‐(2 R, 4S, 2′R), cis‐(2 R, 4S, 2′S), cis‐(2S, 4 R, 2′R), and cis‐(2S, 4 R, 2′S) in a1/1/1/1 ratio of four stereoisomers (two enantiomeric pairs) for the latter [9]. In addition, the clinical effectiveness of azole antifungals is constantly eroded by the long latency time necessary to establish reliable therapeutic responses, which, however, does not cope with the high variability of genomes and quick reproduction rates typical of fungi/yeasts [10, 11]. As a result, azole resistance leads to the constant growth of fungal‐promoted infections in humans, animals, and plants and governs the need for more effective therapeutic tools [12]. In this study, we sought to make use of our know‐how relative to the validation of the fungal‐expressed metalloenzyme carbonic anhydrases (CAs, E.C. 4.2.1.1) target [13, 14, 15, 16, 17] by merging the prototypic primary sulfonamide CA inhibitory (CAI) moiety with a clinically used azole drug. The primary aim of our work is to explore the chemical feasibility of our strategy and to prove antifungal effectiveness, also in comparison to the parent drugs, of our final compounds. Specifically, we focused our attention on the chemically modifiable KTZ drug by adopting standard medicinal chemistry derivatization strategies.
2. Results and Discussion
2.1. Design and Synthesis of Compounds
We aimed to develop an efficient synthetic approach for obtaining KTZ‐based CAI compounds through elongation of the antifungal molecular structure. For instance, we sought to make use of the unprotected piperazine distal nitrogen to establish covalent bonds with a variety of scaffolds incorporating the prototypic CAI moiety of the primary sulfonamide type [18]. For such purposes, the commercially available cis‐(2 R,4S)‐( + ) KTZ 1 enantiomer was deacetylated under basic conditions to afford the valuable synthetic intermediate 2 in a 91% yield on a multigram gram‐scale reaction (Scheme 1).
Scheme 1.

Synthesis of the reactive ketoconazole intermediate 2.
All the final compounds reported in this study were obtained by direct coupling of 2 with ad hoc and freshly synthesized intermediates as reported in Schemes 2, 3, 4, 3. (N.B. The synthesis and characterization of intermediates are reported in the Supporting Information file). In particular, the ureidic connection to afford 7, 9a–d, and 10a–b was obtained by nucleophilic additions of 2 with the appropriate phenyl carbamates 3, 5a–d, and 6a–b, respectively. On the other hand, the thioureido moiety in 24a–d, 25, and 26 was positioned by reacting 2 with the corresponding isothiocyanates 20a–d, 21, and 22, respectively (Schemes 2, 3).
Scheme 2.

(A, B) Synthesis of the ketoconazole derivatives 7, 9a–d, 10a–b, 24a–d, 25, and 26.
Scheme 3.

Synthesis of intermediates 12 and 27, and of the final compound 28.
Scheme 4.

(A, B) Synthesis of the ketoconazole derivatives 35a–c, 36a–c, 37a–c, 38, 39, 48a–d, and 49a–d.
Figure 3.

Viability assays on human keratinocytes HaCaT cells expressed as % survival ± SD % at three different concentrations (256.0, 16.0, and 0.5 µg/mL) and ketoconazole (KTZ) as the reference compound. NC, negative control (Y = 77%) and TR, threshold of survival (Y = 65%).
Additionally, we explored the synthetic feasibility of benzoxazolyl‐containing compounds with the intent of elongating the CAI‐directed warhead by means of a valuable heteroaromatic scaffold. At first, intramolecular cyclization of the commercially available ortho‐amino phenol 11 to afford the key derivative 2‐oxo‐benzoxazolyl 12 was operated, which in turn was exposed to carbon disulfide (CS2) reaction conditions with the intent of positioning the reactive 2‐thioxo‐moiety as in 27 (Scheme 3).
Although thionation attempts on 12 were unsuccessful, to our surprise, the desired intermediate 27 was obtained in a 64% yield by direct treatment of 11 with CS2 and N‐(3‐dimetilaminopropyl)‐N′‐etilcarbodiimide hydrochloride (EDC. HCl). Then, the final compound 28 was obtained in a 26% yield by the addition of 2 on 27 under basic conditions for triethylamine (Et3N).
The nucleophilic features of 2 toward the halo‐substituted benzyl/alkyl derivatives 30a–c, 31a–c, 32a–c, 33, and 34 yielded the final 35a–c, 36a–c, 37a–c, 38, and 39, respectively, as shown in Scheme 4A.
The piperazine moiety in 2 was successfully investigated to perform 1,4‐conjugate additions on acrylate intermediates 43a–b, 44a–d, and 45a–d to yield 37b–c, 48a–d, and 49a–d, respectively (Scheme 4B). It is noteworthy that derivatives 37b and 37c were obtained through both synthetic strategies in Scheme 4A,B with comparable recovery yields. Unfortunately, it was not feasible to obtain 50 under the various reaction conditions explored.
All the final KTZ‐CAIs compounds reported herein were purified by silica gel column chromatography using the appropriate eluting mixtures, followed by trituration or recrystallization as needed (see Section 4). Full characterization was conducted by means of solution 1H‐ and 13C‐NMR. Elemental analyses account for a purity grade ≥ 96%.
2.2. CA In Vitro Enzymatic Assay
All the final compounds were screened in vitro using the stopped‐flow CO2 hydration assay [19] to assess their inhibitory activity and selectivity profiles on the fungal β‐CAs expressed from the Malassezia globosa (MgCA), Malassezia pachydermatis (MpaCA), and Malassezia restricta (MreCA) genus. Human (h) CAs I, II, IX, and XII were assumed to be off‐target isoforms. All the obtained data are reported in Table 1 as K I values and are compared to the primary sulfonamide drug acetazolamide (AAZ) as the reference compound.
Table 1.
Inhibition data of compounds 7, 9a–d, 10a–b, 24a–d, 25, 26, 28, 35a–c, 36a–c, 37a–c, 38, 39, 48a–d, and 49a–d on fungal MgCA, MpaCA, MreCA, hCAs I, II, IX, and XII. AAZ was considered as the reference drug [19].
| K I (nM)a | |||||||
|---|---|---|---|---|---|---|---|
| Cmp | MgCA | MpaCA | MreCA | hCA I | hCA II | hCA IX | hCA XII |
| 7 | 69497 | 513.7 | 206.2 | 5912 | 5847 | 4.4 | 63.1 |
| 9a | 43809 | 601.9 | 90.5 | 1622 | 1864 | 15.1 | 39.6 |
| 9b | 5489 | 516.7 | 613.9 | 337.6 | 71.8 | 32.9 | 8.2 |
| 9c | 5633 | 821.2 | 683.9 | 437.2 | 58.9 | 13.9 | 6.4 |
| 9d | 3333 | 291.6 | 281.7 | 84.0 | 563.4 | 6.0 | 9.5 |
| 10a | 7578 | 4202 | 892.6 | 870.2 | 390.0 | 38.0 | 44.0 |
| 10b | 5045 | 702.1 | 360.6 | 896.5 | 533.9 | 41.2 | 7.4 |
| 24a | 7000 | 4944 | 339.2 | 862.9 | 605.0 | 47.7 | 72.5 |
| 24b | 6866 | 841.7 | 462.7 | 871.7 | 258.0 | 48.7 | 9.2 |
| 24c | 8405 | 604.2 | 924.5 | 328.2 | 392.9 | 40.1 | 8.0 |
| 24d | 4482 | 610.9 | 825.2 | 861.0 | 736.6 | 4.4 | 8.2 |
| 25 | 6058 | 930.7 | 620.6 | 871.5 | 882.6 | 41.0 | 9.0 |
| 26 | 6027 | 2390 | 338.2 | 58.3 | 17.3 | 151.8 | 62.3 |
| 28 | 4570 | 380.9 | 680.8 | 944.0 | 8.3 | 28.3 | 47.2 |
| 35a | 4249 | 803.3 | 683.6 | 352.5 | 26.5 | 46.8 | 8.2 |
| 35b | 4604 | 695.0 | 701.7 | 4767 | 620.0 | 368.6 | 64.4 |
| 35c | 4473 | 646.6 | 318.3 | 5515 | 55.7 | 47.9 | 7.5 |
| 36a | 5429 | 577.0 | 288.2 | 2662 | 180.0 | 311.5 | 5.9 |
| 36b | 4062 | 559.7 | 451.1 | 3418 | 1196 | 41.9 | 6.2 |
| 36c | 6124 | 677.9 | 815.8 | 2986 | 938.8 | 43.9 | 8.1 |
| 37a | 5344 | 5931 | 398.2 | 497.4 | 21.4 | 346.3 | 61.0 |
| 37b | 6484 | 891.7 | 691.9 | 9238 | 798.6 | 49.1 | 9.1 |
| 37c | 7435 | 1927 | 280.2 | 4351 | 92.6 | 331.4 | 9.4 |
| 38 | 5810 | 553.8 | 473.1 | 382.0 | 122.0 | 224.8 | 9.3 |
| 39 | 5538 | 627.8 | 390.5 | 817.8 | 77.1 | 76.0 | 6.3 |
| 48a | 5562 | 679.4 | 403.5 | 82.1 | 49.1 | 30.6 | 3.7 |
| 48b | 6479 | 830.4 | 472.4 | 769.7 | 20.7 | 48.4 | 31.1 |
| 48c | 7208 | 721.5 | 416.6 | 835.8 | 167.9 | 445.7 | 9.4 |
| 48d | 8036 | 630.3 | 471.4 | 363.9 | 44.6 | 306.3 | 56.1 |
| 49a | 56446 | 459.3 | 410.9 | 315.1 | 288.5 | 13.8 | 5.5 |
| 49b | 6855 | 733.0 | 337.3 | 96.1 | 195.6 | 272.1 | 67.9 |
| 49c | 8184 | 877.6 | 470.2 | 5.3 | 7.2 | 284.6 | 7.9 |
| 49d | 4391 | 2583 | 767.9 | 777.0 | 247.4 | 2167 | 55.9 |
| AAZ | 40000 | 620.0 | 100.0 | 250.0 | 12.1 | 25.8 | 5.7 |
Mean from three different assays, using the stopped flow technique (errors were in the range of ± 5–10% of the reported values).
Overall, the data in Table 1 accounted for the hCA isoforms preferentially inhibited over the fungal ones from the panel of compounds reported in this study.
-
i.
Deeper insight revealed that ureido 7, 9a–d, and 10a–b were slightly active toward MgCA, with K Is spanning between 3333 and 69497 nM. Conversely, the affinity for the remaining fungal β‐CAs was increased up to medium nanomolar values. For instance, K Is for the MpaCA ranged between 291.6 and 4202 nM. A slight regioisomeric effect was observed for the 9a/9b regioisomeric pair, with the 4‐substituted being a 1.2‐fold more effective inhibitor and almost equipotent to derivative 7 (i.e., K Is of 513.7 and 516.7 nM for 7 and 9b, respectively). Elongation of the alkyl chain in 9b up to two carbon atoms (i.e., 9d) was beneficial for the inhibition potency, as the lowest inhibition value was obtained for the MpaCA among the ureido‐containing compounds (i.e., K I of 291.6 nM for 9d). Significant regioisomeric effects on the kinetics were reported for the ureido derivatives 10a and 10b, respectively, with the latter being a 6.0‐fold more effective MpaCA inhibitor when compared with its pair (K Is of 4202 and 702.1 nM for 10a and 10b, respectively). Similar values were obtained for the MreCA isoform (i.e., K Is between 206.2 and 892.6 nM). A strong regioisomeric effect was reported for 9a, which inhibited the MreCA at a 90.5 nM concentration, and was thus 6.8‐fold stronger than its 4‐substituted counterpart 9b (K I of 613.9 nM). Replacement of the ureido moiety in 9a–d with the thiourea instead, to afford 24a–d, did not significantly affect the in vitro kinetic profiles for the fungal CAs. Again, MgCA was the least inhibited, with K I values ranging between 4482 and 8405 nM (Table 1). As for the MpaCA, a very slight regioisometic effect was observed for 24c and 24d (K Is of 604.2 and 610.9 nM, respectively), whereas elongation of the tether was far more evident on the kinetics as reported for 24a, b, and d (K Is of 4944, 841.7, and 610.9 nM for 24a, b, and d respectively). A similar trend for 24a–d was also reported on the MreCA, with K I values spanning between 339.2 and 924.5 nM (Table 1). Further elongation of the scaffold as in compounds 25 and 26 yielded K I values for the fungal‐expressed CAs in agreement with the compounds mentioned previously. Data in Table 1 clearly show that MgCA was poorly inhibited from both 25 and 26, with K I values of 6058 and 6027 nM, respectively. The same compounds showed good selectivity for the remaining fungal CAs. For instance, 25 was 2.6‐fold more potent than 26 on the MpaCA (K Is of 930.7 and 2390 nM, respectively), whereas the opposite trend, although with lower intensity, was observed for the MreCA (K Is of 620.6 and 338.2 nM, respectively). As for the benzoxazolyl‐containing 28, preferential inhibition for the MpaCA isoform over Mg‐ and MreCAs was reported (K Is of 380.9, 4750, and 680.8 nM, respectively). Close inhibition potencies were obtained for derivatives 35a–c on MgCA and were between 4604 and 4473 nM (Table 1). Elongation of the alkyl chain in 35a–c yielded a progressive increase in the compound's effectiveness for the MpaCA isoform (i.e., K Is of 803.3, 695.0, and 646.6 nM for 35a–c, respectively). Marked effects on kinetics were reported for the same compounds on the MreCA, as the longest derivative 35c was 2.1‐fold more effective than its progenitor 35a. A close matching trend was also obtained on comparison of 35c with 35b (K Is of 701.7 and 318.3 nM for 35b and 35c, respectively). The introduction in 35a–c of a sulfonyl‐based connection as in 36a–c induced high variability among the kinetic data in Table 1, although the preferential inhibition for the Mpa‐ and MreCAs was retained. Derivative 36b was the most effective inhibitor for MgCA, with a K I value of 4062 nM, thus being 1.3‐ and 1.5‐fold more potent than 36a and 36c, respectively (i.e., K Is of 5429 and 6124 nM for 36a and 36c, respectively). An identical kinetic trend was reported for the MpaCA isoform, although with limited differences (K Is of 577.0, 559.7, and 677.9 nM for 36a–c, respectively). As for the MreCA, compound 36a was the most effective, with a K I value of 288.2 nM. Derivatives 36b and 36c showed K Is of 451.1 and 815.8 nM, respectively (Table 1). Among amido derivatives 37a–c, high kinetic variability was reported. For instance, MgCA was inhibited in a concentration range spanning between 7435 and 5344 nM. Better results were obtained for the remaining fungal‐expressed MpaCA and MreCA isoforms. Elongation of derivative 37a to afford 37b enhanced the inhibition potency by up to 6.7‐fold (i.e., K Is of 5391 and 891.7 nM, respectively). A strong regioisomeric effect was observed among 37b and 37c (i.e., K Is of 891.7 and 1927 nM for 37b and 37c, respectively). As for the MreCA, elongation of the chain significantly reduced the inhibition potency by 1.7‐fold (i.e., K Is of 398.2 and 691.9 nM for 37a and 37b, respectively). Also, for this isoform, a strong regioisomeric effect was reported, as compound 37c was 2.5‐fold more effective inhibitor than its isomer 37b (i.e., K Is of 691.9 and 280.2 nM for 37b and 37c, respectively). Both derivatives 38 and 39 showed similar kinetic profiles for the fungal‐expressed CAs and were all in agreement with the previously discussed compounds. Methoxy 39 was the most effective inhibitor for MreCA, with a K I value of 390.5 nM (Table 1). Derivatives 48a–d showed very few differences within each isoform. For instance, MgCA was inhibited in a concentration range of 5562–8036 nM, MpaCA was inhibited in a concentration range of 630.3–830.4 nM, and MreCA was inhibited in a concentration range of 403.5–472.4 nM, thus indicating that the substituents incorporated did not significantly affect the binding of the ligand within the catalytic cleft (Table 1). Quite interestingly, introduction of the 1,4‐disubstituted diazepane moiety as in 49a–d yielded better discrimination of the K I values among the series (Table 1). Desymmetrization of the CAI‐directed phenyl ring in 49a to yield the 3‐chlorophenyl derivative 49b led to an 8.2‐fold increase in the K I associated value for the MgCA isoform (i.e., K Is of 56446 and 6855 nM for 49a and 49b, respectively). The inhibition potency for MgCA decreased slightly by up to 1.2‐fold by substitution of the chloro in 49b with a fluorine instead to afford derivative 49c (i.e., K Is of 6855 and 8184 nM for 49b and 49c, respectively). Further improvement of the inhibition potency against MgCA was reported for the 3,5‐difluorosubstituted derivative 49d, which showed a K I value of 4391 nM (Table 1). A different kinetic trend was reported for MpaCA, with 49a being the most effective inhibitor, with a K I value of 459.3 nM. All further manipulations on 49a to afford 49b–d induced a sequential increase of the MpaCA associated K I values of up to 2583 nM (Table 1). As for MreCA, introduction in 49a of chloro (49b) and fluoro (49c) halogens yielded opposite and equivalent effects on the ligand's inhibition potencies. As reported in Table 1, the former was more effective inhibitor by 1.2‐fold and the latter was a less effective inhibitor by 1.4‐fold when compared with unsubstituted 49a (i.e., K Is of 410.9, 337.3, and 470.2 nM for 49a, 49b, and 49c, respectively).
-
ii.
Compounds reported herein were profiled in vitro on hCAs I, II, IX, and XII, and the data in Table 1 clearly accounted for the transmembrane CAs being preferentially inhibited over the constitutive and largely expressed I and II isoforms. Among ureido derivatives 9a‐d, the K I associated values for the IX and XII isoforms were within the low nanomolar range (Table 1). The most effective hCA IX inhibitor was 9d, with a K I of 6.0 nM, whereas its shorter derivative 9c was the most potent ligand for the XII isoform with a close matching K I value (i.e., 6.4 nM). It is worth noting that all the 4‐substituted arylsulfonamides 9b–d showed similar potencies for hCA XII, whereas such a kinetic trend was not retained for the IX isoform (Table 1). As for the remaining isoforms, compounds 9a–d were medium‐high‐nanomolar‐range inhibitors. Also, in this case, compounds 9c and 9d were highly selective for hCA II and I, respectively (i.e., K Is of 84.0 and 58.9 nM for hCA I and II, respectively). The introduction in 9a–d of the thioureido ureido group, as in 24a–d, significantly affected the kinetic trend among the hCAs tested, although preferential inhibition for the membrane IX/XII over the cytosolic I/II was retained. For instance, 24d, which is the thioureido derivative of 9b, was the most potent hCA IX inhibitor, with a K I value of 4.4 nM, and all the remaining compounds were up to 10.8‐fold less effective (i.e., K Is spanning between 40.1 and 47.7 nM; Table 1). Closely matching K Is were reported for 24b–d, with values spanning between 8.0 and 9.2 nM, whereas derivative 24a was a 72.5 nM hCA XII inhibitor. As for the hCAs I/II, compounds 24a–d were high nanomolar inhibitors (Table 1). Insertion of the 1,4‐triazolyl moiety as in 10a and 10b retained significant discrimination for hCAs I/II over IX/XII, with narrow differences in the K I values due to the regioisomeric effect. It is worth noting that derivative 10b was the most potent inhibitor of hCA XII, with K I of 7.4 nM, and therefore close to the reference AAZ of 5.7 nM. Interestingly, SAR was reported for 25 and 26. For instance, derivative 25 was selective for the hCA XII isoform at a 9.0 nM concentration. A radical change in isoform selectivity was reported for 26 (i.e., K I of 17.3 nM for hCA II). As for the heterocyclic derivative 28, predominant inhibition of the housekeeping hCA II was observed (K I of 8.3 nM), followed by the tumor‐associated hCAs IX and XII, which showed K I values of 28.3 and 47.2 nM, respectively (Table 1).
Among the amide‐containing compounds 35a–c, preferential selectivity for the hCA XII isoform was reported for compounds 35a and 35c, with K I values of 8.2 and 7.5 nM, respectively, whereas the benzyl intermediate 35b was a medium nanomolar inhibitor (i.e., K I 64.4 nM). A similar kinetic trend was reported for the hCA IX isoform, although the associated K I values were in the medium to high nanomolar range (Table 1). Derivative 35b was poorly effective on hCAs I and II too, with K Is of 4767 and 620.0 nM, respectively. As for the shortest 35a, a 26.5 nM inhibition value was reported for hCA II, whereas it was poorly effective on the hCA I isoform (i.e., K I of 352.5 nM). Finally, the longest 35c was a high nanomolar hCA I inhibitor with a K I value of 5515 nM; it was effective on the hCA II isoform at a 55.7 nM concentration, and thus almost equipotent to the IX isozyme. A diverse kinetic trend on the hCAs considered in this study was obtained for sulfonyl derivatives 36a–c. As reported in Table 1, the entire series was highly selective and potent toward the hCA XII isoform, with K I values progressively increased from 5.9 nM for 36a up to 8.1 nM for 36c (Table 1). As for hCA IX, a 4.1‐fold enhancement in the inhibition potency was obtained when the sulfonyl ester connecting moiety in 36a was replaced with sulfonamide instead to afford 36b (i.e., K Is of 311.5 and 41.9 nM for 36a and 36b, respectively), whereas slight elongation of the alkyl tether (i.e., 36c) did not induce significant effects on kinetics (Table 1). The remaining hCAs I/II were scarcely inhibited from 36a–c, with K I values in the high nanomolar range (Table 1). A clearer kinetic trend was obtained for the amido containing 37a–c. Elongation of the alkyl tether in 37a of a single carbon unit to afford 37b led to loss of effectiveness for hCA II (i.e., K Is of 21.4 and 798.6 nM for 37a and 37b, respectively) and significant gain of inhibition potency for the hCAs IX and XII isoforms (Table 1). For instance, 37b was a 7.05‐fold more effective hCA IX inhibitor when compared with its shorter counterpart 37a (i.e., K Is of 346.3 and 49.1 nM for 37a and 37b, respectively); as for the hCA XII, it showed a potency gain of 6.7‐fold (i.e., K Is of 61.0 and 9.1 nM for 37a and 37b, respectively). Regioisomeric effects on kinetics were clearly observed for 37b and 37c on all hCAs considered, except for the isoform XII (i.e., K Is of 9.1 and 9.4 nM for 37b and 37c, respectively). Both compounds 38 and 39 retained high selectivity and nanomolar potency for the hCA XII isoform, with K I values of 9.3 and 6.3 nM, respectively. As for the remaining hCAs isoforms, it is worth noting that 39 was an almost equipotent inhibitor of the hCA II and IX isoforms, with K I values of 77.1 and 76.0 nM, respectively. Heterogenic SAR was reported for the piperazine‐containing derivatives 48a‐d and depending upon the introduction of various moieties at the CAI arylsulfonamide warhead. For instance, the unsubstituted derivative 48a was a selective and potent inhibitor for the hCA XII isoform, with a K I value of 3.7 nM. A progressive decrease in the inhibition values was obtained for 48a on hCAs I, II, and IX, respectively (i.e., K Is of 82.1, 49.1, and 30.6 nM for hCAs I, II, and IX, respectively). Introduction of the chloro atom at the 3‐position of the phenyl moiety, as in 48b, led to a significant reduction of the ligand affinity for the hCA I, IX, and XII isoforms between 1.6‐ and 9.1‐fold (i.e., K Is of 769.7, 48.4, and 31.1 nM for hCAs I, IX, and XII, respectively). Compound 48b was beneficial only for isozyme II, as a 2.4‐fold inhibition enhancement was obtained (i.e., K Is of 49.1 and 20.7 nM for 48a and 48b, respectively). Substitution of the chloro atom in 48b with the bulky –CF3 moiety afforded the high potent and selective hCA XII inhibitor 48c (i.e., K I of 9.4 nM). Conversely, the fluoro derivative 48d showed a remarkable increase in the K I values and loss of selectivity toward all of the isoforms tested (Table 1). As for the 1,4‐disubstituted diazepane derivatives 49a–d, progressive enhancement of the inhibition potency was reported for the unsubstituted 49a on hCAs I, II, IX, and XII, with K I values of 315.1, 288.5, 13.8, and 5.5 nM, respectively. Introduction of the chloro atom at the 3‐position (i.e., 49b) led to a reduction of the K I values for hCAs I and II isoforms, whereas the opposite trend was observed for the IX and XII isozymes, although all the values were in the medium‐high nanomolar range (Table 1). Change of the chloro in 49b with a fluoro instead to afford 49c further enhanced the inhibition potency for hCAs I, II, and XII up to low nanomolar K I values (i.e., K Is of 5.3, 7.2, and 7.9 nM for hCAs I, II, and XII, respectively), whereas a slight increase in the K I value was reported for the hCA IX isoform (i.e., K I of 284.6 nM). Fluoro disubstitution, as in 49d, led to a decrease in the affinity of the ligand toward all hCAs considered in this study (Table 1).
Overall, the compounds synthesized in this study showed interesting in vitro activity toward the Malassezia spp. CAs, with K I values in the low‐/sub‐micromolar ranges. Specifically, Mpa‐ and MreCA were preferentially inhibited in terms of their hydrase activity over the MgCA isoform.
2.3. Antifungal Assays on Malassezia spp
All synthesized compounds were tested in vitro using the microdilution assay to evaluate the minimum inhibitory concentration (MIC) on M. pachydermatis DSM 6172, M. furfur ATCC 14521, and M. globosa ATCC MYA 4612 reference strains. The data obtained are reported in Table 2 and were compared with the reference drug KTZ.
Table 2.
In vitro evaluation of the compounds using the minimum inhibitory concentration (MIC) assay on Malassezia spp. (i.e., M. pachydermatis DSMZ 6172, M. furfur ATCC 14521, and M. globosa ATCC MYA 4612). The drug ketoconazole (KTZ) was used as the reference. MIC values were expressed as average MIC (µg/mL) of three experiments, with three replicates each ± standard deviation (SD).
| MIC (µg/mL ± SD) M. pachydermatis DSMZ 6172 | MIC (µg/mL ± SD) M. furfur ATCC 14521 | MIC (µg/mL ± SD) M. globosa ATCC MYA 4612 | |
|---|---|---|---|
| 7 | 0.083 ± 0.04 | 4.0 ± 0 | > 256 ± 0 |
| 9a | 3.6 ± 3.25 | 6.0 ± 2.31 | ≤ 0.5 ± 0.00 |
| 9b | ≤ 0.5 ± 0 | 5.0 ± 2.0 | 2.0 ± 0 |
| 9c | 0.6 ± 0.25 | 2.5 ± 1.0 | 1.0 ± 0 |
| 9d | 0.6 ± 0.25 | 7.0 ± 2.0 | 2.0 ± 0 |
| 10a | 0.33 ± 0.14 | 256 ± 0 | 26.7 ± 9.24 |
| 24a | ≤ 0.5 ± 0 | 4.0 ± 0 | 3.0 ± 1.15 |
| 24b | ≤ 0.5 ± 0 | 4.0 ± 0 | 2.0 ± 0 |
| 24c | ≤ 0.5 ± 0 | 10.0 ± 4.0 | 1.0 ± 0 |
| 24d | ≤ 0.5 ± 0 | 4.0 ± 0 | 2.0 ± 0 |
| 25 | 1.0 ± 0 | 16.0 ± 0 | > 256 ± 0 |
| 26 | 0.0625 ± 0 | 8.0 ± 0 | > 256 ± 0 |
| 28 | 0.0078 ± 0 | 0.067 ± 0 | > 256 ± 0 |
| 35a | 0.005 ± 0.002 | 2.0 ± 0 | 128 ± 0 |
| 35b | 0.5 ± 0.43 | 4.0 ± 0 | > 256 ± 0 |
| 35c | 1.0 ± 0 | 2.0 ± 0 | 128 ± 0 |
| 36a | 0.0625 ± 0 | 0.25 ± 0 | > 256 ± 0 |
| 36b | 0.0625 ± 0 | 2.0 ± 0 | 64 ± 0 |
| 36c | 0.0625 ± 0 | 2.0 ± 0 | 16 ± 0 |
| 37a | 0.0625 ± 0 | 1.0 ± 0 | 4 ± 0 |
| 37b | 0.0625 ± 0 | 4.0 ± 0 | 3.3 ± 1.15 |
| 37c | 0.031 ± 0 | 4.0 ± 0 | 4.3 ± 3.5 |
| 38 | 0.013 ± 0.004 | 0.25 ± 0 | 13.3 ± 4.62 |
| 39 | 0.125 ± 0 | 2.0 ± 0 | 128 ± 0 |
| 48a | 0.25 ± 0 | 8.0 ± 0 | > 256 ± 0 |
| 48b | 0.031 ± 0 | 4.0 ± 0 | 8 ± 0 |
| 48c | 0.0625 ± 0 | 4.0 ± 0 | 5.3 ± 2.3 |
| 48d | 0.0625 ± 0 | 8.0 ± 0 | 106.7 ± 36.9 |
| 49a | 0.0625 ± 0 | 10.7 ± 4.62 | 13.3 ± 4.62 |
| 49b | 0.031 ± 0 | 2.0 ± 0 | 4 ± 0 |
| 49c | 0.031 ± 0 | 1.0 ± 0 | > 256 ± 0 |
| 49d | 0.05 ± 0.02 | 4.0 ± 0 | > 256 ± 0 |
| KTZ | 0.0312 ± 0 | 0.25 ± 0 | 0.25 ± 0 |
As reported above, relevant data were obtained for the M. pachydermatis DSMZ 6172 strain, as the majority of compounds screened were highly effective growth inhibitors on this yeast, with associated MIC values below the 1.0 µg/mL threshold value (Table 2). Compound 9a was the least potent among the entire series (i.e., MIC of 3.6 ± 3.25 µg/mL), and an almost identical trend was observed for 9b–d, 24a–d, and 35b, as their MICs were between 0.5/0.6 µg/mL (Table 2). Better results were obtained for compounds 10a (i.e., MIC 0.33 ± 0.14 µg/mL), 48a (i.e., MIC 0.25 ± 0 µg/mL), and 39 (i.e., MIC 0.125 ± 0 µg/mL). It is noteworthy that the benzoxazolyl derivative 28 and the shortest amido 35a were the most effective compounds in inhibiting M. pachydermatis, being far more effective than the reference KTZ (i.e., MICs of 0.0078 ± 0, 0.005 ± 0.002, and 0.0312 ± 0 µg/mL, respectively). Derivative 38 was also of interest, with a MIC value of 0.013 ± 0.004 µg/mL. All the remaining derivatives showed comparable potencies, with associated MIC values being between 0.0625 and 0.031 µg/mL and therefore in the same range of the reference KTZ (Table 2).
Far higher MIC concentrations were obtained when the compounds were tested on the M. furfur ATCC 14521 strain, as most of the values were between 1 and 4 µg/mL, and 10a was the only ineffective derivative (i.e., MIC of 256 µg/mL). Slightly lower effectiveness was reported for derivatives 9a, 9d, 24c, 25, 26, 48a, 48d, and 49a, with MICs spanning between 5 and 16 µg/mL (Table 2). Among the compounds tested, 36a and 38 were equipotent to the reference KTZ (Table 2), whereas 28 was 2.6‐fold less effective (i.e., MICs of 0.67 µg/mL). Derivative 23 was the most effective in the series, being 2.0‐fold more potent than KTZ (i.e., MICs of 0.125 ± 0 and 0.25 ± 0 µg/mL, respectively).
A clearer MIC trend was obtained for the M. globosa ATCC MYA 4612 strain. For instance, derivatives 7, 10a, 25, 26, 28, 35b, 36a, 48a, 49c, and 49d were ineffective, with MIC values > 256 ± 0 µg/mL (Table 2). Compounds 35a, 35c, and 39 were the first to show MIC effectiveness at a 128 ± 0 µg/mL concentration, followed by 48d, 36b, and 10a, which showed a marked decrease to values of 106.7 ± 36.9, 64.0 ± 0, and 26.7 ± 9.24 µg/mL, respectively. Compounds 36c, 38, and 49a were almost all equipotent growth inhibitors (i.e., MICs of 16.0 ± 0 and 13.3 ± 4.62 µg/mL), followed by derivative 23 (i.e., MIC 10.7 ± 4.6 µg/mL). An interesting profile was observed for the remaining compounds. For instance, 48b was effective at 8.0 ± 0 µg/mL, whereas for 48c, the effective concentration reduced to 5.3 ± 2.3 µg/mL. Compounds 37a–c and 49b were all equipotent, with MICs ranging between 3.3 ± 1.15 and 4.3 ± 3.5 µg/mL. Particularly relevant were the data obtained from 9a–d and 24a–d, which were all between 0.5 and 3.0 µg/mL. Compound 9a was the most effective growth inhibitor on M. globosa among the entire series, with a value of 0.5 ± 0 µg/mL (Table 2). Overall, the compounds obtained in this study were less effective as M. globosa ATCC MYA 4612 growth inhibitors when compared with the reference drug KTZ, which showed a MIC value of 0.25 ± 0 µg/mL, thus being 13.2‐fold more potent than the best‐performing compound 37b (i.e., MIC 3.3 ± 1.15 µg/mL).
The most effective compounds were assessed for their effects on human (h) keratinocytes HaCaT cells, considering the values reported by the reference drug KTZ (Figure 2).
Figure 2.

Viability assays on human keratinocytes HaCaT cells expressed as % of cell survival. NC, negative control (Y = 77%) and TR, threshold of survival (Y = 65%).
Overall, the cell viability of human cells was high in the presence of KTZ derivatives, and among them, the most tolerated compound was 48c. Derivative 25 was associated with cell survival values that were consistently above the threshold limit. Higher percentages of cell survival were reported for the compounds screened at the highest concentration; in some cases, an increase in the cell population was observed when compared with the negative control with 1% DMSO. This aspect needs to be investigated in depth to assess whether the data obtained by the MTT test can be traced back to an increased cell proliferation that could be quantified through a cell counting kit (CCK‐8) [20].
Data in Figure 2, 3 show cell survival percentages closely matching those of KTZ when the compounds were tested at 0.5 µg/mL, whereas an increase in the concentration up to 16.0 µg/mL (i.e., 32‐fold higher than the effective dose against M. pachydermatis) led to an increase in the viability, with no marked differences for the reference KTZ. Relevant differences were observed at a 256.0 µg/mL concentration (i.e., 512‐fold higher than the MIC value), as the KTZ induced significantly low keratinocyte survival (i.e., 10.50% ± 0.36), whereas all tested compounds showed just a slight decrease, with percentages ranging from 60% to 90%. On the basis of the data analysis reported above, it is possible to state that although compounds are found to be less effective than KTZ in some cases, they are clearly highly compatible with human cells at high dosages and this endows them with high therapeutic indexes.
2.4. Sterol Analysis
Based on previous experiments, 9a–d, 35a, 38, 49b, and 49c were selected to assess the ability to inhibit the Malassezia spp. CYP51 expressed enzyme by evaluating any disruption in the biosynthesis of ergosterol. Despite our efforts, it proved difficult to grow Malassezia spp. (i.e., M. globosa and M. pachydermatis), and therefore, we performed our experiments on Candida albicans instead by administering 9a–d, 35a, 38, 49b, 49c, and the reference drug KTZ at the final concentrations listed in Table 3, in agreement with experimentally determined KTZ MIC values [21] and MICs for M. globosa.
Table 3.
Final concentrations of compounds 9a‐d, 35a, 38, 49b, 49c, and KTZ used to treat wild‐type Candida albicans.
| Compound | Concentration (µg/mL) |
|---|---|
| 9a | 0.06 |
| 9b | 0.24 |
| 9c | 0.12 |
| 9d | 0.24 |
| 35a | 0.48 |
| 38 | 0.48 |
| 49b | 0.48 |
| 49c | 0.48 |
| KTZ | 0.03 |
Each culture was shaken at 30°C for 48 h, followed by sterol extraction and GC‐MS identification by comparison of the main peaks (i.e., > 1% of the dominant peak) with the NIST database spectra available online (Figure 4, Supporting Information S2: Table S1) [22]. Percentage sterolic profiles (% of the total sterol extracted) were determined by calculating the peak areas determined from each identified peak in relation to the total areas of all identified peaks (Supporting Information S2: Table S2).
Figure 4.

GC‐MS traces of the sterol content for each Candida albicans culture treated with 9a–d, 35a, 38, 49b, 49c, and KTZ as the reference drug.
Overall, the GC‐MS traces showed profiles with significant accumulation of 14α‐methylated sterol derivatives in C. albicans wild‐type strains treated with selected compounds and thus confirmed that the KTZ moiety merged within the synthesized molecules retained its mechanism of action. Interestingly, GC‐MS traces and sterolic percentages for C. albicans treated with 9a–d and 35a were almost equal, with derivative 9b being slightly more effective than the others (i.e., up to 1.6‐fold) in determining the accumulation of lanosterol (i.e., 16.6%). Better performances were obtained for 35a, which, in addition to lanosterol (16.0%), induced significant accumulation of the 14α‐methylated derivative 4,4‐dimethylcholesta‐8, 24‐dien‐3‐ol (i.e., 10.8%), and was associated with the lowest ergosterol content among all (73.2%). Compounds 49b and 49c were identical in determining sterolic profiles and contents and were not too dissimilar to the GC‐MS trace for 9b (Figure 2, 3). Derivative 38 was particularly effective in inhibiting the CYP51 enzyme in C. albicans, as ergosterol accounted for just 21.5% of the total sterol content, and it induced significant accumulation of 4,4‐dimethylcholesta‐8, 24‐dien‐3‐ol (i.e., 5.9%), lanosterol (41.6%), and 4,14‐dimethyl‐9,19‐cycloergost‐24(28)‐en‐3‐ol acetate (28.8%). It is necessary to point out that the results reported here and based on the C. albicans model should be considered as a general reference method that takes into account additional discrepancies among the strains, such as the metabolism of sterol.
3. Conclusions
Herein, we report the design and synthesis of compounds with the antifungal KTZ pharmacophoric moiety connected by means of alkyl/aryl spacers to a warhead‐directed CAI of the primary sulfonamide type. The main aim of this study is to present experimental evidence for next‐generation antifungal agents able to inhibit both yeast‐expressed 14 α‐demethylase (CYP51) and CA enzymes.
The library of compounds reported herein was developed using a rational approach based on elongation of the KTZ piperazine distal nitrogen conveniently modified with the moieties of interest. No major difficulties were encountered during the synthetic and/or the purification processes up to gram scales, thus proving the sustainability of the approach.
All final compounds showed effective inhibition of the relevant Malassezia spp. expressed CAs (i.e., MgCA, MpaCA, and MreCA), with experimental K I values in the low‐sub‐micromolar range. Although hCAs were far more susceptible to inhibition (i.e., K Is in the low nanomolar range), all compounds had low cytotoxicity, with an overall threshold survival on HaCaT cells of 65% at an 8 µg/mL concentration. MIC values clearly showed that the tested compounds did not have exceptional antifungal efficacy against Malassezia spp. strains when compared with KTZ. Nevertheless, a remarkable reduction in cellular toxicity compared with KTZ is quite evident. The low cytotoxic profile of the compounds reported here makes them exploitable at higher concentrations and possibly also for systemic administration for the management of difficult‐to‐treat fungal infections as a replacement for KTZ. Such an application would be quite beneficial to eliminate the typical time‐ and dose‐dependent side effects of this molecule (i.e., suppression of testosterone synthesis) [23]. Data reported in this study allow us to reasonably speculate that the compounds tested are inhibitors of the 14α‐demethylase enzyme, and suppress ergosterol synthesis along with accumulation of 14α‐methylated sterol derivatives within the yeast membranes. The net effect of this process is an additional contribution to reduce the growth of Malassezia spp., as observed in our experiments. For clarity, our data are indicative of retention of the KTZ sterol inhibition in reference to C. albicans. This model, although scientifically relevant for our purposes, does not consider any differences in the metabolism of cholesterol between fungal strains.
The preferential isoform selectivity of our compounds for the fungal strain‐expressed carbonic anhydrases (CAs) over human CAs does not represent a limitation for further development, since Malassezia spp. are typically located on superficial cutaneous areas in humans and pets. Overall, we are confident that the compounds reported in this study are effective antifungal agents endowed with high safety profiles, which make them more appropriate for the management of infections promoted by fungal/yeast by means of topical administration.
4. Experimental
4.1. Chemistry
4.1.1. General
Anhydrous solvents and all reagents were purchased from Sigma‐Aldrich, Alfa Aesar, and Fluorochem (Milan‐Italy). All reactions involving air‐ or moisture‐sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringe techniques to transfer solutions. Nuclear magnetic resonance (1H‐NMR, 13C‐NMR) spectra were recorded using a Bruker Advance III 400 MHz spectrometer in DMSO‐d 6 . Chemical shifts are reported in parts per million (ppm) and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublets; bs, broad singlet; ap s, apparent singlet; ap d, apparent doublet; ap t, apparent triplet; and ap q, apparent quartet. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Analytical thin‐layer chromatography (TLC) was carried out on Merck silica gel F‐254 plates. Flash chromatography purifications were performed on Merck silica gel 60 (230–400 mesh ASTM) as the stationary phase, and methanol/dichloromethane (MeOH/DCM) or ethyl acetate/hexane (EtOAc/Hex) were used as eluents. The solvents used in Mass Spectra (MS) measurements were acetone, acetonitrile (Chromasolv grade), and 56 mQ H2O 18 MΩ, obtained from Millipore's Simplicity system (Milan‐Italy). The mass spectra were obtained using a Varian 1200 L triple quadrupole system (Palo Alto, USA) equipped by the Electrospray Source (ESI) operating in both positive and negative modes. Stock solutions of analytes were prepared in acetone at 1.0 mg/mL and stored at 4°C. Working solutions of each analyte were freshly prepared by diluting stock solutions in a mixture of mQ H2O/ACN 1/1 (v/v) up to a concentration of 1.0 µg/mL. The mass spectra of each analyte were acquired by introducing, via a syringe pump at 10 L/min, the working solution. RawQdata were collected and processed by Varian Workstation Vers. 6.8.
The InChI codes of the investigated compounds, together with some biological activity data, are provided as the Supporting Information.
4.1.2. Synthesis of 1‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)Methyl]‐2‐(2,4‐Dichlorophenyl)‐1,3‐Dioxolan‐4‐yl]Methoxy}Phenyl)Piperazine (2)

In a round‐bottom flask, KTZ 1 (2.0 g; 3.78 mmol; 1.0 eq.) was suspended in MeOH (25 mL). The reaction mixture was stirred at reflux temperature until complete dissolution. Then, aqueous NaOH 20% w/v (5–6 mL) was added. The reaction mixture was stirred at reflux temperature for 12 h. A control via TLC was performed to ensure complete consumption of the starting materials. The reaction was cooled to room temperature, diluted with H2O, and a white precipitate was formed, filtered off under vacuum, washed with H2O, and dried in air. No further purification was needed. White solid 91% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.73 (1H, d, J = 2.08 Hz), 7.60 (1H, d, J = 8.44 Hz), 7.51 (1H, s), 7.49 (1H, dd, J = 8.54 Hz; J = 2.15 Hz), 7.05 (1H, s), 6.89 (2H, d, J = 9.12 Hz), 6.85 (1H, s), 6.81 (2H, d, J = 9.12 Hz), 4.56 (2H, q, J = 11.25 Hz), 4.37 (1H, m, NH exchange with D2O), 3.90 (1H, t, J = 5.01 Hz), 3.67 (2H, m), 3.54 (1H, dd, J = 10.20 Hz; J = 5.09 Hz), 2.95 (4H, t, J = 4.80 Hz), 2.85 (4H, t, J = 4.78 Hz), 2.21 (1H, s); 13C NMR (CDCl3, 100 MHz): 152.3, 146.8, 138.8, 135.9, 134.7, 133.0, 131.3, 129.5, 128.6, 127.2, 121.1, 118.1, 115.3, 108.0, 74.9, 67.8, 67.6, 51.8, 51.3, 46.3; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C24H26Cl2N4O3 488.1382, found 488.1386; Elemental analysis, calculated: C, 58.90; H, 5.36; N, 11.45; found: C, 58.92; H, 5.39; N, 11.43.
4.1.3. General Procedure for the Synthesis of Ureido Derivatives 7, 9a–d, and 10a,b
1‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazine (2) (1.0 eq.) and the appropriate carbamate (3–6) (1.0 eq.) were dissolved in acetonitrile (4 mL). The reaction mixture was stirred at reflux temperature for 12 h. A control via TLC was performed to ensure complete consumption of the starting materials. The reaction was quenched with H2O and the precipitate was filtered off under vacuum, washed with Et2O and H2O, and then dried in air. Purification via silica gel flash chromatography was performed to afford the pure products.

N‐[(R)‐5‐Sulfamoyl‐2,3‐dihydro‐1H‐inden‐1‐yl]‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl}piperazine‐1‐carboxamide (7): White solid 64% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.73 (1H, s, NH, exchange with D2O), 7.69 (2H, s), 7.60 (1H, d, J = 8.44 Hz), 7.52 (1H, s), 7.50 (1H, d, J = 9.00 Hz), 7.43 (1H, d, J = 8.36 Hz), 7.35 (2H, bs, SO2NH 2, exchange with D2O), 7.06 (2H, d, J = 5.52 Hz), 6.96 (2H, d, J = 8.80 Hz), 6.85 (2H, d, J = 7.48 Hz), 6.82 (1H, s), 5.33 (1H, q, J = 8.26 Hz), 4.57 (2H, q, J = 11.32 Hz), 4.38 (1H, m), 3.90 (1H, t, J = 7.50 Hz), 3.68 (2H, m), 3.54 (5H, m), 3.03 (4H, m), 2.99 (1H, m), 2.86 (1H, m), 2.45 (1H, m), 1.96 (1H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 158.3, 152.9, 147.8, 147.2, 146.6, 143.5, 139.4, 136.1, 135.4, 133.3, 131.5, 131.0, 128.5, 128.2, 125.7, 125.6, 122.2, 118.7, 116.0, 108.6, 75.5, 68.7, 67.6, 65.8, 56.0, 51.5, 50.6, 44.5, 34.2, 30.5; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C34H36Cl2N6O6S 727.6580, found 727.6584; Elemental analysis, calculated: C, 56.12; H, 4.99; N, 11.55; found: C, 56.16; H, 5.03; N, 11.51.

N‐(3‐Sulfamoylphenyl)‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carboxamide (9a): White solid 48% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 8.99 (1H, s, NH, exchange with D2O), 8.08 (1H, s), 7.73 (2H, s), 7.61 (1H, d, J = 8.28 Hz), 7.52 (1H, s), 7.46 (3H, m), 7.35 (2H, bs, SO2NH 2, exchange with D2O), 7.06 (1H, s), 6.99 (2H, d, J = 8.36 Hz), 6.84 (3H, m), 4.57 (2H, q, J = 11.23 Hz), 4.38 (1H, m), 3.90 (1H, t, J = 7.14 Hz), 3.65 (6H, m), 3.55 (1H, m), 3.08 (4H, m); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 155.6, 153.0, 146.5, 145.2, 141.9, 136.1, 135.4, 133.3, 131.6, 131.0, 129.8, 128.5, 128.2 123.1, 122.0, 119.6, 118.7, 117.3, 116.0, 108.6, 75.5, 68.7, 67.6, 51.5, 50.6, 44.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C31H32Cl2N6O6S 687.5930, found 687.5934; Elemental analysis, calculated: C, 54.15; H, 4.69; N, 12.22; found: C, 54.17; H, 4.72; N, 12.25

N‐(4‐Sulfamoylphenyl)‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carboxamide(9b): White solid 53% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 9.02 (1H, s, NH, exchange with D2O), 7.72 (2H, m), 7.68 (2H, d, J = 8.88 Hz), 7.61 (1H, d, J = 8.48 Hz), 7.52 (1H, s), 7.50 (1H, dd, J = 8.55 Hz; J = 1.99 Hz), 7.21 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.99 (2H, d, J = 8.96 Hz), 6.86 (2H, s), 6.83 (1H, s), 6.78 (1H, d, J = 8.32 Hz), 4.57 (2H, q, J = 11.20 Hz), 4.38 (1H, m), 3.90 (1H, t, J = 7.50 Hz), 3.68 (6H, m), 3.56 (1H, dd, J = 10.22 Hz; J = 5.27 Hz), 3.09 (4H, m); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 155.5, 153.0, 146.5, 136.2, 135.4, 133.3, 131.6, 131.0, 130.3, 128.2, 127.3, 119.7 119.5, 118.8, 116.0, 108.7, 75.5, 68.7, 67.7, 51.5, 50.6, 44.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C31H32Cl2N6O6S 687.5930, found 687.5936; Elemental analysis, calculated: C, 54.15; H, 4.69; N, 12.22; found: C, 54.11; H, 4.67; N, 12.20.

N‐[(4‐Sulfamoylphenyl)methyl]‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carboxamide (9c): White solid 44% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.79 (2H, d, J = 8.24 Hz), 7.73 (1H, d, J = 2.00 Hz), 7.61 (1H, d, J = 8.44 Hz), 7.52 (1H, s, NH, exchange with D2O), 7.50 (1H, d, J = 8.58 Hz; J = 2.07 Hz), 7.47 (2H, d, J = 8.24 Hz), 7.34 (2H, bs, SO2NH 2, exchange with D2O), 7.32 (1H, m), 7.06 (1H, s), 6.96 (2H, d, J = 9.00 Hz), 6.86 (1H, s), 6.83 (2H, d, J = 8.96 Hz), 4.57 (2H, q, J = 11.22 Hz), 4.38 (1H, m), 4.34 (2H, d, J = 5.40 Hz), 3.90 (1H, t, J = 7.50 Hz), 3.68 (2H, m), 3.55 (1H, t, J = 5.10 Hz), 3.51 (4H, m), 3.02 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 158.2, 152.9, 146.6, 146.1, 143.2, 131.5, 131.0, 130.2, 128.2, 126.4, 119.7, 118.7, 116.1, 116.0, 108.6, 75.5, 68.7, 67.6, 65.8, 51.5, 50.5, 44.4; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C32H34Cl2N6O6S 701.6200, found 701.6215; Elemental analysis, calculated: C, 54.78; H, 4.88; N, 11.98; found: C, 54.80; H, 4.93; N, 12.01.

N‐[2‐(4‐Sulfamoylphenyl)ethyl]‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carboxamide (9 d): White solid 58% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.78 (2H, t, J = 8.09 Hz), 7.72 (1H, d, J = 1.82 Hz), 7.61 (1H, d, J = 8.47 Hz), 7.51 (1H, s, NH, exchange with D2O), 7.49 (1H, dd, J = 8.53 Hz; J = 1.85 Hz), 7.42 (2H, d, J = 8.12 Hz), 7.32 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.95 (2H, d, J = 8.97 Hz), 6.84 (3H, t, J = 7.80 Hz), 6.77 (1H, d, J = 5.76 Hz), 4.57 (2H, q, J = 11.14 Hz), 4.38 (1H, m), 3.90 (1H, t, J = 7.50 Hz), 3.68 (2H, m), 3.56 (1H, dd, J = 9.82 Hz; J = 4.92 Hz), 3.45 (4H, m), 3.31 (2H, q, J = 6.56 Hz), 2.99 (4H, m), 2.85 (2H, t, J = 7.26 Hz); 13C NMR (DMSO‐d 6 , 100 MHz): 158.2, 152.9, 146.6, 145.0, 142.8, 136.1, 135.4, 131.5, 130.3, 130.0, 128.2, 126.6, 119.7, 118.7, 116.1, 116.0, 108.6, 75.5, 68.7, 67.6, 51.5, 50.6, 44.4, 42.4, 36.6; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C33H36Cl2N6O6S 715.6470, found 715.6473; Elemental analysis, calculated: C, 55.39; H, 5.07; N, 11.74; found: C, 55.41; H, 5.03; N, 11.72

N‐{[(1‐(4‐Sulfamoylphenyl)‐1H‐1,2,3‐triazol‐4‐yl)methyl]}‐4‐{4‐((2 R,4S) − 2 − [(1H−imidazol−1−yl)methyl]−2 − (2,4−dichlorophenyl)−1,3−dioxolan−4−yl)methoxyphenyl}piperazine‐1‐carboxamide (10a): Light brown solid 49% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 8.73 (1H, s, NH, exchange with D2O), 8.18 (2H, d, J = 8.44 Hz), 8.04 (2H, d, J = 8.44 Hz), 7.73 (1H, s), 7.61 (1H, d, J = 8.40 Hz), 7.57 (2H, bs, SO2NH 2, exchange with D2O), 7.51 (1H, s), 7.48 (1H, m), 7.29 (1H, t, J = 4.95 Hz), 7.05 (1H, s), 6.96 (2H, d, J = 8.72 Hz), 6.85 (1H, s), 6.82 (2H, d, J = 8.48 Hz), 4.57 (2H, q, J = 11.27 Hz), 4.42 (2H, d, J = 4.88 Hz), 4.37 (1H, t, J = 5.52 Hz), 3.90 (1H, t, J = 7.40 Hz), 3.67 (2H, m), 3.54 (1H, t, J = 5.26 Hz), 3.50 (4H, m), 3.02 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 161.1, 152.6, 146.7, 140.5, 139.5, 136.1, 135.4, 133.3, 132.8, 131.5, 131.0, 130.4, 128.5, 122.0, 121.0, 119.2, 118.0, 117.0, 116.0, 108.6, 75.5, 68.7, 67.6, 51.5, 50.6, 44.7, 43.2, 35.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C34H35Cl2N9O6S 768.6710, found 768.6718; Elemental analysis, calculated: C, 53.13; H, 4.59; N, 16.40; found: C, 53.15; H, 4.62; N, 16.37.

N‐[(1‐(3‐Sulfamoylphenyl)‐1H‐1,2,3‐triazol‐4‐yl)methyl]‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carboxamide (10b): Light brown solid 14% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 8.71 (1H, s, NH, exchange with D2O), 8.41 (1H, s), 8.19 (1H, d, J = 7.96 Hz), 7.93 (1H, d, J = 7.84 Hz), 7.83 (1H, t, J = 7.96 Hz), 7.73 (1H, d, J = 1.88 Hz), 7.62 (2H, bs, SO2NH 2, exchange with D2O), 7.59 (1H, s), 7.51 (1H, s), 7.49 (1H, dd, J = 8.72 Hz; J = 1.92 Hz), 7.29 (1H, t, J = 5.24 Hz), 7.05 (1H, s), 6.95 (2H, d, J = 9.00 Hz), 6.83 (3H, t, J = 8.52 Hz), 4.57 (2H, q, J = 11.40 Hz), 4.43 (2H, d, J = 5.08 Hz), 4.37 (1H, t, J = 5.72 Hz), 3.90 (1H, t, J = 7.48 Hz), 3.67 (2H, m), 3.55 (1H, t, J = 5.14 Hz), 3.50 (4H, m), 3.02 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 161.1, 152.6, 146.7, 140.5, 139.5, 136.1, 135.4, 133.3, 132.8, 131.5, 131.0, 130.4, 128.5, 128.2, 122.7, 122.0, 121.0, 119.2, 118.0, 117.0, 116.0, 108.6, 75.5, 68.7, 67.6, 51.5, 50.6, 44.7, 43.2, 35.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C34H35Cl2N9O6S 768.6710, found 768.6706; Elemental analysis, calculated: C, 53.13; H, 4.59; N, 16.40; found: C, 53.10; H, 4.57; N, 16.42.
4.1.4. General Procedure for the Synthesis of Thioureido Derivatives 24a–d, 25, and 26
1‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazine (2) (1.0 eq.) and the appropriate isothiocyanate (19–22) (1.0 eq.) were dissolved in acetonitrile (4 mL). The reaction mixture was stirred at room temperature for 12 h. A control via TLC was performed to ensure complete consumption of the starting materials. The reaction was quenched with H2O, and the precipitate was filtered off under vacuum, washed with Et2O and H2O, and then dried in air. Purification via silica gel flash chromatography was performed to afford the pure products.

N‐[(4‐Sulfamoylphenyl)methyl]‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carbothioamide (24a): White solid 77% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 8.45 (1H, t, NH, exchange with D2O, J = 5.52 Hz), 7.79 (2H, d, J = 8.28 Hz), 7.72 (1H, d, J = 2.08 Hz), 7.60 (1H, d, J = 8.48 Hz), 7.52 (1H, s), 7.50 (3H, m), 7.33 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.96 (2H, d, J = 9.12 Hz), 6.85 (2H, d, J = 1.92 Hz), 6.82 (1H, s), 4.89 (2H, d, J = 5.28 Hz), 4.57 (2H, q, J = 11.16 Hz), 4.37 (1H, m), 4.00 (4H, m), 3.90 (1H, t, J = 5.01 Hz), 3.68 (2H, m), 3.55 (1H, dd, J = 10.23 Hz; J = 5.24 Hz), 3.11 (4H, m); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 182.2, 153.0, 146.1, 144.8, 139.4, 131.5, 131.0, 129.3, 129.1, 128.5, 128.2, 122.6, 122.0, 118.5, 116.1, 108.6, 68.7, 67.6, 65.8, 51.5, 50.2, 48.9; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C32H34Cl2N6O5S2 717.6810, found 717.6816; Elemental analysis, calculated: C, 53.55; H, 4.78; N, 11.71; found: C, 53.52; H, 4.80; N, 11.74.

N‐(2‐(4‐Sulfamoylphenyl)ethyl)‐4‐{4‐[((2 R,4S)‐2‐((1H‐imidazol‐1‐yl)methyl)‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carbothioamide (24b): White solid 80% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 7.96 (1H, t, NH, exchange with D2O, J = 5.24 Hz), 7.79 (2H, d, J = 8.32 Hz), 7.73 (1H, d, J = 2.12 Hz), 7.61 (1H, d, J = 8.44 Hz), 7.52 (1H, s), 7.50 (1H, dd, J = 8.47 Hz; J = 2.10 Hz), 7.45 (2H, d, J = 8.32 Hz), 7.34 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.96 (2H, d, J = 9.12 Hz), 6.85 (2H, d, J = 4.40 Hz), 6.82 (1H, s), 4.57 (2H, q, J = 11.24 Hz), 4.38 (1H, m), 3.93 (4H, m), 3.89 (1H, m), 3.76 (2H, m), 3.68 (2H, m), 3.55 (1H, dd, J = 10.23 Hz; J = 5.25 Hz), 3.07 (4H, m), 2.99 (2H, t, J = 7.46 Hz); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 182.1, 153.0, 139.5, 136.2, 135.4, 135.4, 133.3, 131.6, 131.0, 128.6, 128.2, 126.7, 124.9, 122.1, 118.5, 108.6, 75.5, 68.7, 67.6, 51.5, 50.2, 49.1, 2.1; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C33H36Cl2N6O5S2 731.7080, found 731.7067; Elemental analysis, calculated: C, 54.17; H, 4.96; N, 11.49; found: C, 54.15; H, 4.99; N, 11.46.

N‐(3‐Sulfamoylphenyl)‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carbothioamide (24c): White solid 91% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 9.68 (1H, s, NH, exchange with D2O), 7.81 (1H, t, J = 1.80 Hz), 7.73 (1H, d, J = 2.11 Hz), 7.62 (1H, s), 7.60 (1H, m), 7.57 (1H, t, J = 1.38 Hz), 7.52 (2H, m), 7.50 (1H, dd, J = 8.62 Hz; J = 2.28 Hz), 7.41 (2H, bs, SO2NH 2, exchange with D2O), 7.06 (1H, s), 6.99 (2H, d, J = 9.12 Hz), 6.86 (2H, s), 6.84 (1H, s), 4.57 (2H, q, J = 11.23 Hz), 4.38 (1H, m), 4.10 (4H, m), 3.90 (1H, t, J = 5.00 Hz), 3.69 (2H, m), 3.56 (1H, dd, J = 10.16 Hz; J = 5.18 Hz), 3.17 (4H, m); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 182.8, 152.9, 146.2, 144.8, 143.3, 133.3, 131.5, 131.0, 128.3, 128.2, 126.4, 118.4, 116.0, 108.6, 75.5, 68.7, 67.6, 51.5, 50.2, 48.9, 48.2; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C31H32Cl2N6O5S2 703.6540, found 703.6547; Elemental analysis, calculated: C, 52.92; H, 4.58; N, 11.94; found: C, 52.95; H, 4.61; N, 11.91.

N‐(4‐Sulfamoylphenyl)‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carbothioamide (24d): White solid 59% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 9.69 (1H, s, NH, exchange with D2O), 7.76 (2H, d, J = 8.64 Hz), 7.73 (1H, s), 7.61 (1H, d, J = 8.40 Hz), 7.54 (1H, m), 7.52 (2H, m), 7.49 (1H, m), 7.32 (2H, bs, SO2NH 2, exchange with D2O), 7.06 (1H, s), 6.99 (2H, d, J = 8.84 Hz), 6.86 (2H, m), 6.84 (1H, m), 4.57 (2H, q, J = 11.22 Hz), 4.38 (1H, m), 4.09 (4H, m), 3.90 (1H, t, J = 7.48 Hz), 3.68 (2H, m), 3.56 (1H, m), 3.17 (4H, m); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 182.2, 152.9, 146.2, 144.7, 143.0, 136.1, 135.4, 133.3, 131.5, 131.0, 130.0, 128.2, 126.7, 118.5, 116.0, 108.6, 75.5, 68.7, 67.6, 65.8, 50.1, 48.0, 47.3, 35.4; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C31H32Cl2N6O5S2 703.6540, found 703.6536; Elemental analysis, calculated: C, 52.92; H, 4.58; N, 11.94; found: C, 52.90; H, 4.57; N, 11.96.
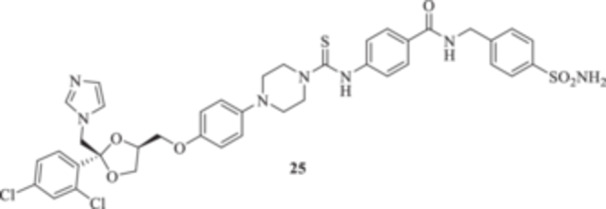
4‐[4‐(4‐(((2 R,4S)‐2‐((1H‐Imidazol‐1‐yl)methyl)‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy)phenyl)piperazine‐1‐carbothioamido]‐N‐[(4‐sulfamoylphenyl)methyl]benzamide (25): Light yellow solid 66% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 9.62 (1H, s, NH, exchange with D2O), 9.08 (1H, t, J = 5.84 Hz), 7.87 (2H, d, J = 8.48 Hz), 7.82 (2H, d, J = 8.16 Hz), 7.72 (1H, d, J = 1.68 Hz), 7.61 (1H, d, J = 8.44 Hz), 7.54 (1H, s), 7.51 (2H, m), 7.47 (3H, ap d, J = 8.24 Hz), 7.34 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.98 (2H, d, J = 8.92 Hz), 6.86 (2H, s), 6.84 (1H, s), 4.57 (4H, q, J = 11.02 Hz), 4.38 (1H, m), 4.09 (4H, m), 3.91 (1H, t, J = 7.40 Hz), 3.69 (2H, ap q, J = 4.77 Hz), 3.58 (1H, dd, J = 10.12 Hz; J = 5.11 Hz), 3.17 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 182.1, 166.9, 153.0, 146.1, 144.8, 143.5, 139.5, 139.4, 136.1, 135.4, 133.3, 131.6, 131.0, 130.2, 128.5, 128.4, 128.2, 126.6, 124.6, 122.0, 118.5, 116.0, 108.7, 75.5, 68.7, 67.6, 51.5, 50.2, 49.0, 43.2, 31.6; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C39H39Cl2N7O6S2 836.8040, found 836.8045; Elemental analysis, calculated: C, 55.98; H, 4.70; N, 11.72; found: C, 56.02; H, 4.73; N, 11.68.

N‐[4‐[(N‐(4‐Sulfamoylphenethyl)sulfamoyl]phenyl]‐4‐{4‐[((2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy]phenyl}piperazine‐1‐carbothioamide (26): Light yellow solid 64% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 9.72 (1H, s, NH, exchange with D2O), 7.77 (1H, s), 7.75 (2H, d, J = 5.12 Hz), 7.73 (2H, t, J = 2.44 Hz), 7.67 (1H, t, J = 5.74 Hz), 7.60 (3H, m), 7.52 (1H, s), 7.50 (1H, dd, J = 8.47 Hz; J = 2.09 Hz), 7.41 (1H, s), 7.39 (1H, s), 7.33 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.98 (2H, d, J = 9.08 Hz), 6.86 (2H, s), 6.84 (1H, s), 4.57 (2H, q, J = 11.12 Hz), 4.38 (1H, m), 4.10 (4H, m), 3.91 (1H, t, J = 5.03 Hz), 3.69 (2H, m), 3.57 (1H, dd, J = 10.24 Hz; J = 5.14 Hz), 3.17 (4H, m), 3.04 (2H, q, J = 6.71 Hz), 2.80 (2H, t, J = 7.10 Hz); 13C NMR (DMSO‐d 6 , 100 MHz): 182.0, 153.0, 146.0, 145.8, 143.9, 143.1, 139.5, 136.1, 135.4, 133.3, 131.6, 131.0, 130.2, 128.5, 128.2, 127.6, 126.6, 124.8, 122.1, 118.5, 116.0, 108.7, 75.5, 68.7, 67.6, 65.9, 51.5, 50.2, 49.1, 44.6, 35.9; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C39H41Cl2N7O7S3 886.8790, found 886.9796; Elemental analysis, calculated: C, 55.82; H, 4.66; N, 11.06; found: C, 55.85; H, 4.64; N, 11.09.

Synthesis of 2‐[4‐(4‐(((2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy)phenyl)piperazin‐1‐yl]benzo[d]oxazole‐5‐sulfonamide (28): 1‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazine (2) (94 mg; 0.19 mmol; 1.0 eq.)k and 2‐thioxo‐2,3‐dihydrobenzo[d]oxazole‐5‐sulfonamide 27 (44.3 mg; 0.19 mmol; 1.0 eq.) were dissolved in DMF (4 mL). Then, triethylamine (33 µL; 0.23 mmol; 1.2 eq.) was added and the reaction mixture was stirred at reflux temperature for 12 h. A control via TLC was performed to ensure complete consumption of the starting materials. The reaction was quenched with H2O, and the product was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and evaporated under reduced pressure. Purification via silica gel flash chromatography was performed, using 3/97 MeOH/DCM. Red‐brown solid 26% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.74 (1H, d, J = 1.60 Hz), 7.72 (1H, d, J = 2.12 Hz), 7.62 (2H, t, J = 8.02 Hz), 7.57 (1H, dd, J = 8.36 Hz; J = 1.80 Hz), 7.52 (1H, s), 7.49 (1H, dd, J = 8.31 Hz; J = 1.77 Hz), 7.35 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 7.01 (2H, d, J = 9.08 Hz), 6.87 (1H, s), 6.85 (2H, d, J = 3.20 Hz), 4.57 (2H, q, J = 11.03 Hz), 4.38 (1H, m), 3.91 (1H, t, J = 5.00 Hz), 3.82 (4H, m), 3.69 (2H, m), 3.58 (1H, dd, J = 10.14 Hz; J = 5.16 Hz), 3.21 (4H, m). 13C NMR (DMSO‐d 6 , 100 MHz): 161.7, 153.1, 151.6, 146.4, 139.5, 136.1, 135.4, 133.3, 132.8, 131.5, 131.0, 128.5, 128.2, 122.0, 121.3, 119.1, 117.5, 116.0, 110.9, 108.6, 75.5, 68.6, 67.6, 51.6, 51.5, 50.5, 45.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C31H30Cl2N6O6S 685.5770, found 685.5776; Elemental analysis, calculated: C, 54.31; H, 4.41; N, 12.26; found: C, 54.28; H, 4.37; N, 12.27.
4.1.5. General Procedure for the Synthesis of Compounds 35a–c, 36a–c, 37a–c, 38, and 39
1‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazine (2) (1.0 eq.), K2CO3 (1.2 eq.), and the appropriate sulfonamide (30–34) (1.0 eq.) were dissolved in acetonitrile (4 mL). The reaction mixture was stirred at reflux temperature for 12 h. A control via TLC was performed to ensure complete consumption of the starting materials. The reaction was quenched with H2O, and the product was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and evaporated under reduced pressure. Purification via silica gel flash chromatography was performed to afford the pure products.

N‐(4‐Sulfamoylphenyl)‐4‐[(4‐{4‐[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]methyl]benzamide (35a): White solid 67% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 10.53 (1H, s, NH, exchange with D2O), 7.96 (4H, m), 7.82 (2H, d, J = 8.64 Hz), 7.68 (1H, d, J = 1.68 Hz), 7.57 (1H, d, J = 8.48 Hz), 7.50 (3H, m), 7.45 (1H, dd, J = 8.46 Hz; J = 1.68 Hz), 7.28 (2H, bs, SO2NH 2, exchange with D2O), 7.01 (1H, s), 6.88 (2H, d, J = 8.92 Hz), 6.82 (1H, s), 6.78 (2H, d, J = 8.84 Hz), 4.53 (2H, q, J = 11.15 Hz), 4.33 (1H, m), 3.86 (1H, t, J = 7.44 Hz), 3.64 (4H, m), 3.52 (1H, dd, J = 10.03 Hz; J = 5.10 Hz), 3.04 (4H, m), 2.57 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 166.9, 152.7, 146.8, 143.5, 143.3, 139.8, 136.3, 135.5, 134.3, 133.4, 131.7, 131.1, 129.9, 128.9, 128.7, 128.3, 127.6, 122.2, 120.8, 118.3, 116.2, 108.8, 75.6, 68.9, 67.8, 62.6, 53.8, 51.6, 50.5, 44.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H38Cl2N6O6S 777.7180, found 777.7184; Elemental analysis, calculated: C, 58.69; H, 4.93; N, 10.81; found: C, 58.71; H, 4.97; N, 10.78.

4‐[(4‐{4‐[[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy]phenyl}piperazin‐1‐yl)methyl]‐N‐[(4‐sulfamoylphenyl)methyl]benzamide (35b): White solid 42% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 9.11 (t, 1H, J = 5.78 Hz, NH, exchange with D2O), 7.90 (2H, d, J = 8.04 Hz), 7.81 (2H, d, J = 8.20 Hz), 7.69 (1H, d, J = 1.88 Hz), 7.58 (1H, d, J = 8.44 Hz), 7.52 (1H, s), 7.50 (2H, d, J = 2.84 Hz), 7.46 (3H, ap d, J = 8.20 Hz), 7.32 (2H, bs, SO2NH 2, exchange with D2O), 7.03 (1H, s), 6.89 (2H, d, J = 9.00 Hz), 6.84 (1H, s), 6.80 (2H, d, J = 8.96 Hz), 4.56 (4H, m), 4.35 (1H, m), 3.88 (1H, t, J = 7.46 Hz), 3.67 (2H, m), 3.60 (2H, s), 3.55 (1H, dd, J = 10.13 Hz; J = 5.14 Hz), 3.37 (2H, s), 3.05 (4H, m), 2.11 (2H, ap s); 13C NMR (DMSO‐d 6 , 100 MHz): 167.1, 152.6, 146.7, 144.7, 143.5, 142.6, 139.4, 136.1, 135.4, 133.8, 133.3, 131.5, 131.0, 129.6, 128.5, 128.4, 128.1, 126.6, 122.0, 118.0, 116.0, 108.6, 75.5, 68.7, 67.6, 62.5, 53.6, 51.5, 50.4, 43.2, 31.5; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C39H40Cl2N6O6S 791.7450, found 791.7458; Elemental analysis, calculated: C, 59.16; H, 5.09; N, 10.61; found: C, 59.12; H, 5.13; N, 10.57.

4‐[(4‐{4‐[[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy]phenyl}piperazin‐1‐yl)methyl]‐N‐(4‐sulfamoylphenethyl)benzamide (35c): White solid 31% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 8.58 (t, 1H, J = 5.44 Hz, NH, exchange with D2O), 7.82 (2H, d, J = 8.12 Hz), 7.79 (2H, d, J = 8.20 Hz), 7.71 (1H, d, J = 1.92 Hz), 7.60 (1H, d, J = 8.48 Hz), 7.51 (1H, s), 7.48 (2H, m), 7.44 (3H, m), 7.33 (2H, bs, SO2NH 2, exchange with D2O), 7.04 (1H, s), 6.90 (2H, d, J = 9.04 Hz), 6.85 (1H, s), 6.81 (2H, d, J = 8.96 Hz), 4.56 (2H, q, J = 11.21 Hz), 4.36 (1H, m), 3.89 (1H, t, J = 7.48 Hz), 3.67 (3H, m), 3.56 (6H, m), 3.21 (1H, d, J = 5.12 Hz), 3.06 (4H, m), 2.96 (2H, t, J = 7.06 Hz), 2.74 (1H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 167.0, 152.6, 146.7, 144.7, 143.0, 142.4, 139.5, 136.1, 135.4, 134.2, 133.3, 131.5, 131.0, 130.0, 129.6, 128.5, 128.2, 128.0, 126.6, 122.0, 118.1, 108.6, 75.5, 68.7, 67.6, 62.5, 54.8, 53.6, 51.5, 50.4, 49.5, 35.7; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C40H42Cl2N6O6S 805.7720, found 805.7728; Elemental analysis, calculated: C, 59.62; H, 5.25; N, 10.43; found: C, 59.65; H, 5.28; N, 10.41.

4‐(Sulfamoyl)phenyl 4‐{[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]methyl}benzenesulfonate (36a): White solid 41% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.89 (2H, d, J = 8.24 Hz), 7.85 (2H, d, J = 8.68 Hz), 7.68 (2H, bs, SO2NH 2, exchange with D2O), 7.65 (1H, s), 7.58 (1H, d, J = 8.48 Hz), 7.49 (1H, s), 7.46 (3H, m), 7.28 (2H, d, J = 8.68 Hz), 7.02 (1H, s), 6.89 (2H, d, J = 9.00 Hz), 6.83 (1H, s), 6.79 (2H, d, J = 9.00 Hz), 4.54 (2H, q, J = 11.14 Hz), 4.34 (1H, m), 3.87 (1H, t, J = 7.48 Hz), 3.66 (4H, m), 3.53 (1H, dd, J = 10.18 Hz; J = 5.18 Hz), 3.40 (1H, ap t, J = 7.02 Hz), 3.05 (4H, m), 2.69 (1H, m), 2.40 (2H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 152.6, 151.8, 147.5, 146.6, 144.0, 139.4, 136.1, 135.4, 133.4, 133.3, 131.5, 131.0, 130.8, 129.2, 128.8, 128.5, 128.2, 126.6, 122.0, 118.1, 108.6, 75.5, 68.7, 67.6, 62.0, 53.6, 51.5, 50.4, 45.4; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C37H37Cl2N5O8S2 814.7500, found 814.7522; Elemental analysis, calculated: C, 54.55; H, 4.58; N, 8.60; found: C, 54.59; H, 4.63; N, 8.59.

N‐[(4‐Sulfamoylphenyl)methyl]‐4‐{[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]methyl}benzenesulfonamide (36b): White solid 50% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 8.26 (1H, s, NH, exchange with D2O), 7.78 (2H, d, J = 8.08 Hz), 7.73 (2H, d, J = 8.16 Hz), 7.69 (1H, d, J = 1.68 Hz), 7.58 (1H, d, J = 8.48 Hz), 7.54 (2H, d, J = 8.12 Hz), 7.49 (1H, s), 7.46 (1H, dd, J = 8.52 Hz; J = 1.67 Hz), 7.42 (2H, d, J = 8.12 Hz), 7.34 (2H, bs, SO2NH 2, exchange with D2O), 7.02 (1H, s), 6.89 (2H, d, J = 8.96 Hz), 6.83 (1H, s), 6.79 (2H, d, J = 8.92 Hz), 4.54 (2H, q, J = 11.16 Hz), 4.34 (1H, m), 4.09 (2H, s), 3.87 (1H, t, J = 7.46 Hz), 3.66 (2H, m), 3.62 (2H, ap s), 3.53 (1H, dd, J = 10.12 Hz; J = 5.12 Hz), 3.39 (1H, ap t, J = 7.10 Hz), 3.06 (4H, m), 2.50 (2H, s), 2.10 (1H, s); 13C NMR (DMSO‐d 6 , 100 MHz): 152.6, 146.6, 144.1, 143.8, 142.8, 140.1, 139.4, 136.1, 135.4, 133.3, 131.5, 131.0, 130.3, 128.8, 128.5, 128.2, 127.4, 126.5, 118.1, 116.0, 108.6, 75.5, 68.7, 67.6, 65.8, 62.2, 53.6, 51.5, 46.5, 16.1; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H40Cl2N6O7S2 827.7930, found 827.7936; Elemental analysis, calculated: C, 55.14; H, 4.87; N, 10.15; found: C, 55.12; H, 4.90; N, 10.17.

4‐[(4‐{4‐[[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy]phenyl}piperazin‐1‐yl)methyl]‐N‐(4‐sulfamoylphenethyl)benzenesulfonamide (36c): White solid 28% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.76 (4H, t, J = 8.44 Hz), 7.72 (2H, m), 7.59 (2H, m), 7.56 (1H, s), 7.51 (1H, bs), 7.48 (1H, dd, J = 8.48 Hz; J = 2.00 Hz), 7.37 (2H, d, J = 8.20 Hz), 7.33 (2H, bs, SO2NH 2, exchange with D2O), 7.04 (1H, s, NH, exchange with D2O), 6.90 (2H, d, J = 9.04 Hz), 6.85 (1H, s), 6.81 (2H, s, J = 9.00 Hz), 4.56 (2H, q, J = 11.16 Hz), 4.36 (1H, m), 3.89 (1H, t, J = 7.48 Hz), 3.67 (2H, m), 3.64 (2H, ap s), 3.55 (1H, dd, J = 10.19 Hz; J = 5.18 Hz), 3.41 (1H, ap q, J = 7.04 Hz), 3.04 (6H, m), 2.79 (2H, t, J = 7.06 Hz), 2.51 (1H, s), 2.12 (1H, s), 1.18 (1H, s); 13C NMR (DMSO‐d 6 , 100 MHz): 152.6, 146.6, 144.0, 143.8, 143.1, 139.8, 136.1, 135.4, 133.3, 131.5, 131.0, 131.3, 130.1, 128.5, 128.2, 127.4, 126.6, 122.0, 118.1, 116.0, 108.6, 75.5, 68.7, 67.6, 65.8, 53.6, 51.5, 50.4, 44.5, 30.5, 16.1; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C39H42Cl2N6O7S2 841.8200, found 841.8212; Elemental analysis, calculated: C, 55.64; H, 5.03; N, 9.98; found: C, 55.65; H, 5.01; N, 9.97.

N‐(4‐Sulfamoylphenyl)‐2‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]acetamide (37a): White solid 27% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 10.15 (1H, s, NH, exchange with D2O), 7.86 (2H, d, J = 8.44 Hz), 7.80 (2H, d, J = 8.64 Hz), 7.73 (1H, d, J = 1.84 Hz), 7.61 (1H, d, J = 8.36 Hz), 7.52 (1H, s), 7.49 (1H, m), 7.30 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.93 (2H, d, J = 8.40 Hz), 6.84 (2H, d, J = 9.40 Hz), 6.81 (1H, s), 4.56 (2H, q, J = 10.71 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.11 Hz), 3.68 (2H, m), 3.55 (1H, m), 3.27 (2H, s), 3.13 (4H, m), 2.71 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 169.0, 152.7, 146.5, 136.2, 135.4, 133.3, 131.6, 131.0, 128.5, 128.2, 127.0, 122.8, 122.1, 118.2, 117.6, 116.0, 114.9, 108.7, 75.5, 68.7, 67.6, 62.1, 53.7, 51.5, 50.8, 48.8; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C32H34Cl2N6O6S 701.6200, found 701.6190; Elemental analysis, calculated: C, 54.78; H, 4.88; N, 11.98; found: C, 54.80; H, 5.01; N, 12.03.

N‐(4‐Sulfamoylphenyl)‐3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanamide (37b): White solid 44% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 10.44 (1H, s, NH, exchange with D2O), 7.78 (4H, ap s), 7.73 (1H, d, J = 2.00 Hz), 7.60 (1H, d, J = 8.44 Hz), 7.51 (1H, s), 7.49 (1H, dd, J = 8.65 Hz; J = 2.14 Hz), 7.28 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.92 (2H, d, J = 9.04 Hz), 6.85 (1H, s), 6.81 (2H, d, J = 9.04 Hz), 4.56 (2H, q, J = 11.19 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.50 Hz), 3.67 (2H, m), 3.54 (1H, dd, J = 10.20 Hz; J = 5.27 Hz), 3.06 (4H, m), 2.73 (2H, t, J = 6.86 Hz), 2.60 (6H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 171.7, 152.6, 146.6, 143.0, 139.4, 139.1, 136.9, 136.1, 135.4, 133.3, 131.5, 128.5, 128.2, 127.6, 122.0, 118.0, 116.0, 108.6, 75.5, 68.7, 67.6, 54.5, 53.4, 51.5, 50.4, 35.1, 30.5; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C33H36Cl2N6O6S 715.6470, found 715.6478; Elemental analysis, calculated: C, 55.39; H, 5.07; N, 11.74; found: C, 55.42; H, 5.03; N, 11.71.

N‐(3‐Sulfamoylphenyl)‐3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanamide (37c): White solid 59% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 10.39 (1H, s, NH, exchange with D2O), 8.21 (1H, s), 7.76 (1H, m), 7.72 (1H, d, J = 2.04 Hz), 7.61 (1H, d, J = 8.48 Hz), 7.53 (1H, s), 7.52 (2H, ap d, J = 1.96 Hz), 7.49 (1H, dd, J = 8.51 Hz; J = 2.04 Hz), 7.40 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (1H, s), 6.92 (2H, d, J = 9.04 Hz), 6.85 (1H, s), 6.81 (2H, d, J = 9.00 Hz), 4.56 (2H, q, J = 11.25 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.52 Hz), 3.67 (2H, m), 3.54 (1H, dd, J = 10.11 Hz; J = 5.19 Hz), 3.06 (4H, m), 2.74 (2H, t, J = 6.86 Hz), 2.59 (6H, m, J = 5.54 Hz); 13C NMR (DMSO‐d 6 , 100 MHz): 171.5, 152.6, 146.7, 140.5, 139.5, 136.1, 135.4, 133.3, 131.5, 131.0, 130.4, 128.5, 128.2, 122.7, 122.0, 121.0, 118.0, 117.0, 116.0, 108.6, 75.5, 68.7, 67.6, 54.6, 53.5, 51.5, 50.4, 35.1, 31.6; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C33H36Cl2N6O6S 715.6470, found 715.6468; Elemental analysis, calculated: C, 55.39; H, 5.07; N, 11.74; found: C, 55.41; H, 5.05; N, 11.77.

4‐[(4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl)methyl]benzenesulfonamide (38): White solid 42% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.83 (2H, d, J = 8.12 Hz), 7.70 (1H, d, J = 1.88 Hz), 7.59 (1H, d, J = 8.48 Hz), 7.55 (2H, d, J = 8.16 Hz), 7.50 (1H, s), 7.47 (1H, dd, J = 8.55 Hz; J = 2.09 Hz), 7.35 (2H, bs, SO2NH 2, exchange with D2O), 7.04 (1H, s), 6.89 (2H, d, J = 9.00 Hz), 6.84 (1H, s), 6.80 (2H, d, J = 9.00 Hz), 4.55 (2H, q, J = 11.20 Hz), 4.36 (1H, m), 3.89 (1H, t, J = 7.49 Hz), 3.66 (4H, m), 3.53 (1H, dd, J = 10.23 Hz; J = 5.11 Hz), 3.41 (1H, q, J = 7.00 Hz), 3.06 (4H, m), 2.98 (1H, ap t, J = 4.78 Hz), 2.89 (1H, ap t, J = 4.68 Hz), 2.51 (1H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 152.6, 146.6, 143.3, 139.4, 136.1, 135.4, 133.3, 131.5, 131.0, 130.0, 128.5, 128.2, 126.6, 122.0, 118.1, 116.0, 108.6, 75.5, 68.7, 67.6, 65.8, 62.2, 53.6, 51.5, 50.4; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C31H33Cl2N5O5S 658.5950, found 658.5948; Elemental analysis, calculated: C, 56.54; H, 5.05; N, 10.63; found: C, 56.57; H, 5.02; N, 10.60.

N‐[2‐Hydroxy‐5‐(sulfamoyl)phenyl]‐2‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]acetamide (39): White solid 40% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 11.04 (1H, bs, OH, exchange with D2O), 9.84 (1H, s, NH, exchange with D2O), 8.73 (1H, s), 7.72 (1H, s), 7.61 (1H, d, J = 8.44 Hz), 7.52 (1H, s), 7.49 (1H, m), 7.43 (1H, d, J = 7.60 Hz), 7.17 (2H, bs, SO2NH 2, exchange with D2O), 7.05 (s, 1H), 6.98 (2H, ap t, J = 8.28 Hz), 6.94 (1H, s), 6.86 (1H, s), 6.84 (2H, d, J = 8.88 Hz), 4.57 (2H, q, J = 10.98 Hz), 4.38 (1H, m), 3.91 (1H, t, J = 7.46 Hz), 3.69 (2H, m), 3.57 (1H, dd, J = 10.04 Hz; J = 5.06 Hz), 3.27 (2H, s), 3.15 (4H, m), 2.75 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 169.0, 152.7, 146.5, 139.5, 136.2, 135.6, 135.4, 133.3, 131.6, 131.0, 128.5, 128.2, 127.0, 122.8, 122.1, 118.2, 116.0, 114.9, 114.2, 108.6, 82.1, 75.5, 68.7, 67.6, 62.1, 55.2, 53.7, 51.5, 50.8; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C32H34Cl2N6O7S 717.6190, found 717.6188; Elemental analysis, calculated: C, 53.56; H, 4.78; N, 11.71; found: C, 53.58; H, 4.76; N, 11.67.
4.1.6. General Procedure for the Synthesis of Compounds 37b,c, 48a–d, and 49a–d
1‐(4‐(((2 R,4S)‐2‐((1H‐Imidazol‐1‐yl)methyl)‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl)methoxy)phenyl)piperazine (2) (1.0 eq.) and the appropriate acryloyl derivative (43–45) (1.0 eq.) were dissolved in methanol (4–5 mL). The reaction mixture was stirred at reflux temperature for 12 h. A control via TLC was performed to ensure complete consumption of the starting materials. The reaction was quenched with H2O, and the precipitate was filtered off under vacuum, washed with Et2O and H2O, and then dried. Purification via silica gel flash chromatography was performed to afford the pure products.
N‐(4‐Sulfamoylphenyl)‐3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanamide (37b): Yield 42%. Experimental as reported above.
N‐(3‐Sulfamoylphenyl)‐3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanamide (37c): Yield 48%. Experimental as reported above.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)piperazin‐1‐yl]benzenesulfonamide (48a): White solid 61% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.73 (1H, s), 7.67 (2H, d, J = 8.64 Hz), 7.61 (1H, d, J = 8.48 Hz), 7.52 (1H, s), 7.50 (1H, d, J = 8.68 Hz), 7.12 (2H, bs, SO2NH 2, exchange with D2O), 7.09 (1H, s), 7.06 (2H, d, J = 6.28 Hz), 6.91 (2H, d, J = 8.88 Hz), 6.86 (1H, s), 6.81 (2H, d, J = 8.76 Hz), 4.56 (2H, q, J = 11.39 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.56 Hz), 3.67 (6H, m), 3.53 (1H, dd, J = 10.00 Hz; J = 4.90 Hz), 3.46 (1H, m), 3.30 (2H, m), 3.04 (4H, m), 2.63 (4H, m), 2.58 (4H, m), 1.34 (1H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 170.8, 153.4, 152.5, 146.7, 136.1, 135.4, 134.0, 133.3, 131.5, 131.0, 128.5, 128.2, 128.0, 122.0, 118.0, 116.0, 114.8, 108.6, 75.5, 68.7, 67.6, 54.7, 53.7 51.5, 50.3, 48.2, 47.8, 45.3, 41.4; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C37H43Cl2N7O6S 784.7540, found 784.7546; Elemental analysis, calculated: C, 56.63; H, 5.52; N, 12.49; found: C, 56.67; H, 5.49; N, 12.47.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)piperazin‐1‐yl]‐3‐chlorobenzenesulfonamide (48b): White solid 49% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.86 (1H, d, J = 2.12 Hz), 7.75 (1H, dd, J = 8.53 Hz; J = 2.08 Hz), 7.73 (1H, d, J = 2.08 Hz), 7.61 (1H, d, J = 8.48 Hz), 7.52 (1H, s), 7.49 (1H, dd, J = 8.48 Hz; J = 2.11 Hz), 7.42 (2H, bs, SO2NH 2, exchange with D2O), 7.33 (1H, d, J = 8.56 Hz), 7.05 (1H, s), 6.92 (2H, d, J = 9.16 Hz), 6.86 (1H, s), 6.81 (2H, d, J = 9.08 Hz), 4.56 (2H, q, J = 11.28 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 5.01 Hz), 3.68 (4H, m), 3.65 (2H, m), 3.54 (1H, dd, J = 10.22 Hz; J = 5.28 Hz), 3.11 (2H, ap s), 3.05 (6H, m), 2.64 (4H, m), 2.59 (4H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 170.8, 152.5, 152.3, 146.7, 139.9, 139.4, 136.1, 135.4, 133.3, 131.5, 131.0, 128.7, 128.5, 128.2, 128.0, 126.6, 122.0, 118.0, 116.0, 108.6, 75.5, 68.7, 67.6, 54.7, 53.7, 51.8, 51.5, 51.3, 50.3, 46.0, 42.0; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C37H42Cl3N7O6S 818.1916, found 818.2004; Elemental analysis, calculated: C, 54.25; H, 5.17; N, 11.97; found: C, 54.28; H, 5.16; N, 11.95.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)piperazin‐1‐yl]‐3‐(trifluoromethyl)benzenesulfonamide (48c): White solid 57% yield; 1H NMR (400 MHz, DMSO‐d 6 ) δ(ppm): 8.12 (1H, s), 8.09 (1H, d, J = 8.52 Hz), 7.73 (2H, m), 7.61 (1H, d, J = 8.47 Hz), 7.53 (2H, bs, SO2NH 2, exchange with D2O), 7.51 (1H, s), 7.49 (1H, dd, J = 8.54 Hz; J = 2.06 Hz), 7.05 (1H, s), 6.92 (2H, d, J = 9.08 Hz), 6.85 (1H, s), 6.81 (2H, d, J = 9.07 Hz), 4.56 (2H, q, J = 11.27 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 5.03 Hz), 3.67 (6H, m), 3.55 (1H, dd, J = 10.08 Hz; J = 5.08 Hz), 3.05 (4H, m), 2.99 (2H, m), 2.94 (2H, m), 2.63 (4H, m), 2.59 (4H, m); 13C NMR (100 MHz, DMSO‐d 6 ) δ(ppm): 170.8, 155.6, 152.6, 146.8, 143.3, 139.6, 138.2, 136.3, 135.5, 133.4, 131.7, 131.1, 128.6, 128.3, 123.7, 122.1, 120.2, 118.1, 116.1, 114.9, 114.7, 108.7, 75.6, 68.8, 67.7, 54.8, 53.8, 51.6, 51.0, 50.4, 45.8, 41.8; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H42Cl2F3N7O6S 852.7522, found 852.7526; Elemental analysis, calculated: C, 53.52; H, 4.96; N, 11.50; found: C, 53.49; H, 4.93; N, 11.47.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)piperazin‐1‐yl]‐3‐fluorobenzenesulfonamide (48d): White solid 70% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.73 (1H, d, J = 2.04 Hz), 7.58 (3H, m), 7.52 (1H, s), 7.49 (1H, dd, J = 8.50 Hz; J = 2.09 Hz), 7.36 (2H, bs, SO2NH 2, exchange with D2O), 7.21 (1H, t, J = 8.70 Hz), 7.05 (1H, s), 6.92 (2H, d, J = 9.12 Hz), 6.86 (1H, s), 6.81 (2H, d, J = 9.08 Hz), 4.56 (2H, q, J = 11.35 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.52 Hz), 3.67 (6H, m), 3.53 (1H, dd, J = 10.11 Hz; J = 5.20 Hz), 3.43 (1H, t, J = 7.00 Hz), 3.20 (3H, d, J = 5.20 Hz), 3.11 (2H, m), 3.05 (4H, m), 2.61 (6H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 170.7, 152.5 (d, J 1 C‐F = 248.2 Hz), 146.7, 143.2 (q, J 3 C‐F = 7.32 Hz), 139.5, 138.1 (q, J 3 C‐F = 5.97 Hz), 136.1, 135.4 (J 1 C‐F = 275.0 Hz), 133.3, 131.5, 131.0, 128.5 (t, J 2 C‐F = 35.45 Hz), 128.2, 123.6, 122.0, 120.0, 118.0, 116.0, 114.5, 108.6, 75.5, 68.7, 67.6, 54.7, 53.7, 51.5 (t, J 2 C‐F = 38.3 Hz), 50.9, 50.3, 45.7 (t, J 2 C‐F = 18.8 Hz), 41.7, 31.6; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C37H42Cl2FN7O6S 802.7444, found 802.7452; Elemental analysis, calculated: C, 55.36; H, 5.27; N, 12.21; found: C, 55.38; H, 5.29; N, 12.18.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)‐1,4‐diazepan‐1‐yl]benzenesulfonamide (49a): White solid 52% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.73 (1H, d, J = 1.96 Hz), 7.62 (2H, t, J = 4.48 Hz), 7.60 (1H, s), 7.52 (1H, s), 7.50 (1H, dd, J = 8.92 Hz; J = 2.32 Hz), 7.05 (1H, s), 7.03 (2H, bs, SO2NH 2, exchange with D2O), 6.88 (5H, m), 6.81 (2H, dd, J = 8.93 Hz; J = 1.76 Hz), 4.57 (2H, q, J = 12.16 Hz), 4.37 (1H, m), 3.89 (1H, t, J = 7.46 Hz), 3.77 (1H, m), 3.65 (6H, m), 3.53 (1H, dd, J = 9.67 Hz; J = 4.73 Hz), 3.43 (1H, s), 3.01 (4H, m), 2.52 (6H, m), 2.12 (6H, s); 13C NMR (DMSO‐d 6 , 100 MHz): 171.3, 152.5, 150.3, 146.7, 139.4, 136.1, 135.4, 133.3, 132.5, 131.5, 131.2, 131.0, 128.5, 128.2, 122.0, 118.0, 116.0, 108.6, 75.5, 68.7, 67.6, 54.8, 53.7, 51.5, 50.6, 50.3, 48.1, 46.9, 45.0, 31.0, 30.7, 30.5; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H45Cl2N7O6S 798.7810, found 798.7828; Elemental analysis, calculated: C, 57.14; H, 5.68; N, 12.27; found: C, 57.18; H, 5.65; N, 12.24.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)‐1,4‐diazepan‐1‐yl]‐3‐chlorobenzenesulfonamide (49b): Light yellow solid 86% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.81 (1H, m), 7.72 (1H, d, J = 2.04 Hz), 7.68 (1H, m), 7.61 (1H, d, J = 8.44 Hz), 7.51 (1H, s), 7.49 (1H, dd, J = 8.51 Hz; J = 2.09 Hz), 7.37 (2H, bs, SO2NH 2, exchange with D2O), 7.33 (1H, d, J = 8.68 Hz), 7.05 (1H, s), 6.90 (2H, m), 6.86 (1H, s), 6.81 (2H, d, J = 9.08 Hz), 4.56 (2H, q, J = 11.30 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.48 Hz), 3.67 (6H, m), 3.55 (1H, m), 3.46 (1H, m), 3.35 (6H, m), 2.61 (6H, m), 2.03 (4H, ap s), 1.95 (1H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 153.5, 153.4, 152.5, 146.7, 139.5, 138.5, 136.1, 135.4, 133.3, 131.5, 131.0, 129.0, 128.5, 128.2, 126.5, 126.3, 122.0, 118.0, 116.0, 108.6, 75.5, 68.7, 67.6, 60.7, 54.8, 53.8, 53.6, 51.5, 48.8, 47.1, 46.4, 44.6, 29.7, 28.2; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H44Cl3N7O6S 832.2230, found 832.2224; Elemental analysis, calculated: C, 54.78; H, 5.32; N, 11.77; found: C, 54.81; H, 5.35; N, 11.74.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)‐1,4‐diazepan‐1‐yl]‐3‐fluorobenzenesulfonamide (49c): White solid 29% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.73 (1H, d, J = 2.04 Hz), 7.61 (1H, d, J = 8.48 Hz), 7.52 (1H, s), 7.49 (2H, dd, J = 8.46 Hz; J = 2.05 Hz), 7.47 (1H, d, J = 5.27 Hz), 7.23 (2H, bs, SO2NH 2, exchange with D2O), 7.10 (1H, q, J = 8.35 Hz), 7.05 (1H, s), 6.90 (2H, t, J = 8.16 Hz), 6.86 (1H, s), 6.81 (2H, m), 4.56 (2H, q, J = 11.38 Hz), 4.37 (1H, m), 3.90 (1H, t, J = 7.50 Hz), 3.67 (5H, m), 3.53 (5H, m), 3.47 (1H, t, J = 5.60 Hz), 3.00 (4H, m), 2.95 (1H, m), 2.52 (4H, m), 2.03 (3H, s), 1.92 (1H, m), 1.84 (1H, m); 13C NMR (DMSO‐d 6 , 100 MHz): 171.6, 152.5, 151.6 (J 1 C‐F = 280.53 Hz), 146.7, 139.4 (d, J 3 C‐F = 6.90 Hz), 136.1, 135.4, 134.2 (J 2 C‐F = 30.5 Hz), 133.3 (d, J 3 C‐F = 6.48 Hz), 131.5, 131.0, 128.5, 128.2, 123.8, 122.0, 118.5, 118.0, 117.5 (d, J 3 C‐F = 4.66 Hz), 113.8 (d, J 2 C‐F = 25.44 Hz), 108.6, 75.5, 68.7, 67.6, 60.6, 54.8 (d, J 3 C‐F = 5.01 Hz), 51.5 (d, J 3 C‐F = 5.34 Hz), 50.3, 49.4, 48.2, 45.0, 28.9, 26.8, 21.6, 15.0; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H44Cl2FN7O6S 816.7714, found 816.7728; Elemental analysis, calculated: C, 55.88; H, 5.43; N, 12.00; found: C, 55.91; H, 5.46; N, 12.03.

4‐[4‐(3‐[4‐(4‐{[(2 R,4S)‐2‐[(1H‐Imidazol‐1‐yl)methyl]‐2‐(2,4‐dichlorophenyl)‐1,3‐dioxolan‐4‐yl]methoxy}phenyl)piperazin‐1‐yl]propanoyl)‐1,4‐diazepan‐1‐yl]‐3,5‐difluorobenzenesulfonamide (49d): White solid 56% yield; 1H NMR (DMSO‐d 6 , 400 MHz): 7.71 (1H, d, J = 1.96 Hz), 7.60 (1H, d, J = 8.48 Hz), 7.51 (1H, s), 7.46 (5H, m), 7.04 (1H, s), 6.90 (2H, d, J = 7.04 Hz), 6.85 (1H, s), 6.80 (2H, d, J = 8.88 Hz), 4.56 (2H, q, J = 11.23 Hz), 4.36 (1H, m), 3.89 (1H, t, J = 7.44 Hz), 3.67 (5H, m), 3.60 (1H, ap t, J = 5.96 Hz), 3.54 (1H, dd, J = 10.12 Hz; J = 5.19 Hz), 3.49 (1H, m), 3.41 (3H, ap q, J = 6.98 Hz), 3.03 (4H, m), 2.61 (6H, m), 2.12 (2H, s), 1.92 (1H, s), 1.82 (1H, s); 13C NMR (DMSO‐d 6 , 100 MHz): 171.7, 171.5, 158.0 (dd, J 1 C‐F = 248.15; 7.94 Hz), 155.4, 152.5, 146.7, 139.5 (t, J 2 C‐F = 12.28 Hz), 138.0, 135.4, 132.8, 131.6, 131.0, 128.5 (d, J 3 C‐F = 9.18 Hz), 122.0, 118.0, 116.0, 111.0 (d, J 3 C‐F = 9.21 Hz), 108.6, 75.5, 68.7, 67.7, 65.9, 54.9, 53.8, 51.5, 50.3, 49.8, 47.3, 47.2, 31.6, 31.1, 30.4; ESI‐HRMS (m/z) calculated for [M+H]+ ion species C38H43Cl2F2N7O6S 834.7618, found 834.7626; Elemental analysis, calculated: C, 54.68; H, 5.19; N, 11.75; found: C, 54.71; H, 5.17; N, 11.79.
4.2. In Vitro CA Inhibition Assay
The CA‐catalyzed CO2 hydration activity measurement was performed on an Applied Photophysics stopped‐flow instrument using phenol red, at a concentration of 0.2 mM, with a pH indicator with 20 mM HEPES (pH 7.5) as the buffer, 20 mM Na2SO4, and following the initial rates of the CA‐catalyzed CO2 hydration reaction for a period of 10–100 s and working at a maximum absorbance of 557 nm. The CO2 concentrations ranged from 1.7 to 17 mM. Enzyme concentrations varied between 5 and 12 nM [24, 25]. For each inhibitor, six traces of the initial 5%−10% of the reaction have been used to determine the initial velocity. The uncatalyzed reaction rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (0.1 mM) were prepared in distilled H2O, and dilutions up to 0.01 nM were prepared. Solutions containing an inhibitor and an enzyme were preincubated for 15 min at room temperature before the assay to allow the formation of the E−I complex. The inhibition constants were obtained by nonlinear least‐squares methods using PRISM 3 and the Cheng–Prusoff equation as reported earlier [26], and represent the mean from at least three different determinations. All CAs were recombinant ones and were obtained in‐house [27, 28].
4.3. Antifungal Activity Evaluation Methods
All the fungal strains were stored at –80°C in cryovials, and then the inoculum preparations for broth microdilution antifungal assays were performed following the CLSI guidelines for antifungal susceptibility testing of yeasts, with some minor modifications (Clinical and Laboratory Standards Institute (CLSI) [29]. Briefly, yeasts M. pachydermatis DSMZ 6172 (CDC 16334) (MP), M. furfur ATCC 14521 (MF), and M. globosa ATCC MYA 4612 (MG) were inoculated into 6 mL of modified RPMI 1640 broth (Gibco, Life Technologies Limited, UK) with the addition of ingredients suggested by Rojas et al. and incubated at 37°C for 48 h for MP and MF, while MG was incubated at 35°C for 72 h [30]. The fungal inoculum was prepared by suspending in phosphate buffer (PB), 10 mM pH 7 four to five colonies of about 1 mm diameter. The fungal suspension was then adjusted to an optical density of 0.5 McFarland, to reach a final concentration in the wells of the plates of 5 × 105 CFU/mL. Tested compounds were dissolved in DMSO at 25.6 mg/mL, and then two‐fold dilutions of the tested compounds were performed to reach final concentrations ranging from 256 to 0.00390 µg/mL. KTZ (purchased from Sigma Aldrich, Saint Louis, MO USA) was dissolved in DMSO at a concentration of 25.6 mg/mL and tested at a final range of 32–0.008 µg/mL. Growth and sterility controls were performed. After 48 h of incubation at 37°C for MP and MF and at 35°C for 72 h for MG, a MIC reading was performed. Quality control strains (C. albicans ATCC 11006, American Type Culture Collection, Manassas, VA, USA, and M. pachydermatis DSM 6172, German Collection of Microorganisms and Cell Cultures GmbH, DSMZ, Braunschweig, DE) were added each day to check the accuracy of the drug dilutions and the reproducibility of the results. For each test, three experiments were performed, with three replicates each. The MIC value of each tested compound against each strain was calculated as the average value of replicates (µg/mL) ± standard deviation (SD).
4.4. Cell Viability Test
Compounds' viability was evaluated on Human Keratinocyte cell line (HaCat) cells (BS CL 168), purchased from Biobanking of Veterinary Resources of Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna (Via Bianchi 9, 25124, Brescia, Italy). In a microtiter 96‐well plate, about 5 × 103 cells/mL per well were incubated for 12 h at 37°C in DMEM medium in a humidified atmosphere with 5% CO2 in the presence of 0.0625–8 μg/mL of the tested compounds and KTZ. Negative controls were performed both with 1% DMSO and with cells and DMEM only. After incubation, 10 μL of MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) (Sigma Aldrich, Saint Louis, MO, USA) was added to each well and incubated at 37°C for 6 h. At the end of the incubation, 100 μL of the solubilization solution (10% SDS in 0.01 M HCl) was added to each well and then incubated for 12 h. The yellow tetrazolium MTT salt is reduced in metabolically active cells to form insoluble purple formazan crystals, which are solubilized by the addition of a detergent. After incubation, plates were read using a spectrophotometer, and the optical density was measured at 540 nm [31].
4.5. Sterol Analysis
4.5.1. Compound Dilution
A volume of 1.0 mL of dimethyl sulfoxide (DMSO) was added to 9a–d, 35a, 38, 49b, 49c, and the reference drug KTZ, which were serially diluted to 1, 100, and 10 µg/mL in Modified Dixon (DM) media and stored at –20°C. A volume of 1.0 L DM adjusted to pH 6 contained 36 g of malt extract, 20 g of desiccated Ox‐Bile, 6 g of peptone, 10 mL of Tween 40, 2 mL of glycerol, and 2 mL of oleic acid.
4.5.2. Culturing of Wild‐Type C. albicans
C. albicans was cultured in 10 mL of YPD media in 60 mL pots at 30°C for 2 days. A volume of 1.0 L of YPD for agar plates contains 2% agar, 10 g of yeast extract, 20 g of peptone, and 20 g of glucose.
4.5.3. Drug‐Treated Growth of C. albicans
Final concentrations of 9a‐d, 35a, 38, 49b, 49c, and the reference drug KTZ were based on the MIC value for KTZ from previous work [21] and the MIC values for M. globosa reported in Table 2. The media were spiked with each drug to obtain the final concentrations and inoculated with 100 µL of culture to obtain 10 mL of culture. The cultures were incubated at 30°C with shaking for 2 days.
4.5.4. Cells Pelleting From C. albicans
A total of 1.0 mL from each culture was transferred to 1.5 mL eppendorph tubes and centrifuged at 3000 rpm for 3 min. The supernatant was removed, and another 1 mL of culture was added and centrifuged at 3000 rpm. This procedure was repeated so that in total, 3 mL of culture was pelleted. The supernatant was removed, and the pellet was resuspended in deionized H2O, centrifuged at 3000 rpm for 3 min. This was repeated once more. The supernatant was removed, and the pellet was stored at −20°C until sterols were extracted.
4.5.5. Sterol Extraction
Each pellet was resuspended in 1.0 mL of MeOH and transferred to glass test tubes. A volume of 3 mL of 60% w/v KOH and MeOH (1/1) was added. A volume of 1.0 mL of 0.5% pyrogallol was added; glass marbles were placed on top of the test tubes and covered with an aluminum foil. The sample was vortexed for 30 s to mix the components, heated in a water bath at 80°C for 2 h, and allowed to cool. Approximately 1.0 mL of n‐hexane was added to each tube, which was then vortexed for 30 s, and allowed to settle to form two layers. The top n‐hexane layer containing the sterols was removed and transferred to a 2 mL glass vial. This was repeated so that approximately 2 mL of the n‐hexane fraction was collected. n‐hexane was dried under a flow of nitrogen, leaving a dried residue of sterols, which was derivatized by adding 400 µL of pyridine and 100 µL of BSTFA, and heating at 70°C on a hot block for 1 h.
4.5.6. GC‐MS Sterol Analysis
Trimethylsilyl (TMS)‐derivatized sterols were analyzed using a Thermo Trace 1300 GC system (Agilent Technologies) coupled to a Thermo ISQ LT‐MS unit and identified with reference to retention times and fragmentation spectra for known standards. A DB‐5MS fused silica column (30 m × 0.25 mm × 0.25 µm film thickness) was used for all GC separations (J&W Scientific). The initial oven temperature was held at 70°C for 4 min, followed by ramping (25°C/min) to a final temperature of 280°C; this temperature was held for 25 min. Samples were analyzed in splitless mode (1 L injection volume) using helium carrier gas and electron impact ionization (ion source temperature, 290°C) and scanning from 50 to 700 atomic mass units (amu). GC‐MS data files were analyzed using Agilent software (MSD Enhanced ChemStation) to generate sterol profiles and for the derivation of integrated peak areas.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1: Mass spectra of the main peaks (> 1% of the dominant peak) were compared with NIST database spectra to identify each component Compounds in green identified with probability < 20%; Compounds in yellow 2nd library hit identified with probability below 20%.
Table S2: The peak area of each identified peak was used to calculate their percentages in relation to the total peak areas of all identified peaks.
Acknowledgments
F.C. was grateful to Ente Cassa di Risparmio di Firenze for partially supporting this work. Project title “Progettazione e Sintesi di Nuovi Agenti per la Cura e la Profilassi di Infezioni Batteriche Resistenti”. CRF 2018.1001. Open access publishing facilitated by Universita degli Studi di Firenze, as part of the Wiley ‐ CRUI‐CARE agreement.
Contributor Information
Andrea Angeli, Email: andrea.angeli@unifi.it.
Fabrizio Carta, Email: fabrizio.carta@unifi.it.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Donato R., Sacco C., Pini G., and Bilia A. R., “Antifungal Activity of Different Essential Oils Against Malassezia Pathogenic Species,” Journal of Ethnopharmacology 249 (2020): 112376. [DOI] [PubMed] [Google Scholar]
- 2. WHO . WHO releases first‐ever list of health‐threatening fungi, Accessed February 18, 2025, https://www.who.int/news/item/25-10-2022-who-releases-first-ever-list-of-health-threatening-fungi. [PMC free article] [PubMed]
- 3. Rhimi W., Theelen B., Boekhout T., Otranto D., and Cafarchia C., “Malassezia Spp. Yeasts of Emerging Concern in Fungemia,” Frontiers in Cellular and Infection Microbiology 10 (2020): 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bond R., Ferguson E. A., Curtis C. F., Craig J. M., and Lloyd D. H., “Factors Associated With Elevated Cutaneous Malassezia Pachydermatis Populations in Dogs With Pruritic Skin Disease,” Journal of Small Animal Practice 3 (1996): 103. [DOI] [PubMed] [Google Scholar]
- 5. Bond R., Morris D. O., Guillot J., et al., “Biology, Diagnosis and Treatment of Malassezia Dermatitis in Dogs and Cats: Clinical Consensus Guidelines of the World Association for Veterinary Dermatology,” Veterinary Dermatology 1 (2020): 75. [DOI] [PubMed] [Google Scholar]
- 6. Rhimi W., Theelen B., Boekhout T., Aneke C. I., Otranto D., and Cafarchia C., “Conventional Therapy and New Antifungal Drugs Against Malassezia Infections,” Medical Mycology 59 (2021): 215–234. [DOI] [PubMed] [Google Scholar]
- 7. Billamboz M. and Jawhara S., “Anti‐Malassezia Drug Candidates Based on Virulence Factors of Malassezia‐Associated Diseases,” Microorganisms 11 (2023): 2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arakaki R. and Welles B., “Ketoconazole Enantiomer for the Treatment of Diabetes Mellitus,” Expert Opinion on Investigational Drugs 19 (2010): 185–194. [DOI] [PubMed] [Google Scholar]
- 9. Kunze K. L., Nelson W. L., Kharasch E. D., Thummel K. E., and Isoherranen N., “Stereochemical Aspects of Itraconazole Metabolism In Vitro and In Vivo,” Drug Metabolism and Disposition 34 (2006): 583–590. [DOI] [PubMed] [Google Scholar]
- 10. Brown G. D., Denning D. W., Gow N. A. R., Levitz S. M., Netea M. G., and White T. C., “Hidden Killers: Human Fungal Infections,” Science Translational Medicine 4 (2012): 165rv13. [DOI] [PubMed] [Google Scholar]
- 11. Fisher M. C., Hawkins N. J., Sanglard D., and Gurr S. J., “Worldwide Emergence of Resistance to Antifungal Drugs Challenges Human Health and Food Security,” Science 360 (2018): 739–742. [DOI] [PubMed] [Google Scholar]
- 12. Fones H. N., Fisher M. C., and Gurr S. J., “Emerging Fungal Threats to Plants and Animals Challenge Agriculture and Ecosystem Resilience,” Microbiology Spectrum 5 (2017): 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Supuran C. T. and Capasso C., “A Highlight on the Inhibition of Fungal Carbonic Anhydrases as Drug Targets for the Antifungal Armamentarium,” International Journal of Molecular Sciences 22 (2021): 4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Supuran C. T., “Emerging Role of Carbonic Anhydrase Inhibitors,” Clinical Science 135 (2021): 1233–1249. [DOI] [PubMed] [Google Scholar]
- 15. Angeli A., Velluzzi A., Selleri S., et al., “Seleno Containing Compounds as Potent and Selective Antifungal Agents,” ACS Infectious Diseases 9 (1905): 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carta F., Angeli A., Selleri S., Supuran C. T., Cabassi C. S., and Spadini C., “Carbamoselenoyl Derivatives as Anti‐Infective Agents,” WO, no. A1, 2023073634.
- 17. Spadini C., Mezzasalma N., Odigie A. E., et al., “Comparative Efficacy of Selenoureido Carbonic Anhydrase Inhibitors and Azole Antifungal Drugs Against Clinical Isolates of Malassezia Pachydermatis,” Veterinary Dermatology 36 (2025): 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. D'Ambrosio K., Di Fiore A., Alterio V., et al., “Multiple Binding Modes of Inhibitors to Human Carbonic Anhydrases: An Update on the Design of Isoform‐Specific Modulators of Activity,” Chemical Reviews 125 (2025): 150–222. [DOI] [PubMed] [Google Scholar]
- 19. Khalifah R. G., “The Carbon Dioxide Hydration Activity of Carbonic Anhydrase,” Journal of Biological Chemistry 246 (1971): 2561–2573. [PubMed] [Google Scholar]
- 20. Cai L., Qin X., Xu Z., et al., “Comparison of Cytotoxicity Evaluation of Anticancer Drugs Between Real‐Time Cell Analysis and CCK‐8 Method,” ACS Omega 4, no. 7 (2019): 12036–12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martel C. M., Parker J. E., Bader O., et al., “Identification and Characterization of Four Azole‐Resistant erg3 Mutants of Candida albicans ,” Antimicrobial Agents and Chemotherapy 54 (2010): 4527–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.NIST Data. Accessed February 18, 2025, https://www.nist.gov/data.
- 23. Van Tyle J. H., “Ketoconazole; Mechanism of Action, Spectrum of Activity, Pharmacokinetics, Drug Interactions, Adverse Reactions and Therapeutic Use,” Pharmacotherapy 6 (1984): 343. [DOI] [PubMed] [Google Scholar]
- 24. D'Agostino I., Zara S., Carradori S., et al., “Antimalarial Agents Targeting Plasmodium Falciparum Carbonic Anhydrase: Towards Dual Acting Artesunate Hybrid Compounds,” Chem Med Chem 1 (2023): e202300267. [DOI] [PubMed] [Google Scholar]
- 25. Baroni C., Bozdag M., Renzi G., et al., “X‐Ray Crystallographic and Kinetic Studies of Biguanide Containing Aryl Sulfonamides as Carbonic Anhydrase Inhibitors,” RSC Medicinal Chemistry 16 (2025): 1633–1640, 10.1039/d4md01018c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yung‐Chi C. and Prusoff W. H., “Relationship Between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50% Inhibition (I50) of an Enzymatic Reaction,” Biochemical Pharmacology 23 (1973): 3099. [DOI] [PubMed] [Google Scholar]
- 27. Berrino E., Michelet B., Vitse K., et al., “Superacid‐Synthesized Fluorinated Diamines act as Selective hCA IV Inhibitors,” Journal of Medicinal Chemistry 67 (2024): 19460–19474. [DOI] [PubMed] [Google Scholar]
- 28. Kilbile J. T., Sapkal S. B., Renzi G., et al., “Lasamide Containing Sulfonylpiperazines as Effective Agents for the Management of Glaucoma Associated Symptoms,” Chem Med Chem 19 (2024): e202400601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clinical and Laboratory Standards Institute (CLSI) . Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts. 4th edition M27. Vol. M27. 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA; 2017.
- 30. Rojas F. D., Sosa M. A., Fernandez M. S., Cattana M. E., Cordoba S. B., and Giusiano G. E., “Antifungal Susceptibility of Malassezia Furfur, Malassezia Sympodialis, and Malassezia Globosa to Azole Drugs and Amphotericin B Evaluated Using a Broth Microdilution Method,” Medical Mycology 52 (2014): 641–646. [DOI] [PubMed] [Google Scholar]
- 31. Donofrio G., Franceschi V., Capocefalo A., Cavirani S., and Sheldon I. M., “Bovine Endometrial Stromal Cells Display Osteogenic Properties,” Reproductive Biology and Endocrinology 6 (2008): 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mass spectra of the main peaks (> 1% of the dominant peak) were compared with NIST database spectra to identify each component Compounds in green identified with probability < 20%; Compounds in yellow 2nd library hit identified with probability below 20%.
Table S2: The peak area of each identified peak was used to calculate their percentages in relation to the total peak areas of all identified peaks.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.