

Abstract
Chimeric antigen receptor (CAR) T cell immunotherapy relies on CAR targeting of tumor-associated antigens; however, heterogenous antigen expression, interpatient variation and off-tumor expression by healthy cells remain barriers. Here we develop synthetic antigens to sensitize solid tumors for recognition and elimination by CAR T cells. Unlike tumor-associated antigens, we design synthetic antigens that are orthogonal to endogenous proteins to eliminate off-tumor targeting and that have a small genetic footprint to facilitate efficient tumor delivery to tumors by lipid nanoparticles. Using a camelid single-domain antibody (VHH) as a synthetic antigen, we show that adoptive transfer of anti-VHH CAR T cells to female mice bearing VHH-expressing tumors reduced tumor burden in multiple syngeneic and xenograft models of cancer, improved survival, induced epitope spread, protected against tumor rechallenge and mitigated antigen escape in heterogenous tumors. Our work supports the in situ delivery of synthetic antigens to treat antigen-low or antigen-negative tumors with CAR T cells.
Chimeric antigen receptor (CAR) T cell therapy for solid tumors remains limited1–3. CAR targeting of tumor-associated antigens (TAA) is particularly challenging4 as heterogenous antigen expression, low antigen density and antigen loss result in poor efficacy and antigen escape5–7. Expression of CAR antigens by healthy tissues also poses safety concerns from on-target off-tumor toxicities8–10. For example, CD19 is expressed by healthy B cells and brain mural cells11 and patients that receive anti-CD19 CAR T cells require lifelong IgG replacement therapy and are at risk for neurotoxicity11. Similarly, CAR T cells designed to target solid tumor TAAs (for example, human epidermal growth factor receptor 2 (HER2) and carbonic anhydrase IX) show cross-reactivity with healthy cells in vital organs3,12, limiting their clinical translation13. Emerging strategies to address the scarcity of tumor antigens include genome-wide activation of endogenous genes using CRISPRa14, combinatorial or universal antigen sensing circuits to improve discrimination of malignant cells from normal tissue15–19 and spatially controlled redirection of CAR T cells against tumor cells that lack the CAR antigen20. To alleviate the need to validate CARs for each target antigen candidate, direct delivery of CD19 by oncolytic virus, GFP by tumor-colonizing bacteria or the fluorescent small molecule FITC by membrane-inserting ligands enabled tumor recognition by a single CAR T cell construct21–24. These efforts underscore the need to reduce the bottleneck of antigen selection and enable CAR T cell recognition of tumors that otherwise lack targetable antigens.
We develop synthetic antigens (syntAgs) to sensitize solid tumors to CAR T cell therapy and bypass tumor antigen discovery. An ideal syntAg would be orthogonal to the human proteome, small and genetically encodable for delivery by viral and nonviral approaches, stably expressed and targetable by single-chain variable fragments (scFvs) for CAR T cell design. Considering these criteria, we repurpose a xenogeneic protein, the antigen-binding fragment of the heavy-chain-only camelid antibodies (VHH), as a syntAg. Its small genetic footprint (~375 bp), thermal stability, protease resistance and pH stability have facilitated numerous in vivo applications25,26, including as neutralizing agents27,28, fusion proteins for imaging29,30, payload carriers for targeted vaccine delivery31,32 and CARs33. We deliver VHH as an mRNA-encoded syntAg using lipid nanoparticles (LNPs) and demonstrate that its expression on the surface of tumor cells results in cytotoxicity by anti-VHH CAR T cells. We show that anti-VHH CAR T cells exhibit low off-target toxicity in cocultures with peripheral blood mononuclear cells (PBMCs) and are well tolerated in naïve C57BL/6J mice up to 100 days following adoptive transfer by serum chemistry analysis. In multiple syngeneic and xenograft models of cancer-bearing VHH-treated tumors, we show that the adoptive transfer of anti-VHH CAR T cells significantly reduced tumor burden, improved survival, induced epitope spread, protected against tumor rechallenge and mitigated antigen escape. Our data show that sensitizing tumors to CAR-mediated cytotoxicity using syntAgs could potentiate antitumor immunity and improve responses against solid tumors.
Results
Expression of mRNA-encoded syntAgs by multiple tumor types
We tested two xenogeneic molecules, SunTag and VHH, as syntAgs for CAR targeting. SunTag is a repeat of the small 19-aa epitope EELLSKNYHLENEVARLKK of the yeast transcriptional factor GCN4, is orthogonal to the human proteome and binds to its cognate scFv with high affinity and specificity34. VHHs are small (~15 kDa) antigen-binding fragments derived from camelid antibodies and are highly stable in vivo25,26,35. We designed syntAg constructs with a membrane anchor for surface expression and retention, a linker to provide flexibility and either a SunTag or VHH antigen domain for CAR T cell recognition (Fig. 1a). We first compared the CD4 transmembrane (TM) domain36 to the glycosylphosphatidylinositol (GPI) anchor from the decay-accelerating factor36–38 as membrane anchors for VHH39 in wild-type (WT) cancer cells by surface staining and flow cytometry following mRNA transfection using jetMESSENGER, a commercial lipoplex formulation (Fig. 1b and Supplementary Fig. 1). Maximum surface expression, as measured by median fluorescence intensity (MFI), was ~10-fold higher for GPI-anchored VHH compared to CD4TM-anchored VHH across the three tumor cell lines (Fig. 1c). Moreover, VHH expression was maintained for 5–12 days for the GPI-anchored construct compared to 2–4 days for CD4TM–VHH (Fig. 1c). Because of higher and sustained expression, we selected GPI as the membrane anchor.
Fig. 1 |. Design of syntAg constructs.
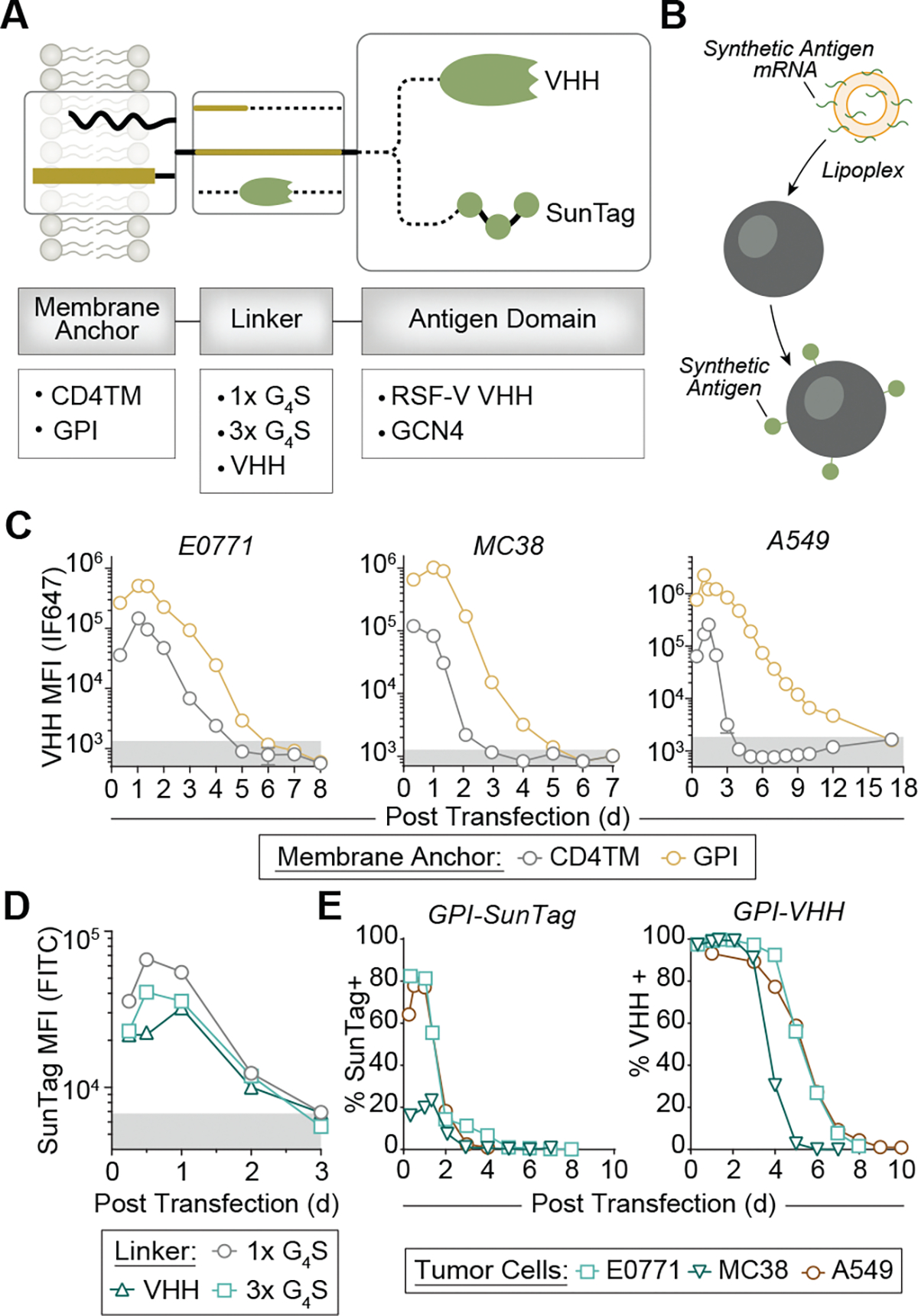
a, mRNA constructs tested consist of a membrane anchor, a linker and a recognition domain. b, These constructs are transfected into tumor cells using the lipoplex jetMESSENGER to achieve syntAg expression. c, Expression kinetics of GPI-anchored and CD4TM-anchored syntAgs consisting of a 1× G4S linker and the respiratory syncytial virus fusion (RSV-F) VHH recognition domain on the surface of indicated tumor cells. Data represent the mean of n = 3 technical replicates from a single experiment. d, Expression kinetics of mRNA constructs with a GPI anchor, SunTag recognition domain and indicated linker domains. Data represent the mean of n = 3 technical replicates from a single experiment. e, Expression kinetics of GPI-anchored SunTag or VHH on the surface of indicated tumor cell lines. Data represent the mean of n = 3 technical replicates from a single experiment.
To evaluate SunTag and VHH as antigen domains, we designed two SunTag constructs, each linked to the GPI anchor by either a 1× repeat of the flexible G4S peptide or a 3× repeat of G4S. As peptides are susceptible to enzymatic or chemical degradation, we hypothesized that fusing SunTag to a protein could enhance its surface expression. Therefore, we designed a third construct where SunTag was fused to the C terminus of VHH. All three GPI-anchored SunTag constructs were indistinguishable from background fluorescence by day 3 after transfection in A549 cells (Fig. 1d), demonstrating that neither linker length nor fusion to VHH affected surface expression of SunTag. Moreover, while VHH-linked SunTag was undetectable by day 3 (Fig. 1d, triangles), the VHH linker remained detectable on 96.7% of cells by flow cytometry 5 days after transfection (Supplementary Fig. 2), showing that VHH surface expression was longer than that of SunTag. To confirm this, we further tested VHH expression by E0771, MC38 and A549 tumor lines and found a half-maximal expression of 4.8 days compared to 1.8 days for 1×G4S-linked SunTag (Fig. 1d). Altogether, the expression of GPI-anchored VHH by murine and human cell lines was sustained for at least 5 days (Fig. 1e and Supplementary Fig. 2). For comparison, the Kb–SIINFEKL peptide major histocompatibility complex (pMHC) has a half-life of ~8 h on the surface of MC38 cells (Extended Data Fig. 1), while the half-life of the HLA-A*02:01–gp100154–162 complex on human immune cells such as dendritic cells, B cells and monocytes is 1.5–22.5 h (ref. 40). On the basis of the sustained expression of VHH (Fig. 1), we selected the VHH syntAg for subsequent validation.
LNP-mediated mRNA delivery of syntAgs to broad tumor types
We next developed an LNP formulation for syntAg delivery to tumors. We evaluated eight LNP formulations of distinct ionizable lipids, cholesterol, phospholipids and PEGylated lipids (Supplementary Fig. 3) loaded with GPI–nLuc-encoding mRNA (Fig. 2a). We screened each formulation on MDA-MB-468 tumor cells and evaluated the luminescence signal by IVIS with and without the addition of apolipoprotein E (ApoE), which adsorb onto LNPs to increase their cellular uptake through the ApoE–low-density lipoprotein receptor pathway (Fig. 2b). Among the LNP formulations tested, transfection with cKK-E12 LNPs resulted in the highest luminescence signal of ~4 × 108 photons per s (Fig. 2b). We expanded our evaluation of cKK-E12 LNPs to deliver VHH mRNA (VHH-LNPs) by testing additional murine (SB28, E0771, MC38 and GL261) and human (MDA-MB-468, A549, DU145 and IMR-32) cancer cells (Fig. 2c,d). VHH surface expression was detected on all cell types at the lowest concentration tested of 0.1 ng of encapsulated mRNA per 2 × 104 cells (Fig. 2d). The addition of ApoE improved transfection efficiency at doses of 0.1–1 ng in seven of eight cell lines and at 5–50 ng for DU145 cells. Increasing the VHH-LNP dose to 100 ng resulted in an expression frequency greater than 84.6% across all cell lines, independent of ApoE, and extended expression for up to 9 days (Fig. 2d and Extended Data Fig. 2). Taken together, these data demonstrate that cKK-E12 LNPs can effectively deliver syntAg-encoding mRNA for its sustained surface expression on a broad range of tumor types.
Fig. 2 |. mRNA-loaded LNPs deliver syntAgs to the surface of tumor cells.
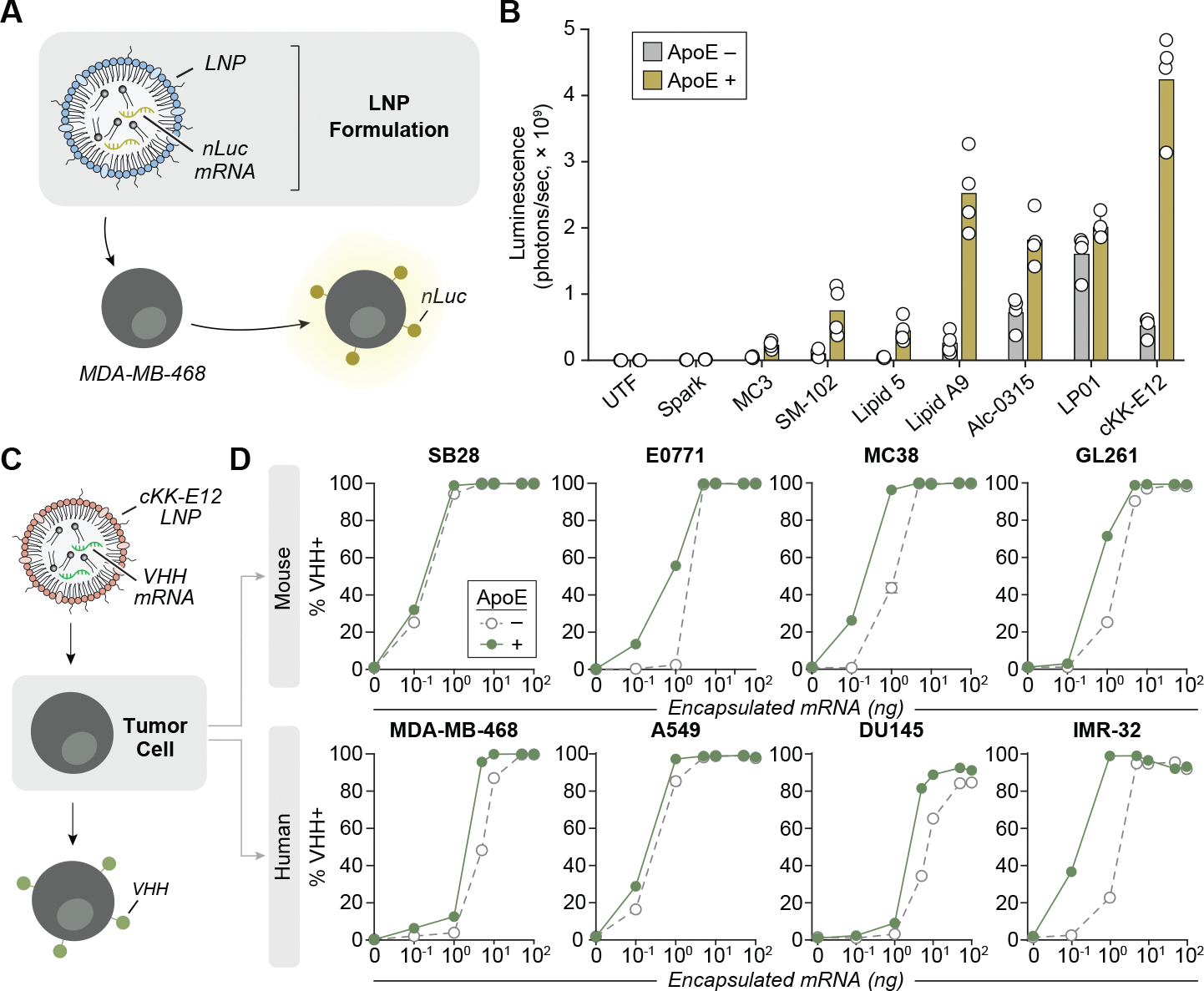
a, Various LNP formulations were loaded with mRNA encoding for GPI-anchored nLuc and coincubated with MDA-MB-468 tumor cells for 18 h before quantifying nLuc expression. b, nLuc expression on MDA-MB-468s after coincubation with various LNP formulations (1 μg ml−1) with or without 1 μg ml−1 ApoE. Luminescence was quantified by IVIS 18 h after transfection. Data represent the mean of n = 4 technical replicates from a single experiment. c, VHH-LNPs were screened against various murine and human tumor cell lines. d, VHH expression was quantified by flow cytometry 16–18 h after transfection at the indicated concentrations of VHH-LNPs with or without 1 μg ml−1 ApoE. Data represent the mean of n = 4 technical replicates from a single experiment.
Anti-VHH CAR T cells recognize and kill VHH-expressing tumor cells
To design a murine CAR to target the VHH syntAg, an anti-VHH scFv sequence was fused to the murine CD8α hinge and TM, CD28 costimulatory and the CD3ζ signaling domains. Similarly, human anti-VHH CARs comprised the anti-VHH scFv but with human sequences for the CD8α hinge and TM, 4–1BB costimulatory and CD3ζ signaling domains (Fig. 3a,b). We also designed anti-SunTag CARs by replacing the VHH-targeting scFv with an anti-GCN4 scFv as a negative control. After viral transduction, both anti-VHH and anti-SunTag CAR constructs were found to be stably expressed on the surface of primary monoclonal (Pmel-1) or polyclonal (C57BL6/J) murine T cells (Supplementary Fig. 4) and human T cells (Fig. 3c). To confirm antigen-dependent activation, we coincubated murine anti-VHH CAR T cells with E0771 breast cancer cells transfected with either VHH or SunTag and observed significant upregulation of activation markers CD25 and CD69 in greater than 94% of CAR T cells expressing the cognate syntAg (Supplementary Fig. 5 and Extended Data Fig. 3). To assess CAR T cell cytotoxicity, anti-VHH CAR T cells or control anti-SunTag CAR T cells produced interferon-γ (IFNγ) at higher levels over WT T cells when cocultured with VHH-expressing or SunTag-expressing tumor cells, respectively (Fig. 3d). By contrast, no IFNγ elevation was detected when cultured alone or with tumor cells expressing a mismatched syntAg (Fig. 3d). IFNγ levels were similar among engineered murine CAR T cells sourced from either Pmel-1 or C57BL6/J mice (Extended Data Fig. 3b).
Fig. 3 |. Anti-VHH CAR T cells recognize and kill tumor cells expressing their cognate syntAgs in murine and human models.

a, Genetic circuit design of the murine and human anti-syntAg CAR. b, Schematic of murine and human CAR constructs expressed on the surface of tumor cells. c, Representative surface expression of anti-SunTag or anti-VHH CARs on primary murine or human T cells following viral transduction. E0771 tumor cells were transfected with VHH or SunTag mRNA and coincubated with either murine anti-SunTag (S) or anti-VHH (V) CARs. Data represent the mean of n = 3 technical replicates from a single experiment. APC, allophycocyanin. d,e, IFNγ secretion (d) and killing (e) by indicated murine T cells were measured following a 24-h coculture at a 2:1 E:T ratio with transfected tumor cells. Data represent the mean of n = 4 technical replicates from a single experiment. f, Killing of untransfected, SunTag-transfected or VHH-transfected A549 tumor cells following coincubation with WT T cells, anti-SunTag CAR T cells or anti-VHH CAR T cells for 24 h quantified by LDH assay. Data represent the mean of n = 3 technical replicates (untransfected and Suntag mRNA) or mean of n = 4 technical replicates (VHH mRNA), each from a single experiment. mCAR, murine CAR.
Next, we assessed anti-VHH CAR T cell killing of VHH-expressing tumor cells. At a 2:1 effector-to-target (E:T) ratio, no cytolytic activity was observed by untransduced (UTD) murine T cells on either SunTag-transfected or VHH-transfected E0771 tumor cells or by CAR T cells cultured on tumor cells expressing a mismatched syntAg. To determine potential for nonspecific toxicity, we cultured anti-VHH and anti-SunTag CAR T cells with untransfected or transfected A549 tumor cells at six E:T ratios ranging from 1:10 to 20:1. We observed no cytolytic activity on untransfected, VHH-transfected or SunTag-transfected tumor cells across all E:T ratios tested (Fig. 3f). When anti-VHH CAR T cells were coincubated with their cognate syntAg, however, we observed increases in cytotoxicity for both murine (63.2%) (Fig. 3e and Extended Data Fig. 3c) and human (Fig. 3f) CAR T cells. Both murine and human VHH CAR T cells killed target cells more potently than their anti-SunTag CAR T cell counterparts (Fig. 3e,f). Murine VHH CAR T cells killed 16.6% more tumor cells at a 2:1 E:T ratio (VHH: 63.2% versus SunTag: 46.6%) and human VHH CAR T cells showed a 1.7-fold lower E:T ratio required to generate a 50% killing response (EC50: VHH = 0.9 versus SunTag = 1.5), consistent with earlier data that showed higher secretion of IFNγ by anti-VHH CAR T cells (Fig. 3d). Collectively, our data show that tumor cells expressing the VHH syntAg can be recognized and killed by both murine and human CAR T cells.
Anti-VHH CAR T cells are well tolerated systemically
We postulated that anti-VHH CAR T cells would exhibit minimal off-tumor toxicity because its target antigen is not natively found in the mouse or human proteome. To test this, we administered primary murine anti-VHH CAR T cells intravenously into immunocompetent and naïve C57BL/6J mice (Fig. 4a). Compared to saline controls or mice receiving an equal number of UTD T cells, we found no significant elevations in serum levels of alanine aminotransferase (ALT) or aspartate transaminase (AST) (Fig. 4b) up to 100 days after transfer nor did we detect elevations in other serum chemistry values, including total protein, urea nitrogen, creatinine, phosphorous and calcium, as measured on day 7 (Extended Data Fig. 4a). Moreover, no significant difference was observed in body weight among naïve mice receiving saline, UTD T cells or anti-VHH CAR T cells (Extended Data Fig. 4b). By contrast, we observed B cell depletion and upregulation of AST and ALT by murine CAR T cells targeting the endogenous antigens CD19 and HER2, respectively, indicative of systemic toxicity (Extended Data Fig. 4c,d). These data demonstrate that murine anti-VHH CAR T cells were well tolerated systemically, with no appreciable abnormalities in renal and liver function, body weight or B cell counts.
Fig. 4 |. Anti-VHH CAR T cells are well tolerated by immunocompetent mice and isolated human PBMCs.
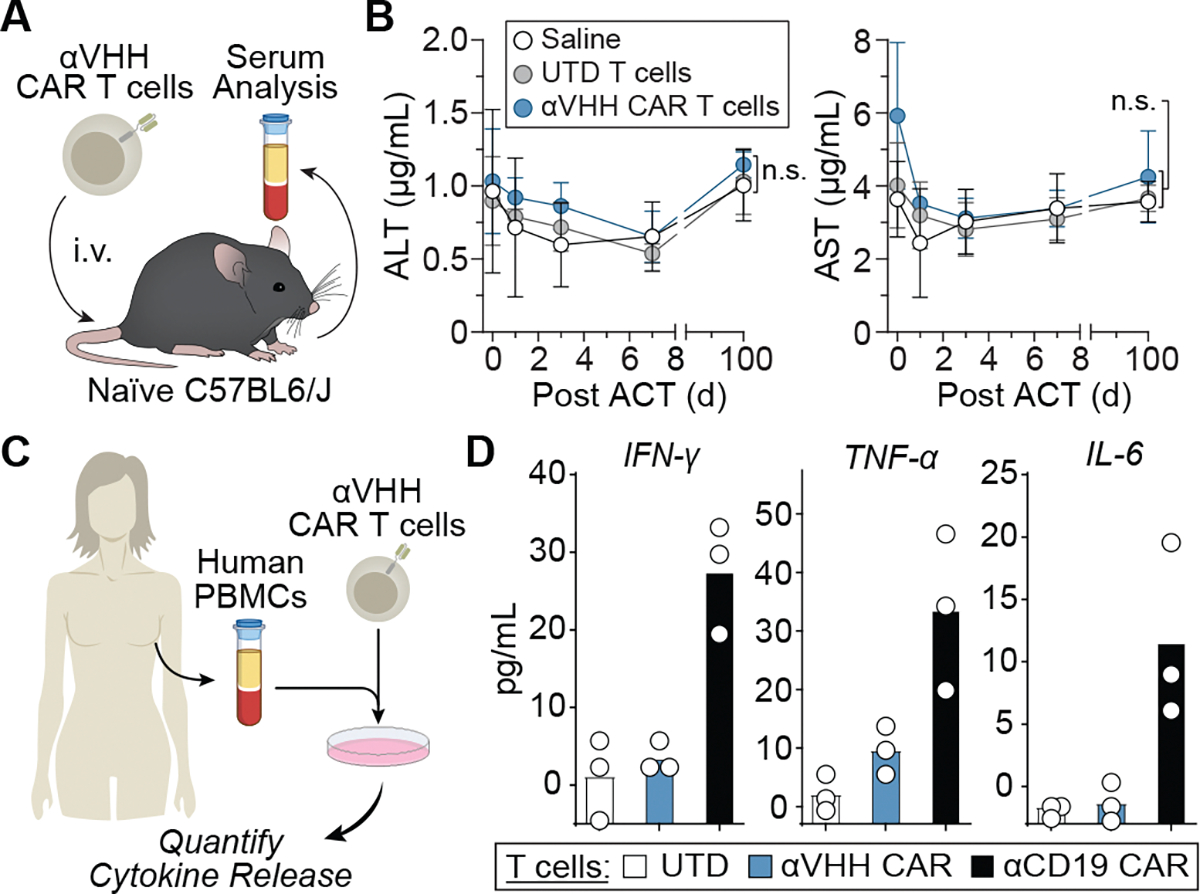
a, Systemic toxicity of anti-VHH CAR T cells was assessed by the measure of liver enzyme ALT and AST levels in serum following systemic administration (intravenously) of indicated T cells. i.v., intravenous. b, Serum analysis of AST and ALT at indicated days after ACT. Statistical analysis was performed using a one-way ANOVA (mean ± s.e.m., n = 5 mice). NS, nonsignificant. c, Human anti-VHH CAR T cells were cocultured human PBMCs from the matching donor. d, IFNγ, TNF and IL-6 secretion by human anti-VHH CAR T or anti-CD19 CAR T cells following a 48-h coculture at a 1:1 E:T ratio with donor-matched PBMCs. Data represent the mean of n = 3 technical replicates from a single experiment.
We next determined off-tumor toxicity of human anti-VHH CAR T cells by quantifying in vitro immunogenicity against donor-matched PBMCs, benchmarking their activity against CAR T cells targeting the CD19 antigen. We coincubated PBMCs from healthy donors with UTD, anti-VHH CAR or anti-CD19 CAR T cells at a 1:1 E:T ratio and measured the release of inflammatory cytokines IFNγ, tumor necrosis factor (TNF) and interleukin 6 (IL-6) after 48 h (Fig. 4c,d). As expected, the coincubation of PBMCs with anti-CD19 CAR T cells led to an increase in all the measured inflammatory cytokines compared to UTD T cells, whereas coincubation with anti-VHH CAR T cells did not lead to detectable cytokine secretion, underscoring improved safety of human CAR T cells targeting VHH rather than CD19.
Anti-VHH CAR T cells enhance antitumor immunity in solid tumors
To evaluate the antitumor efficacy of anti-VHH CAR T cells, we first compared the tumor growth kinetics of E0771 and MC38 tumor cells stably transduced to express VHH and found that VHH expression did not alter tumor growth compared to their WT counterparts (Extended Data Fig. 5). Similarly, because anti-VHH CAR T cells target a xenogeneic protein not found in murine tissue, we did not expect that the adoptive transfer of anti-VHH CAR T cells alone would alter tumor growth in the absence of target antigen. To test this, we compared tumor growth kinetics of WT E0771 tumors treated with UTD or anti-VHH CAR T and found no changes to tumor growth compared to saline-treated mice (Extended Data Fig. 6). These data show that neither VHH expression nor anti-VHH CAR T cell treatment alone is sufficient to reduce tumor burden.
We compared the antitumor efficacy of anti-VHH CAR T cells against anti-HER2 CAR T cells as a benchmark (Extended Data Fig. 7a). We coincubated MDA-MB-468 breast cancer cells transduced with both the VHH and HER2 antigen (468-HV) with UTD, anti-VHH CAR or anti-HER2 CAR T cells at a 2:1 E:T ratio. We observed no cytolytic activity by UTD T cells and an increase in cytotoxicity by both anti-VHH (99.3%) and anti-HER2 (98.3%) CAR T cells (Extended Data Fig. 7b). Notably, the potency of anti-VHH CAR T cells was indistinguishable from anti-HER2 CARs. Following adoptive cell transfer in cohorts of mice bearing 468-HV tumors (Extended Data Fig. 7c), we observed tumor clearance in mice treated with either anti-HER2 or anti-VHH CAR T cells (UTD versus anti-VHH CAR, ***P < 0.001; UTD versus anti-HER2 CAR, †††P < 0.001) but not in control mice treated with UTD T cells, demonstrating that the efficacy of VHH-targeting CARs is comparable to a conventional CAR targeting an endogenous antigen.
To evaluate the potential for antigen spread, we adoptively transferred murine anti-VHH CAR T cells into immunocompetent mice bearing syngeneic MC38 colon adenocarcinoma tumors that were stably transduced to express VHH. Compared to control anti-SunTag CAR T cells, transfer of anti-VHH CAR T cells significantly reduced tumor burden (Fig. 5a,b); within 18 days of adoptive cell transfer, tumor volumes within the control group grew ~9-fold on average, whereas those treated with anti-VHH CAR T cells grew ~2.5-fold. Flow cytometric analysis of the tumor, spleen and the ipsilateral and contralateral lymph nodes revealed that anti-VHH CAR T cell treatment increased the number of CD8+ tumor-infiltrating lymphocytes (TILs) compared to treatment with control CAR T cells (**P < 0.01; Fig. 5c). Additionally, CAR+ CD8 T cells comprised a higher frequency of the CD8+ TILs within mice receiving anti-VHH CAR T cells (55% versus 22%, **P < 0.01; Fig. 5c). To determine whether CAR targeting of VHH could prime endogenous immunity against a different tumor antigen, we quantified the percentage of endogenous T cells reactive against Reps1, an H-2Db-restricted mutant neoepitope in MC38 cells41. We observed a significant increase in CAR-negative, Reps1-reactive CD8+ T cells (*P < 0.05; Fig. 5c and Supplementary Fig. 6) in the tumor and in the ipsilateral lymph nodes (*P < 0.05; Fig. 5d), which was not observed in animals given control CAR T cells (Fig. 5d). These data support that CAR-mediated killing of VHH+ tumors primes endogenous T cells against tumor antigens and promotes epitope spread.
Fig. 5 |. Adoptive transfer of anti-VHH CAR T cells into mice with VHH-expressing tumors delays tumor growth, enhances infiltration of tumor-reactive T cells and promotes endogenous antitumor immunity.

a, MC38-VHH tumors were treated with murine anti-VHH CAR T cells and monitored for tumor growth. T cells were isolated from tumors, spleens and lymph nodes on day 18 for downstream analysis. s.c., subcutaneous; LN, lymph node; TCR, T cell receptor. b, Tumor growth curves following ACT. Statistical analysis was performed using a two-way ANOVA (mean ± s.e.m., n = 4 mice). **P = 0.0043 and ****P < 0.0001. c, Count and frequency of indicated T cell populations isolated from the tumor. Statistical analysis was performed using an unpaired one-sided Student’s t-test (mean displayed, n = 4 tumors for control CAR T cells and n = 5 tumors for anti-VHH CAR T cells). Left, **P = 0.0013; middle, **P = 0.0016; right, *P = 0.0492. d, Representative flow plot (n = 5 for anti-VHH CAR T cells and n = 4 for control CAR T cells) of endogenous (CAR−) T cell expression of the Reps1 T cell receptor in tdLNs and the frequency of this population in the ipsilateral and contralateral draining lymph nodes. Statistical analysis was performed using an unpaired two-sided Student’s t-test (mean displayed, n = 3 independent lymph nodes for contralateral control CAR T cell, n = 4 independent lymph nodes for ipsilateral control CAR T cell and contralateral anti-VHH CAR T cell and n = 5 independent lymph nodes for ipsilateral anti-VHH CAR T cell. *P = 0.0374. NS, nonsignificant. Each lymph node was obtained from an individual mouse. e, E0771-VHH tumors were treated with murine anti-VHH CAR T cells. f, Individual traces of tumor growth curves of E0771-VHH tumor-bearing mice treated with control CAR T cells (black) or anti-VHH CAR T cells (blue) (n = 4 mice). g, Next, 45 days after initial treatment, cured mice (n = 3) were rechallenged with WT E0771 tumor cells. Statistical analysis was performed using a two-way ANOVA (mean ± s.e.m., n = 5 mice). ****P < 0.0001. h, Survival curves of tumor-bearing mice following treatment. Statistical analysis was performed using a log-rank (Mantel–Cox) test (n = 4 mice). **P = 0.0019.
We further sought to evaluate the antitumor efficacy of anti-VHH CAR T cells against triple-negative breast cancer (TNBC), which lacks estrogen and progesterone receptors and HER2 to make them unresponsive to endocrine and anti-HER2 therapies42,43. We tested anti-VHH CAR T cells in mice bearing E0771 tumors, a TNBC originally isolated from a spontaneous mammary tumor in a female C57BL/6J mouse44. Following adoptive transfer of anti-VHH CAR T cells (Fig. 5e), three of four mice treated with anti-VHH CAR T cells were complete responders (CRs) for at least 100 days after adoptive T cell therapy (ACT) (blue traces, Fig. 5f), whereas all mice that received control CARs reached endpoint criteria (tumor volume > 1,000 mm3) within 30 days of treatment (black traces, Fig. 5f). To determine whether treatment could initiate epitope spread and provide protection against the WT tumor, either naïve mice or CRs were rechallenged with WT VHH-negative E0771 tumor cells in the contralateral mammary fat pad 45 days after initial therapy (Fig. 5e). While naïve mice grew tumors that reached the endpoint in 28–32 days, CRs rechallenged with VHH-negative E0771 tumors were resistant to tumor growth (****P < 0.0001; Fig. 5g). Additionally, the survival of rechallenged mice was significantly extended by at least 70 days over mice treated with control CAR T cells (**P < 0.01; Fig. 5h).
SyntAg treatment triggers potent antitumor responses
We next sought to explore whether in situ delivery of syntAgs by LNPs could sensitize WT tumors to CAR T cell cytotoxicity. We evaluated the ability of anti-VHH CAR T cells to kill TNBC tumor cells transfected with VHH-LNPs in vitro (Fig. 6a). At doses up to 100 ng of VHH-LNP, we observed no cytolytic activity by UTD murine or human T cells on E0771 and MDA-MB-468 tumor cells, respectively (Fig. 6b). However, when anti-VHH CAR T cells were coincubated with VHH-LNP-transfected target cells, we observed an increase in cytotoxicity against both E0771 and MDA-MB-468 cells at doses above 1 ng (Fig. 6b). To validate cKK-E12 LNPs for tumor transfection, we loaded LNPs with mRNA encoding a GPI-anchored nLuc reporter to enable analysis of tissues by luminescence imaging. Thus, 24 h after intratumoral injection, tumors treated with nLuc-LNPs showed an 83-fold increase in luminescence compared to tumors treated with LNPs loaded with control mRNA (****P < 0.001; Extended Data Fig. 8a). Low levels of luminescence signal were detected in the spleen and tumor-draining lymph node (tdLN), whereas negligible signals were detected from the other major organs. We also measured tumor luminescence following multiple intratumoral injections of nLuc-LNPs and found that a second dose 4 days following the initial injection increased the luminescence signal, demonstrating the ability to boost mRNA delivery with repeated injections (**P < 0.01; Extended Data Fig. 8b).
Fig. 6 |. LNP-mediated syntAg delivery sensitizes tumors to CAR-mediated cytoxicity in models of TNBC.

a, Murine and human tumor cells were transfected with VHH-LNPs for 18 h before coincubation with anti-VHH CAR T cells. b, Cytotoxicity of E0771 and MDA-MB-468 tumor cells quantified by LDH assay following a 20-h coincubation with either murine or human UTD or anti-VHH CAR T cells, respectively, at various VHH-LNP doses. Data represent the mean of n = 4 technical replicates from a single experiment. c, Mice bearing WT E0771 tumors were treated intratumorally with saline, Fluc-LNP or VHH-LNP followed by adoptive transfer of anti-VHH CAR T cells. m.f.p., mammary fat pad; i.t., intratumoral. d, Left, MFI of VHH expression on E0771 tumor cells 24 h after LNP treatment. Right, representative flow plot of three independent mice from the same population. Statistical analysis was performed using an unpaired two-sided Student’s t-test (mean ± s.e.m., n = 3 tumors, each from individual mice). *P = 0.0480. FSC-A, forward scatter area. e, Tumor growth curves of WT E0771 tumor-bearing mice treated intratumorally with saline, Fluc-LNP or VHH-LNP and anti-VHH CAR T cells on days indicated by a dashed line (days 0 and 7). Statistical analysis was performed using a mixed-effects analysis with Tukey correction for multiple comparisons (mean ± s.e.m., n = 5 mice for Fluc-LNP and VHH-LNP treatments and n = 6 mice for saline treatment). *P = 0.0204 and ****P < 0.0001. f, Survival curves of tumor-bearing mice following two (n = 5 mice for Fluc-LNP and VHH-LNP treatments and n = 6 mice for saline treatment) or five (n = 5 mice) doses of indicated LNPs and CARs. g, Mice bearing WT MDA-MB-468 tumors were treated intratumorally with either saline or VHH-LNP followed by adoptive transfer of anti-VHH CAR T cells. h, Tumor growth curves of WT MDA-MB-468 tumor-bearing mice treated with saline, VHH-LNP or VHH-LNP + anti-VHH CAR T cells on days indicated by a dashed line (days 0 and 7). Statistical analysis was performed using a two-way ANOVA (mean ± s.e.m., n = 4 mice for VHH-LNP + CAR T cells and n = 5 mice for all other cohorts). ****P < 0.0001. hCAR, human CAR.
To validate the expression of VHH by tumors following intratumoral injections of VHH-LNPs, we next injected WT tumors intratumorally with 5 μg of VHH-LNPs and quantified the MFI of dissociated tumor cells. The MFI of VHH-LNP-treated tumor cells was 33-fold greater compared to cells obtained from saline-injected tumors (*P < 0.05), corresponding to a mean frequency of 26.1% VHH+ tumor cells (Fig. 6c,d). Notably, VHH-LNP injection at this dose did not alter tumor growth (Extended Data Fig. 9a) nor did the administration of anti-VHH CAR T cells to Fluc-LNP treated mice (Fig. 6e). By contrast, two treatment doses of both systemically administered anti-VHH CAR T cells and an intratumoral injection of VHH-LNPs led to a significant delay in tumor growth in a murine model of TNBC (****P < 0.001; Fig. 6e) and an improvement in median survival from 21–24 days in control cohorts to 35 days in syntAg-treated mice. By increasing the number of syntAg treatments from two to five doses, we observed a further improvement in median survival to 61 days (Fig. 6f and Extended Data Fig. 9b). Next, to evaluate LNP-mediated syntAg treatment of a murine model of human TNBC, we administered VHH-LNP followed by adoptive transfer of human anti-VHH CAR T cells into MDA-MB-468 tumor-bearing NSG mice (Fig. 6g). Control cohorts treated with saline or VHH-LNPs alone grew from ~80 mm3 to an average tumor volume of ~600 mm3 58 days after initial treatment, while the tumor burden in mice that received VHH-LNPs and anti-VHH CAR T cells decreased to 40 mm3 (****P < 0.001; Fig. 6h). Taken together, these data demonstrate that LNP-mediated delivery of mRNA-encoded VHH sensitizes WT tumors to immune recognition, leading to improved antitumor responses in vivo.
SyntAg treatment enhances control of heterogeneous tumors
Unlike certain hematologic malignancies where endogenous antigens such as CD19 and B cell maturation antigen are expressed on most tumor cells45–47, the expression of targetable antigens in solid tumors is highly heterogenous48–50. Targeting endogenous antigens can exert selective pressure on tumors, promoting antigen-negative relapse that is refractory to initial therapy51,52. We, therefore, sought to evaluate whether LNP-mediated delivery of the VHH syntAg combined with anti-VHH CAR T cell treatment could improve therapeutic outcomes over conventional CARs in preclinical models of heterogenous tumors. Before in vivo assessment, we compared the ability of VHH-LNP to transfect both epidermal growth factor receptor variant III (EGFRvIII)+ and EGFRvIII− SB28 tumor cells, a murine glioblastoma cell line characterized by an immunogenicity profile similar to human glioblastoma multiforme53. Following in vitro transfection with 100 ng of VHH-LNP, we observed high surface expression of VHH as quantified by flow cytometry 18 h after transfection in both EGFRvIII+ and EGFRvIII− tumor cells (EGFRvIII+, 99.9% VHH+; EGFRvIII−, 99.3% VHH+). VHH expression among EGFRvIII+ and EGFRvIII− tumors were indistinguishable from one another (Extended Data Fig. 10a).
Next, we inoculated immunocompetent mice with a tumor comprising 10% EGFRvIII+ and 90% EGFRvIII− SB28 cells (Extended Data Fig. 10b), a heterogenous antigen composition comparable to that of patients with glioblastoma eligible for EGFRvIII CAR T cell therapy in a recent clinical study54. Once SB28 tumors with heterogenous EGFRvIII composition reached ~100 mm3, a 5-μg dose of VHH-LNP was administered intratumorally. Consistent with in vitro transfection, VHH-LNP treatment led to a significant increase in VHH expression on SB28 tumors in vivo (52.0% VHH+, ***P < 0.001; Extended Data Fig. 10c,d) while the same treatment did not alter EGFRvIII expression, which remained at 7.8% for both treated and untreated groups. Using murine anti-human EGFRvIII CAR T cells as a benchmark, we treated tumor-bearing mice intratumorally with either saline or 5 μg of VHH-LNP followed by adoptive transfer of either VHH-targeting or EGFRvIII-targeting murine CAR T cells once a week for a total of three treatments (Extended Data Fig. 10e). Compared to untreated mice, we did not observe a significant increase in survival in groups receiving VHH-LNPs alone or EGFRvIII CAR T cells (Extended Data Fig. 10f). All mice treated with EGFRvIII CAR T cells reached endpoint criteria (tumor volume > 1,000 mm3) within 29 days after treatment with a median survival of ~27 days, whereas mice that received both VHH-LNPs and anti-VHH CAR T cells exhibited a significant improvement in survival (**P < 0.01), with median survival improved by at least 48 days.
We next evaluated syntAg treatment on a heterogenous model of breast cancer. We inoculated a heterogenous mixture of MDA-MB-468 tumor cells where 15% of the tumor expressed human HER2 (Fig. 7a). Following tumor engraftment, mice were treated intratumorally with either saline or 5 μg of VHH-LNP followed by adoptive transfer of either VHH-targeting or HER2-targeting human CAR T cells once a week for a total of three treatments (Fig. 7b,c). VHH-LNP treatment alone did not alter tumor growth compared to untreated mice. On the contrary, treatment with VHH-LNPs and anti-VHH CAR T cells led to a significant decrease in tumor burden 16 days following the initiation of treatment, which continued to decrease until no palpable tumors were detected by day 34. Treatment of the heterogenous tumors with CAR T cells targeting the endogenous HER2 antigen, however, did not clear the tumor. To determine whether antigen escape contributed to the lack of tumor clearance, we assessed HER2 expression within remaining tumors. Compared to control cohorts that maintained expression of HER2 on the surface of tumor cells (untreated, 23.8%; VHH-LNP, 22.1 %), mice treated with anti-HER2 CAR T had a significant decrease in the frequency of HER2+ tumor cells to 1.1% (*P < 0.05; Fig. 7d,e). Collectively, our data indicate that syntAg treatment has the potential to mitigate antigen escape in tumors with heterogenous expression of endogenous antigens.
Fig. 7 |. LNP-mediated syntAg treatment augments antitumor immunity in preclinical models of heterogenous tumors and mitigates outgrowth of antigen-negative tumor cells.

a, Mice bearing a heterogenous tumor mixture comprising 85% HER2− and 15% HER2+ MDA-MB-468 tumor cells were first treated with intratumorally with either saline or VHH-LNP followed by adoptive transfer of anti-VHH CAR T cells or anti-HER2 CAR T cells. b,c, Tumor growth curves (b) and individual spider plots (c) of heterogenous MDA-MB-468 tumors treated intratumorally with saline or VHH-LNP and systemically with indicated T cells on days indicated by a dashed line (days 0, 7 and 14). Statistical analysis was performed using a mixed-effect analysis with Tukey correction for multiple comparisons (mean ± s.e.m., n = 4 mice for WT and anti-HER2 CAR T cell treatments and n = 5 mice for LNP + WT and LNP + anti-VHH CAR T cell treatments). Saline + WT versus LNP + anti-VHH CAR T cell, **P = 0.0050 on day 16, ***P = 0.0008 on day 20 and ****P < 0.0001 for days 25–34; LNP + WT versus anti-HER2 CAR T cell, *P = 0.0337 for day 27, **P = 0.0010 for day 31 and ****P < 0.0001 for day 34. d, Tumors were isolated and evaluated for HER2 expression using flow cytometry. Data represent flow plots from n = 4 tumors, each from individual mice. e, Quantification of endogenous antigen (HER2) tumor composition at endpoint. Statistical analysis was performed using a multiple unpaired one-sided t-test (mean ± s.e.m., n = 4 tumors, each from individual mice). *P = 0.0427 for HER2 CAR versus LNP + WT and *P = 0.0267 for HER2 CAR versus saline + WT. H, HER2; V, VHH.
Discussion
The scarcity of tumor-specific antigens limits the safety and efficacy of CAR T cell therapies for solid tumors. We developed syntAgs to sensitize solid tumors for recognition and elimination by CAR T cells. Unlike TAAs, we designed a syntAg VHH that is orthogonal to the human proteome and has a small genetic footprint for efficient delivery to tumors by LNPs. We found that a GPI membrane anchor linked to a CAR recognition domain led to stable VHH surface expression by tumor cells following mRNA transfection. To deliver VHH to tumors in vivo, we tested LNP formulations comprising distinct ionizable lipids and found that particles formulated with cKK-E12 successfully transfected all tumor cells tested of both mouse and human origin. We also designed and validated both human and murine anti-VHH CAR T cells, which showed potent antigen-dependent activation, cytokine production and cytotoxicity upon recognition of surface-expressed VHH on tumor cells in vitro. Importantly, anti-VHH CAR T cells exhibited potent antitumor activity in vivo against VHH+ tumors in syngeneic models of colorectal and TNBC, which led to the recruitment of endogenous tumor-reactive cells, resistance to tumor rechallenge and extended survival. We further demonstrated that LNP-mediated delivery of VHH combined with anti-VHH CAR T cells led to the recognition and killing of tumors in both syngeneic and xenograft tumor models of glioblastoma and TNBC, which reduced tumor burden, improved survival and mitigated antigen escape in heterogenous tumors.
The majority of tumor antigens under evaluation as CAR targets are also expressed at low levels in healthy tissue. Consequently, on-target off-tumor toxicity with CAR T cells remains a primary safety concern including severe respiratory distress, hearing loss, vestibular dysfunction, liver toxicity and neurotoxicity11,12,55–58. As a syntAg derived from camelids, VHH is not expressed by healthy tissue. Our data demonstrate that murine anti-VHH CAR T cells are well tolerated in vivo, as measured by serum chemistry and body weights of non-tumor-bearing mice. Moreover, we show that the coincubation of human anti-VHH CAR T cells with donor-matched PBMCs does not elicit expression of cytokines related to cytokine release syndrome, unlike CAR T cells targeting CD19. Nonetheless, because of differences in the mouse and human proteome and the limitations of preclinical models, murine studies and in vitro toxicity assays may not fully recapitulate CAR T cell cytotoxicity in humans59; thus, as with any new CAR T cell construct under consideration, future studies are warranted to assess the potential off-tumor activity of anti-VHH CAR T against healthy human tissue. If future assays were to reveal measurable levels of reactivity that we did not observe in the studies we performed in this manuscript, several strategies from the field of CAR T cell engineering can be used for mitigation, including selecting an alternative VHH-targeting scFv2,60, optimizing other components of the CAR (that is, hinge, TM and intracellular domains)2, integrating with biocircuits designed to increase tumor specificity such as protease-regulated CARs61 or the tumor-localized control of CAR expression using thermally sensitive promoters20. Notably, however, the compact size, low immunogenicity and stability of VHHs shown in numerous preclinical studies25,26,28–33,35 and its US Food and Drug Administration (FDA) approval as a neutralizing agent against the von Willebrand factor27 further support that this class of molecules has the potential for clinical translation.
In addition to antigen identification, each CAR construct requires independent optimization for parameters such as affinity62,63, linker lengths and intracellular domains51,64. Streamlining CAR design with a single optimized CAR construct that could be used for multiple types of tumors could accelerate clinical testing. Other strategies to deliver T cell targets to tumor cells including antibody-mediated delivery of immunodominant viral epitopes65, intratumoral or systemic delivery of sfGFP using tumor-colonizing bacteria (Escherichia coli Nissle 1917)24, intratumoral injection of membrane anchoring FITC21 and local delivery of CD19 using oncolytic virus, for subsequent treatment with their cognate CAR T cells22,23. In our studies, we observed that CAR T cell clearance of tumors expressing the VHH syntAg primed endogenous T cells against neoantigens, as indicated by the increased number of Reps1 tetramer-positive T cells in the tumor and draining lymph nodes in the syngeneic MC38 murine model of colon adenocarcinoma. We also observed indication of epitope spread in mice bearing TNBC tumors where CRs to anti-VHH CAR T cell therapy were resistant to rechallenge to WT tumor cells, indicating the establishment of antitumor immunity against tumor antigens that were not targeted by CAR T cells. We envision that intratumoral delivery of VHH could be used to kickstart antitumor immunity, analogous to in situ vaccination strategies such as intratumoral delivery of toll-like receptor agonists, radiation and cytokines that use the tumor to prime or enhance immune responses66–69. Outside of LNPs, an increasing number of FDA-approved viral (for example, adeno-associated viruses or the oncolytic virus T-VEC70) and nonviral (for example, polymeric nanoparticles71,72) gene delivery vehicles are amenable for intratumoral or locoregional delivery. Whereas viral vectors are limited by preexisting or treatment-induced immunity73, LNPs can be readministered to increase the expression of the syntAg where a single dose may not be sufficient to generate durable antitumor responses. Treatment efficacy may also be boosted by using circular RNA74 or self-amplifying mRNA75 to extend VHH expression or through codelivery of VHH mRNA with adjuvants, cytokines or costimulatory molecules that promote a proinflammatory tumor microenvironment76. While nonviral vectors lack cell type specificity, nonspecific delivery to cells within the tumor microenvironment may prove advantageous, such as through the depletion of immunosuppressive stromal cells. With emerging high-throughput approaches, it is also possible to further optimize LNP formulations for improved delivery and tumor tropism in vivo77. Altogether, our studies provide support for the design and use of syntAgs to promote CAR-mediated tumor cytotoxicity for solid tumors with otherwise limited antigen targets.
Methods
Oversight and ethics
All research performed complied with relevant ethical regulations. Animal protocols were approved by the Georgia Tech Institutional Animal Care and Use Committee (IACUC). The use of recombinant DNA molecules or synthetic nucleic acid molecules was approved by the Georgia Tech Institutional Biosafety Committee. PBMCs were collected from peripheral blood of healthy volunteers above the age of 18 years and above 110 lbs, under a protocol approved by the Georgia Tech and Emory University Institutional Review Boards. Written informed consent was obtained from all participants of this study. Samples were immediately deidentified before downstream processing.
Statistics and reproducibility
Power analyses were performed using G*Power 3.1.9.6 (HHU) on the basis of effect sizes determined by previous studies. Differences in animal survival (Kaplan–Meier survival curves) were analyzed using a two-sided Mantel–Cox test. For statistical analysis of two groups, an unpaired Student’s t-test was applied. For grouped analyses, we performed a two-way analysis of variance (ANOVA). Statistical analysis was performed using GraphPad Prism (version 10.3.1). Data are presented as the mean and s.d. or s.e.m. (error bars) as defined in the legends. Differences were considered significant when the P values were <0.05. Data distribution was assumed to be normal but this was not formally tested. Mice were inoculated with tumors and then randomized before allocation to experimental groups. For the remaining studies, randomization was not relevant, as these experiments were conducted on uniform biological materials, such as commercially sourced cell lines. The investigators were not blinded to group allocation during experiments and outcome analysis. Blinding was not feasible for both in vitro and in vivo studies, as these experiments were conducted by individual investigators who were aware of the experimental groups. Additionally, blinding was not considered relevant, as the data analysis was performed quantitatively (for example, flow cytometry and ELISA) and did not involve subjective, qualitative assessments. Mice were randomly assigned to experimental groups but the experiments were not randomized and the investigators were not blinded to group allocation or outcome assessment. No data were excluded from the analyses. Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Statistical analysis
Appropriate statistical analyses were performed using GraphPad Prism (*P < 0.05, **P < 0.01, *** P < 0.001 and ****P < 0.0001). Central values represent the mean and error bars depict the s.d. or s.e.m. as defined. Flow cytometry data were analyzed using FlowJo X. Power analyses were performed using G*Power 3.1.9.6 (HHU).
Animals
Female C57BL/6J and NSG mice (6–12 weeks old) were used. C57BL/6J mice were purchased from the Jackson Laboratory (000664). B6-HER2 mice were purchased from Jackson Laboratory (010562), bred in house and used at 2–6 months of age. Tumor dimensions were measured with calipers in three dimensions and are reported as an ellipsoidal volume. Following our approved IACUC protocol, a maximal tumor size of 1.5 cm in any direction was not exceeded in any experiment.
Cell culture and generation of cell lines
Cell lines used, including their sources, are provided in Supplementary Table 1. HEK293T and MC38 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. E0771 and SB28 cells were cultured in RPMI-1640 with 10% FBS and 1% penicillin–streptomycin. SB28 cells were maintained in RPMI-1640 supplemented with 10% FBS, 1% penicillin–streptomycin, 1% HEPES, 1% Glutamax, 1% nonessential amino acids, 1% sodium pyruvate and 0.5 mM β-mercaptoethanol. VHH-positive cell lines were generated by lentiviral transduction of tumor cells with the VHH gene driven by the EF1α core promoter in a LeGO-C lentiviral backbone. VHH+ cells were single-cell-sorted using the BD Fusion in the Georgia Institute of Technology’s Cellular Analysis Core using the BD FACSDIVA v8 software (BD Biosciences). Similarly, E0771-Thy1.1 and MC38-Thy1.1 were generated by lentiviral transduction of the Thy1.1 gene, single-cell-sorted and stably maintained using Blasticidin. All cells were cultured at 37 °C in 5% CO2.
Construction of CAR T cells
REmAb protein sequencing with WILD analysis (Rapid Novor) was performed on the MonoRab rabbit anti-camelid VHH antibody to obtain the anti-VHH scFv sequence. The human CAR is composed of a human CD8α signal peptide, antigen-specific scFv (anti-GCN4 (ref. 34) or anti-VHH scFv), CD8α hinge and TM domains and the 4–1BB and CD3ζ intracellular domains. The human anti-CD19 CAR (US9499629B2) was kindly provided by K. Roy (Georgia Institute of Technology) and comprises a anti-CD19 scFv, CD8α hinge and TM domain and the 4–1BB and CD3ζ signaling domains. The human anti-HER2 CAR (US20180326032A1) was kindly provided by S. Priceman (City of Hope) and comprises anti-HER2 scFv, IgG4 hinge and CD8α TM domains and 4–1BB and CD3ζ signaling sequences. The murine CARs are composed of a mouse CD8 signal peptide, antigen-specific scFv (anti-GCN4 (ref. 34), anti-VHH, 2173 (anti-human EGFRviii)78 or trastuzumab-derived scFv (anti-human HER2)), CD8α hinge and TM domains and the CD28 and CD3ζ intracellular domains. The CAR constructs coexpress an eGFP reporter using the T2A sequence and were generated using custom DNA fragments (Eurofins Genomics). Human CAR constructs were cloned into a lentiviral vector using the BsiWI and NheI restriction sites while murine CAR constructs were cloned into the pMKO.1 retroviral vector using the EcoRI and NotI restriction sites.
In vitro transcription of syntAg mRNA
Plasmid templates were linearized with NotI-HF overnight at 37 °C, purified the following day by sodium acetate precipitation and subsequently rehydrated with nuclease-free water. In vitro transcription was performed using the HiScribe T7 kit (New England Biolabs) following the manufacturer’s instructions, using complete substitution of uridine for N1-methylpseudouridine-5′-triphosphate. The RNA product was treated with DNase for 30 min and purified using lithium chloride precipitation. RNAs were capped using guanylyl transferase and 2′-O-methyltransferase (Aldevron) to generate a Cap 1 structure, purified by lithium chloride precipitation, treated with alkaline phosphatase (New England Biolabs) and repurified. The concentration of the purified mRNA was measured using a Nanodrop and stored at −80 °C at 1–4 mg ml−1. Purified RNA product was analyzed by gel electrophoresis to ensure purity.
LNP synthesis
Lipid components were mixed in ethanol according to the molar ratios described in Supplementary Fig. 3 and mRNA was diluted in 100 mM pH 5 sodium acetate buffer to a concentration of 0.1 mg ml−1. LNPs were synthesized by mixing lipid components and mRNA solutions using NanoAssemblr Ignite (Precision Nanosystems) at a lipid-to-mRNA mass ratio of 20:1, volumetric ratio of 1:3 and total flow rate of 12 ml min−1. The formulated LNPs were buffer-exchanged into PBS by repeated centrifugation using 10-kDa Amicon Ultra-15 centrifugal filter unit at 2,000g to reach at least 300-fold dilution of the formulation buffer. The size of the LNPs was measured using a Malvern Zetasizer. LNPs were stored in sucrose (8% w/v) and kept at −80 °C until use.
To quantify the mRNA concentrations and encapsulation efficiency (EE), Invitrogen Quant-it RiboGreen RNA assay kit was used following the manufacturer’s protocol. Samples were analyzed using a fluorescence microplate reader (excitation, 485 nm; emission, 530 nm). The mRNA EE was calculated using the following equation:
Evaluation of syntAg expression kinetics following in vitro transfection
Expression of mRNA-encoded syntAg constructs was first evaluated by in vitro transfection into target cell lines using the lipoplex jetMESSENGER mRNA transfection reagent following the manufacturer’s protocol. Expression kinetics were analyzed using flow cytometry by staining for SunTag expression using an anti-GCN4 antibody or for VHH expression using an anti-camelid VHH antibody. Samples were collected using the Accuri C6 Plus flow cytometer and its software (version 1.0.27.1) at indicated time points. Subsequent assessment of VHH expression kinetics by LNP-mediated delivery was evaluated by loading VHH mRNA into various LNP formulations and coincubating with tumor cells at indicated doses in 96-well plates with or without 1 μg ml−1 ApoE in complete medium. Expression levels and kinetics were analyzed using flow cytometry with Northern Lights (Cytek) following staining and analyzed using SpectroFlo (version 3.2.1).
In vivo kinetics of expression
Evaluation of in vivo LNP kinetics was conducted using nLuc-loaded or Fluc-loaded LNPs. C57BL6/J mice were inoculated with 1 × 106 MC38 or 5 × 105 E0771 tumor cells. When tumors reached ~100 mm3, 5 μg of nLuc-LNPs or Fluc-LNPs were injected intratumorally. Luciferase expression was quantified by IVIS imaging at indicated time points using appropriate substrates (nLuc using Nano-Glo(R) in vivo substrate and Fluc using d-luciferin potassium salt, prepared following the manufacturer’s instructions). Then, 4 days after injection, indicated cohorts received a second nLuc-LNP or Fluc-LNP (5 μg) injection to evaluate luminescence by IVIS.
Lentiviral and retroviral production
Retrovirus was made by cotransfection with pCL-Eco and pMKO.1 retroviral vectors encoding murine CARs in HEK293T cells using TransIT-293. Supernatant was collected 48 h later, filtered through a 0.45-μm syringe filter and concentrated using Retro-Concentin virus precipitation solution following the manufacturer’s protocol before immediate use for transduction of primary murine T cells. Lentivirus was produced by cotransfection of lentiviral expression plasmids with psPAX2 and pMD2.G using TransIT-LT1 into HEK293T cells. Supernatant was collected after 48 h, filtered through a 0.45-μm syringe filter, concentrated using PEG-it virus precipitation solution following the manufacturer’s protocol and stored at −80 °C until use.
Primary murine CAR T cell production
Cells from Pmel-1, P14 and C57BL/6 mouse spleens were isolated by gently dissociating the tissues using frosted glass slides, centrifuged at 1,000g for 5 min and resuspended in 1× red blood cell (RBC) lysis buffer for 5 min at 4 °C. Then, 1× PBS was added to quench the lysis reaction and cells were centrifuged and resuspended in murine T cell medium (mTCM; RPMI, 10% FBS, 1% penicillin–streptomycin, 1× nonessential amino acids, 1× sodium pyruvate, 50 μM β-mercaptoethanol and 100 IU per ml recombinant human (rh) IL-2) and passed through a 40-μm cell strainer. Pmel-1 cells were cultured in the presence of 1 μM human gp100 (Pmel-1). CD3+ cells from C57BL/6 mice were isolated using the StemCell EasySep mouse T cell isolation kit following the manufacturer’s protocol. After isolation, C57BL/6 CD3+ T cells were resuspended at a concentration of 1 × 106 cells per ml in mTCM supplemented with 5 μg ml−1 anti-CD28 antibody (BD) and cultured on an anti-CD3 antibody-coated plate (3 μg per well). Then, 2 days after activation, cells were collected, washed and resuspended at 1 × 106 cells per ml in concentrated retroviral supernatant supplemented with 100 IU per ml hIL-2 and 8 μg ml−1 polybrene. Spinfection was performed in a U-bottom 96-well plate at 2,000g for 90 min at 32 °C. Transduced cells were maintained at 1 × 106 cells per ml in complete mTCM supplemented with 100 IU per ml hIL-2 and passaged daily until use on days 5–6. Murine CD19 CAR expression was evaluated by staining with Alexa Fluor 647–F(ab′)2 goat anti-mouse IgG at a 1:800 dilution. For the murine VHH, SunTag and hHER2 CARs, expression was evaluated by surface staining with biotinylated antigen (biotinylated VHH, biotinylated GCN4 synthesized in house at 100 μg ml−1 and biotinylated hHER2 10 μg ml−1) followed by a secondary stain with streptavidin–APC at a 1:500 dilution. For staining, biotinylated SunTag was synthesized on Rink Amide ProTide (LL) resin using CEM Liberty blue, including Fmoc deprotection in piperidine, amino acid coupling in N,N′-diisopropylcarbodiimide and Oxyma Pure and N-terminal biotinylation by biotin p-nitrophenyl ester in the presence of Oxyma Pure. The peptide was cleaved off resin in trifluoroacetic acid, precipitated in diethyl ether, dried overnight and resuspended at 10 mg ml−1 in water for storage at −20 °C. Additionally, surface expression of CD3, CD4 and CD8 was quantified by flow cytometry after staining T cells with anti-CD4 and anti-CD8 antibodies as described above.
Primary human CAR T cell production
Peripheral blood was drawn from healthy human donors. PBMCs were isolated using Lymphoprep density gradient medium and a SepMate 15-ml tube, according to the manufacturer’s instructions. CD3+ cells were then isolated using the EasySep human CD3-positive selection kit II and activated using Dynabeads at a 3:1 bead-to-cell ratio. Activated cells were cultured in complete human T cell medium (hTCM; X-vivo 10, 5% human AB serum, 10 mM N-acetyl l-cysteine and 55 μM β-mercaptoethanol supplemented with 50 U per ml rhIL-2) for 24 h at 37 °C in 5% CO2. Concentrated lentivirus was added to a 24-well suspension culture plate coated with Retronectin according to the manufacturer’s instructions and centrifuged at 1,200g for 90 min at 37 °C. Following centrifugation, activated human T cells in hTCM supplemented with 100 U per ml of hIL-2 were added to each well and the plate was spun at 1,200g for 60 min at 37 °C. Cells were incubated on the virus-coated plate for 24 h before expansion. Then, 7 days after activation, T cells were supplemented with Dynabeads at a 1:1 bead-to-cell ratio. The beads were removed on day 9. Cells were maintained at a concentration of 0.7 × 106–2 × 106 cells per ml until use on days 10–14.
Biodistribution study
Organs were isolated 24 h following intratumoral injection of nLuc-LNPs and immediately placed on ice-cold 1× PBS−/− until imaging. Organs were treated with the Nano-Glo live cell assay system initially diluted 20× in provided buffer and then further diluted 50× in 1× PBS−/− before use. Before IVIS imaging, organs were dipped in the diluted substrate for 5 min at 37 °C. Excess liquid was removed by tapping the tissue on a KimWipe before placing on black construction paper for imaging.
Liver function analysis
Naive C57BL6/J mice received PBS, UTD T cells or 5 × 106 CAR T cells. Then, 7 days following administration, blood was drawn through the jugular vein, collected in serum separator tubes and sent out for blood chemistry testing at Antech Diagnostics. To assess the longitudinal toxicity CAR T cells, peripheral blood samples were collected at each time point into BD Microtainer tubes and left undisturbed for 30 min before centrifugation at 2,500g for 10 min. Serum was stored at −80 °C until analysis. Collected samples were thawed and evaluated for AST and ALT levels using AST and ALT ELISA kits following the manufacturer’s instructions.
PBMC toxicity assay
PBMCs were isolated from whole blood using Lymphoprep and SepMate PBMC isolation tubes following the manufacturer’s protocol. Isolated PBMCs were cocultured with human anti-VHH CAR T or anti-CD19 CAR T cells of the matching donor at 1:1 ratio for 48 h. Supernatant was collected after incubation and assayed for human IFNγ, TNF and IL-6 with ELISA kits according to the manufacturer’s protocols.
In vitro cytotoxicity assays
Tumor cells were transfected in a 96-well plate with 100 ng of either VHH or SunTag mRNA using the jetMESSENGER transfection reagent kit following manufacturer instructions. For LNP-mediated transfections, tumor cells were treated with 1–100 ng of VHH-LNP. Transfected target cells were then cocultured with UTD, anti-VHH CAR or anti-SunTag CAR T cells at the indicated E:T ratio. Supernatant was collected after incubation and assayed for IFNγ and cytotoxicity with species-matched IFNγ ELISA kit or using a lactase dehydrogenase (LDH) release assay, respectively, according to the manufacturer’s instructions.
Comparison of murine CAR T cells sourced from either Pmel-1 or C57BL6/J splenocytes was conducted using MC38-Rluc or MC38-VHH-Rluc. Tumor cells were seeded in a 96-well plate at 2 × 104 cells per well. UTD or CAR T cells were added at a 1:1 E:T ratio and cytotoxicity was assessed by luminescence. To do so, ViviRen live cell substrate was added to wells 24 h after coculture and luminescence was quantified by IVIS Spectrum CT. The supernatant from the coculture was collected and IFNγ levels were quantified using an IFNγ mouse ELISA kit according to the manufacturer’s protocol.
Validating VHH CAR functionality in vivo against tumors constitutively expressing VHH
C57BL6/J mice were inoculated with 1 × 106 MC38-VHH, 5 × 105 E0771-VHH or 5 × 105 E0771 WT tumor cells. For E0771 experiments, cells were implanted intradermally in the left mammary fat pad (fourth). For MC38 experiments, cells were implanted subcutaneously into the left flank. Tumor burden, quantified as 0.52 × length × width × depth, was monitored until the average tumor volume was approximately 100 mm3 before initiating treatment. Mice were sublethally irradiated with 500 cGy and 5 × 106 CAR-transduced Pmel-1 splenocytes were adoptively transferred through the tail vein. rhIL-2 was administered intraperitoneally twice daily for 3 days. Mice were classified as CRs or partial responders or as having progressive disease or stable disease according to the RECIST criteria79. On day 18 after ACT, MC38-VHH tumors and lymph nodes were isolated for flow cytometry analysis. For E0771-VHH experiments, CRs were rechallenged 45 days after initial treatment with 5 × 105 syntAg-negative (E0771 WT) tumor cells in the right mammary fat pat (fourth).
Flow cytometry analysis of TILs
Antibodies used, with vendor information and dilutions, are provided in Supplementary Table 2. Unless otherwise noted, all antibodies were used at a 1:100 dilution. Single-cell suspensions from spleens and lymph nodes were prepared by homogenizing tissue between the frosted glass slides. Homogenized cells were passed through a 40-μm cell strainer and depleted of red blood cells using 1× RBC lysis buffer. Less than 1 g of MC38-VHH tumors were dissociated using the mouse tumor dissociation kit and gentleMACS dissociator. TILs were isolated from the single-cell suspension using a density gradient with Percoll centrifugation medium and DMEM supplemented with 10% FBS and 1% penicillin–streptomycin at a 44:56 volume ratio. The panel used for staining was as follows: anti-CD8–BV786 (53–6.7) and anti-Thy1.1–BV711 (OX-7). Cell viability was assessed by staining with the live/dead fixable Aqua dead cell stain kit following the manufacturer’s instructions. To detect VHH CAR expression, biotinylated VHH and streptavidin–Alexa Fluor 488 were used. Reps1-reactive T cells were detected by Reps1 pMHC tetramer staining. To ensure signal specificity, samples were stained with Reps1 pMHC tetramers separately conjugated to streptavidin–APC and streptavidin–BV421. For surface staining, cells were blocked with anti-Fc receptor anti-CD16/CD32 and then stained in FACS buffer (1× Dulbecco’s PBS, 2% FBS, 1 mM EDTA and 25 mM HEPES). Fixation was performed using eBioscience intracellular fixation and permeabilization buffer set following the manufacturer’s instructions. Counting beads were added to each sample of stained cells before analysis by the LSR Fortessa Flow Cytometer (BD).
Tetramer production
For tetramer staining, tetramers were generated from pMHC monomers and streptavidin. Biotinylated Reps1 pMHC monomers were diluted in exchange buffer (20 mM Tris-HCl and 150 mM NaCl, pH 7.0) at 0.15 mg ml−1. APC-conjugated or BV421-conjugated streptavidin was added to the diluted pMHC monomers in four small aliquots to achieve a final 4:1 pMHC-to-streptavidin molar ratio. Each aliquot was added to the diluted pMHC monomers every 5 min for a total of four times. After each addition, the pMHC–streptavidin mixture was mixed thoroughly and incubated on ice for 5 min. Then, 15 min after the last addition, the pMHC tetramer was centrifuged at 16,000g for 1 min and the supernatant was collected for cell staining at 1:50 dilution (0.2 μg per sample).
SyntAg treatment by LNP-mediated delivery of VHH
C57BL6/J mice were inoculated with 5 × 105 E0771 WT tumor cells intradermally in the left mammary fat pad (fourth). When tumors reached ~75–150 mm3, mice were sublethally irradiated with 500 cGy and injected intratumorally with 5 μg of Fluc-LNP or VHH-LNP. On the same day, 5 × 106 anti-VHH CAR T cells were administered intravenously. rhIL-2 was administered intraperitoneally twice daily for 3 days. Alternatively, NSG mice were inoculated with 5 × 106 MDA-MB-468 WT tumor cells in the left flank. When tumors reached ~50–100 mm3, mice were injected intratumorally with 5 μg of either VHH-LNP with 3 mg ml−1 ApoE4 (Peprotech, 350–04) or PBS. On the same day, 5 × 106 anti-VHH CAR T cells were administered intravenously.
To evaluate syntAg treatment of heterogenous tumors, C57BL6/J mice were inoculated with 5 × 105 SB28 (10% EGFRviii+) tumor cells subcutaneously in the left flank. Before the first treatment, mice were sublethally irradiated with 500 cGy and injected intratumorally with 5 μg of either Fluc-LNP or VHH-LNP. On the same day, 5 × 106 anti-VHH CAR T cells were administered intravenously. rhIL-2 was administered intraperitoneally twice daily for 3 days. To evaluate tumor expression of VHH, tumors from a subset of mice were isolated 24 h after LNP injection, dissociated using a mouse tumor dissociation kit and stained using anti-VHH-iFluor 647 and the live/dead fixable near-infrared dead cell stain kit. Analysis was performed using the Northern Lights flow cytometer (Cytek).
Similarly, to evaluate syntAg treatment of human tumors, NSG mice were inoculated with 5 × 106 MDA-MB-468 (15% HER2+) tumor cells resuspended in 100 μl of PBS−/− and injected subcutaneously in the left flank. When tumors reached a volume of ~100 mm3, mice were injected intratumorally with 5 μg of VHH-LNP with 3 mg ml−1 ApoE4. On the same day, 5 × 106 anti-VHH CAR T cells were administered intravenously, repeated as indicated. To evaluate tumor composition after treatment, tumors were isolated at the endpoint (day 34), dissociated using a human tumor dissociation kit and stained using the following panel: anti-human β2-microglobulin–PE, anti-VHH–iFluor 647 and live/dead fixable near-infrared dead cell stain kit. Analysis was performed using the Northern Lights flow cytometer (Cytek).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Kinetics of expression of Kb-SIINFEKL pMHC complex.

Surface expression kinetics of the Kb-SIINFEKL pMHC complex on the surface of MC38 tumor cells following peptide pulsing with the SIINFEKL peptide for one hour at room temperature. Mean of n = 3 technical replicates from a single experiment.
Extended Data Fig. 2 |. Kinetics of expression of LNP mediated delivery of VHH synthetic antigen to tumor cells.

VHH expression on MDA-MB-468 (left) and E0771 (right) following VHH-LNP treatment at the indicated doses. VHH mean fluorescent intensity (MFI) quantified by flow cytometry at indicated days after transfection. Mean of n = 4 technical replicates from a single experiment.
Extended Data Fig. 3 |. Characterization of murine CAR T cells sourced from either pmel-1 or C57BL6/J mice.

(a) Staining of indicated murine T cell population with activation markers CD25 and CD69 following co-incubation with ST- or VHH-expressing E0771 tumor cells. Data is representative of 3 technical replicates from a single experiment. (b) IFN-γ secretion by Pmel-1 or C57BL/6J derived αVHH CAR T cells quantified following a 24 hr coculture with MC38-VHH tumor cells at a 1:1 E:T ratio. Mean of n = 3 technical replicates from a single experiment. (c) Cytotoxicity of Pmel-1 and C57BL/6 derived αVHH CAR T cells were assessed following a 24 hr coculture with MC38-VHH at a 1:1 E:T ratio. Mean of n = 3 technical replicates from a single experiment.
Extended Data Fig. 4 |. Murine αVHH CAR T cells are well tolerated by immunocompetent mice.
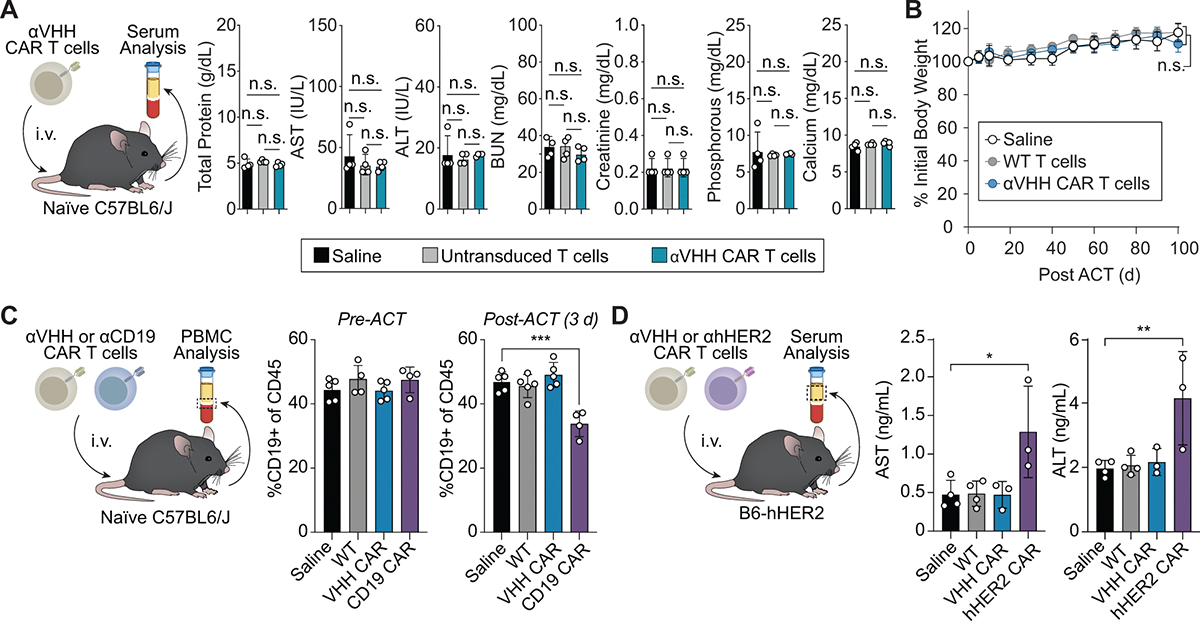
(a) Blood serum analysis 7 d post i.v. administration of αVHH CAR T cells, untransduced T cells, or saline into naïve C57BL6/J mice. One-way ANOVA, mean ± s.d., n = 4 mice.; n.s. = not significant. (b) Body weight measurements following i.v. administration of αVHH CAR T cells, wild-type T cells, or saline into naïve C57BL6/J mouse. Two-way ANOVA, mean ± s.d., n = 5 mice per cohort; n.s. = not significant. (c) PBMC analysis before and 3 days after ACT of saline, WT, αVHH CAR, or αCD19 CAR T cells into C57BL6/J mice. One-way ANOVA; mean ± s.e.m., n = 4 mice for αCD19 CAR treated mice, n = 5 mice in all other cohorts; n.s. = not significant ***p = 0.0002. (d) Blood serum analysis 3 d post i.v. administration of saline, WT, αVHH CAR, or αhHER2 CAR T cells into naïve B6-HER2 mice. One-way ANOVA; mean ± s.e.m., n = 3 mice for hHER2 and VHH treatments, n = 4 mice for Saline and WT treatments.; *p = 0.01, **p = 0.0065.
Extended Data Fig. 5 |. VHH expression alone does not alter tumor growth.
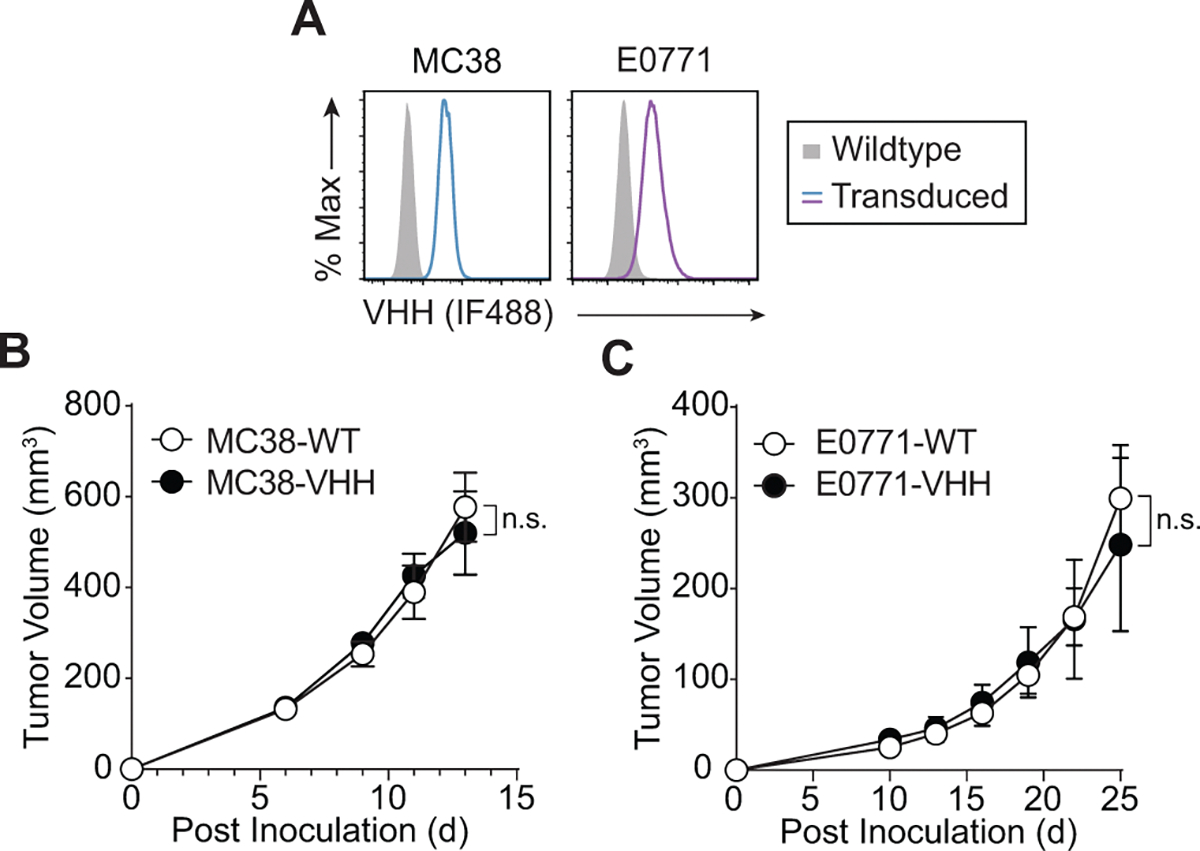
(a) VHH expression on wildtype or transduced MC38 and E0771 tumor cells. Data is representative of 2 independent experiments. (b) Tumor growth curves of wildtype MC38 (MC38-WT) or MC38 cells transduced to stably express VHH (MC38-VHH) without adoptive cell transfer of αVHH CAR T cells. n = 4 mice inoculated with MC38-WT and n = 3 mice inoculated with MC38-VHH. Two-way ANOVA, mean ± s.e.m. is depicted; n.s. = not significant (c) Tumor growth curves of wildtype E0771 (E0771-WT) or E0771 cells transduced to stably express VHH (E0771-VHH) without adoptive cell transfer of αVHH CAR T cells n = 4 mice inoculated with E0771-WT and n = 6 mice inoculated with E0771-VHH; Two-way ANOVA, mean ± s.e.m. is depicted; n.s. = not significant.
Extended Data Fig. 6 |. Adoptive transfer of αVHH CAR T cells to mice bearing wildtype tumors does not alter tumor growth.

Tumor growth curves of mice bearing wiltdype E0771 tumors treated intravenously with either saline, 5 × 106 untransduced (UTD) or αVHH CAR T cells. Two-way ANOVA, mean ± s.e.m., n = 5 mice; n.s. = not significant.
Extended Data Fig. 7 |. The antitumor activity αVHH CAR T cells is comparable to αHER2 CAR T cells against VHH + HER2+ tumors.
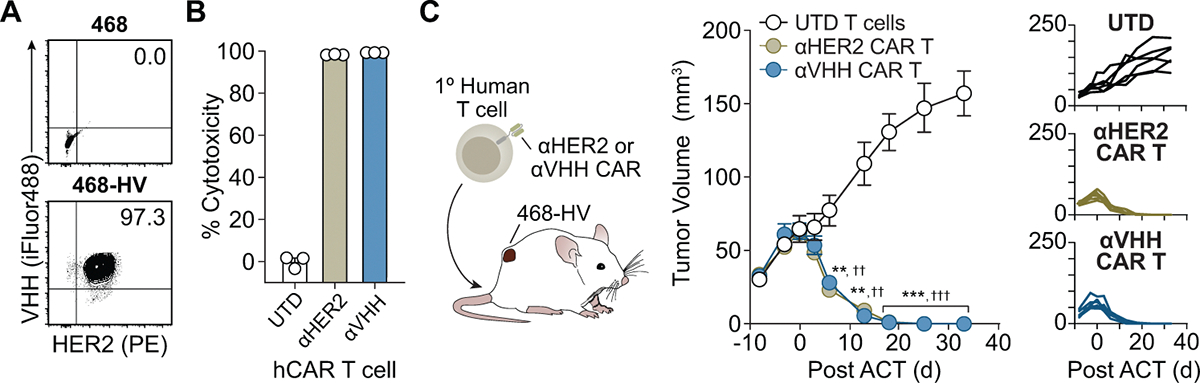
(a) Representative flow plots of HER2 and VHH expression on either wildtype or 468-HV tumor cells. Data is representative from a single experiment, repeated 2 times with similar results. (b) 468-HV tumor cells were cocultured with either human untransduced (UTD), αHER2 CAR, or αVHH CAR T cells at a 2:1 E:T ratio and assessed for cytotixicity after 24 hrs (Mean of n = 3 technical replicates) and (c) growth curves of 468-HV tumors following treatment with the UTD, αHER2 CAR, or αVHH CAR T cells. (Two-way ANOVA, mean ± s.e.m., n = 6 mice per cohort, αVHH (**p = 0.0080 on day 6, **p = 0.0015 on day 13, ***p = 0.0003 on day 18, ***p = 0.0005 on day 25, ***p = 0.0003 on day 33) and αHER2 (††p = 0.0054 on day 6, ††p = 0.0016 on day 13, †††p = 0.0003 on day 18, †††p = 0.0005 on day 25, †††p = 0.0003 on day 33) CAR T cell treatments are compared to UTD.
Extended Data Fig. 8 |. In vivo biodistribution of functional mRNA delivery mediated by CKK-LNPs following intratumoral injection.
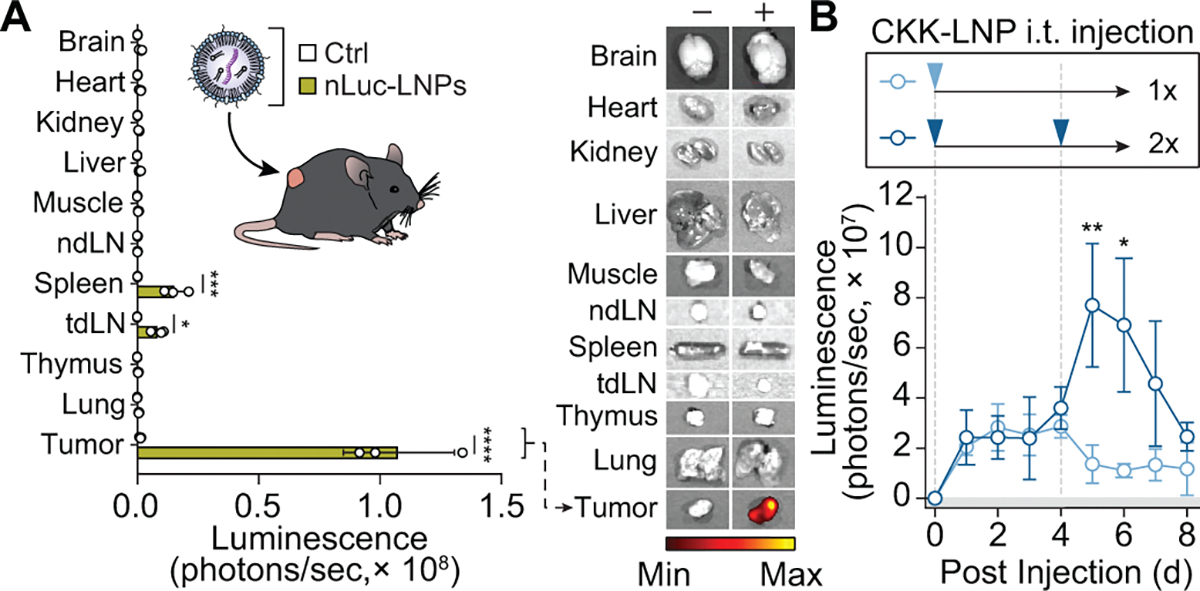
(a) In vivo biodistribution following saline or a 2 μg intratumoral injection of nLuc mRNA-loaded LNPs (nLuc-LNPs) as measured by IVIS 24 hrs following injection. Luminesence in organs quantified (left) and representative (of n = 3 organs, each from individual mice) images are displayed on the right. mean ± s.d., multiple unpaired t test, n = 3 organs, each from individual mice; *p = 0.0397, ***p = 0.00047, ****p < 0.0001. (b) In vivo kinetics of nLuc expression following one (light blue) or two (dark blue) intratumoral injections of 5 μg nLuc-LNPs. Transfection kinetics quantified by IVIS. Two-way ANOVA, mean ± s.e.m., n = 4 mice; *p = 0.0196, **p = 0.0080.
Extended Data Fig. 9 |. αVHH CAR T cells reduce tumor burden of VHH LNP injected tumors.

(a) Tumor growth curves of wildtype E0771 (E0771-wt) tumors transfected with VHH-LNPs (CKK-E12) by intratumoral injection of 5 μg mRNA-loaded LNPs without adoptive cell transfer of αVHH CAR T cells. Two-way ANOVA, mean ± s.e.m. is depicted; n = 6 saline injected mice and n = 5 for VHH-LNP injected mice.; n.s. = not significant. (b) Tumor growth curves of wildtype E0771 tumor-bearing mice treated with intratumorally with either saline, Fluc-LNP or VHH-LNP and αVHH CAR T cells on days indicated by a dashed line (D0, D7, D14, D21, and D28). Two-way ANOVA, mean ± s.e.m., n = 5 mice. Saline vs. VHH-LNP: *p = 0.0176, ***p = 0.0006, ****p < 0.0001.
Extended Data Fig. 10 |. LNP-mediated synthetic antigen treatment augments antitumor immunity in a preclinical model of heterogenous glioblastoma tumors.

(a) EGFRvIII- (vIII-) or EGFRvIII+ (vIII+) SB28 tumor cells were transfected with 100 ng of VHH-LNP and evaluated for VHH expression by flow cytometry after 18 hrs, mean of n = 5 technical replicates from a single experiment (b) Mice bearing a heterogenous tumor mixture comprised of 90% vIII- and 10% vIII+ SB28 tumor cells were treated with saline or VHH-LNP followed by adoptive transfer of indicated CAR T cells (c) 24 hrs following intratumoral injection VHH-LNP (5 μg), SB28 tumors were dissociated and analyzed by flow cytometry for EGFRvIII (left) and VHH (right) expression, one-way unpaired T test, mean ± s.d., n = 3 dissociated tumors, each from individual mice; ***p = 0.0008, n.s.=nonsignficiant. (d) Representative flow plots of VHH expression on SB28 tumors treated with either saline or VHH-LNP. Data is representative of 3 tumors each isolated from one mouse (e) Tumor growth curves of heterogenous SB28 tumor-bearing mice treated with intratumorally with saline or VHH-LNP and systemically with either WT, αVHH, or αEGFRvIII CAR T cells days indicated days by dashed lines (D0, D7, & D14) n = 5 untreated and VHH-LNP + αVHH CAR treated mice, n = 6 mice with αEGFRvIII CAR T cell treatment, n = 7 mice with VHH-LNP + saline treatment. (f) Survival curves from (e) of heterogenous SB28 tumor-bearing mice following synthetic antigen treatment, log-rank (Mantel–Cox) test; **p = 0.0019 comparing EGFRvIII CAR treatment with LNP + αVHH CAR.
Supplementary Material
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43018-025-00968-5.
Acknowledgements
We thank D. R. Meyers (Emory) and C. D. Sago for helpful insights. This work was funded by the National Institutes of Health (NIH) Director’s New Innovator Award (DP2-HD091793), NIH Director’s Pioneer Award (DP1-CA280832), National Cancer Institute (R01CA273878), National Institute of Biomedical Imaging and Bioengineering (R01EB032822), National Center for Advancing Translational Sciences (UL1TR000454), Shurl and Kay Curci Foundation and NIH Shared Instrumentation Grant (1S10OD016264–01A1) and was partially performed at the Georgia Tech Institute for Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (NSF; grant ECCS-1542174). L.G. was supported by the Alfred P. Sloan Foundation and the NIH GT BioMAT Training Grant under award number 5T32EB006343. L.G., A.S. and S.N.D. were supported by the NSF Graduate Research Fellowship under grant number DGE-1451512. A.H.Z. was supported by the NIH Kirschstein National Research Service Award program under award number F31-CA271803. C.A.T. was supported by the NIH Cell and Tissue Engineering training program under grant number 5T32GM145735. F.S. was supported by a postdoctoral fellowship from the Wallace H. Coulter Department of Biomedical Engineering and the College of Engineering at Peking University. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests
G.A.K. reports equity or consulting roles for Sunbird Bio, Port Therapeutics, Send Biotherapeutics and Ridge Biotechnologies. The terms of this arrangement were reviewed and approved by Georgia Tech in accordance with its conflict-of-interest policies. L.G., A.H.Z., D.V., P.J.S., C.S.C., H.J.L., C.A.T. and G.A.K. are listed as inventors on a patent application related to the results of this paper, titled ‘Synthetic Antigens as Chimeric Antigen Receptor (CAR) Ligands and Uses Thereof’ (US20230390335A1). The patent applicant is the Georgia Tech Research Corporation.
Extended data is available for this paper at https://doi.org/10.1038/s43018-025-00968-5.
Data availability
All raw IVIS images for Extended Data Fig. 8 are available from the Georgia Institute of Technology Research Repository (repository@library.gatech.edu) upon publication of this paper. The remaining data are available within the article and Supplementary Information or upon reasonable request to the corresponding author. Source data are provided with this paper.
References
- 1.Lim WA & June CH The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rafiq S, Hackett CS & Brentjens RJ Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 17, 147–167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou AJ, Chen LC & Chen YY Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 20, 531–550 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Martinez M & Moon EK CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 10, 128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majzner RG & Mackall CL Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Majzner RG et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 10, 702–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamieh M et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wudhikarn K et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 10, 79 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahas G et al. Persistent cytopenias after chimeric antigen receptor t-cell immunotherapy for CD19+ aggressive lymphoma: a single institution experience. Biol. Blood Marrow Transplant. 25, S180 (2019). [Google Scholar]
- 10.Fried S et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 54, 1643–1650 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Parker KR et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell 183, 126–142.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramakrishna S, Barsan V & Mackall C Prospects and challenges for use of CAR T cell therapies in solid tumors. Expert Opin. Biol. Ther. 20, 503–516 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Wang G et al. Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nat. Immunol. 20, 1494–1505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho JH, Collins JJ & Wong WW Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426–1438.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roybal KT et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohmueller JJ, Ham JD, Kvorjak M & Finn OJ mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 7, e1368604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee YG et al. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. 79, 387–396 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Landgraf KE et al. convertibleCARs: a chimeric antigen receptor system for flexible control of activity and antigen targeting. Commun. Biol. 3, 296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller IC et al. Enhanced intratumoural activity of CAR T cells engineered to produce immunomodulators under photothermal control. Nat. Biomed. Eng. 5, 1348–1359 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang AQ et al. Universal redirection of CAR T cells against solid tumours via membrane-inserted ligands for the CAR. Nat. Biomed. Eng. 7, 1113–1128 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park AK et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 12, eaaz1863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aalipour A et al. Viral delivery of CAR targets to solid tumors enables effective cell therapy. Mol. Ther. Oncolytics 17, 232–240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vincent RL et al. Probiotic-guided CAR-T cells for solid tumor targeting. Science 382, 211–218 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunz P et al. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci. Rep. 8, 7934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingram JR, Schmidt FI & Ploegh HL Exploiting nanobodies’ singular traits. Annu. Rev. Immunol. 36, 695–715 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Peyvandi F et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 374, 511–522 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Tiwari PM et al. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 9, 3999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Movahedi K et al. Nanobody-based targeting of the macrophage mannose receptor for effective in vivo imaging of tumor-associated macrophages. Cancer Res. 72, 4165–4177 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Keyaerts M et al. Phase I study of 68Ga-HER2-nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J. Nucl. Med. 57, 27–33 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Nchinda G et al. The efficacy of DNA vaccination is enhanced in mice by targeting the encoded protein to dendritic cells. J. Clin. Invest. 118, 1427–1436 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Idoyaga J et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl Acad. Sci. USA 108, 2384–2389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie YJ et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl Acad. Sci. USA 116, 7624–7631 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS & Vale RD A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hussack G, Hirama T, Ding W, Mackenzie R & Tanha J Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS ONE 6, e28218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinaud F & Dahan M Targeting and imaging single biomolecules in living cells by complementation-activated light microscopy with split-fluorescent proteins. Proc. Natl Acad. Sci. USA 108, E201–E210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du Y, Pattnaik AK, Song C, Yoo D & Li G Glycosylphosphatidylinositol (GPI)-anchored membrane association of the porcine reproductive and respiratory syndrome virus GP4 glycoprotein and its co-localization with CD163 in lipid rafts. Virology 424, 18–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nickells MW, Alvarez JI, Lublin DM & Atkinson JP Characterization of DAF-2, a high molecular weight form of decay-accelerating factor (DAF; CD55), as a covalently cross-linked dimer of DAF-1. J. Immunol. 152, 676–685 (1994). [PubMed] [Google Scholar]
- 39.Rossey I et al. Potent single-domain antibodies that arrest respiratory syncytial virus fusion protein in its prefusion state. Nat. Commun. 8, 14158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zehn D, Cohen CJ, Reiter Y & Walden P Extended presentation of specific MHC–peptide complexes by mature dendritic cells compared to other types of antigen-presenting cells. Eur. J. Immunol. 34, 1551–1560 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Yadav M et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Yin L, Duan J-J, Bian X-W & Yu S-C Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 22, 61 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dees S, Ganesan R, Singh S & Grewal IS Emerging CAR-T cell therapy for the treatment of triple-negative breast cancer. Mol. Cancer Ther. 19, 2409–2421 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Johnstone CN et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Model. Mech. 8, 237–251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Wei G & Liu D CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 1, 36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dogan A et al. B-cell maturation antigen expression across hematologic cancers: a systematic literature review. Blood Cancer J. 10, 73 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Viardot A, Locatelli F, Stieglmaier J, Zaman F & Jabbour E Concepts in immuno-oncology: tackling B cell malignancies with CD19-directed bispecific T cell engager therapies. Ann. Hematol. 99, 2215–2229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerdes MJ et al. Emerging understanding of multiscale tumor heterogeneity. Front. Oncol. 4, 366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Restle D et al. Organ-specific heterogeneity in tumor-infiltrating immune cells and cancer antigen expression in primary and autologous metastatic lung adenocarcinoma. J. Immunother. Cancer 11, e006609 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen N, Li X, Chintala NK, Tano ZE & Adusumilli PS Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 51, 103–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sterner RC & Sterner RM CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11, 69 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Sayes N, Vito A & Mossman K Tumor heterogeneity: a great barrier in the age of cancer immunotherapy. Cancers 13, 806 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Genoud V et al. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 7, e1501137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Rourke DM et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seaman BJ et al. Audiovestibular dysfunction associated with adoptive cell immunotherapy for melanoma. Otolaryngol. Head Neck Surg. 147, 744–749 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamers CH et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther. 21, 904–912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nomura N et al. Prostate specific membrane antigen (PSMA) expression in primary gliomas and breast cancer brain metastases. Cancer Cell Int. 14, 26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gust J, Taraseviciute A & Turtle CJ Neurotoxicity associated with CD19-targeted CAR-T cell therapies. CNS Drugs 32, 1091–1101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruella M & June CH Predicting dangerous rides in CAR T cells: bridging the gap between mice and humans. Mol. Ther. 26, 1401–1403 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richman SA et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res. 6, 36–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Labanieh L et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 185, 1745–1763.e22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu X et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hernandez-Lopez RA et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science 371, 1166–1171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh N et al. Single chain variable fragment linker length regulates CAR biology and T cell efficacy. Blood 134, 247 (2019). [Google Scholar]
- 65.Millar DG et al. Antibody-mediated delivery of viral epitopes to tumors harnesses CMV-specific T cells for cancer therapy. Nat. Biotechnol. 38, 420–425 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marabelle A, Tselikas L, de Baere T & Houot R Intratumoral immunotherapy: using the tumor as the remedy. Ann. Oncol. 28, xii33–xii43 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Melero I, Castanon E, Alvarez M, Champiat S & Marabelle A Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol. 18, 558–576 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hong WX et al. Intratumoral immunotherapy for early-stage solid tumors. Clin. Cancer Res. 26, 3091–3099 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hammerich L, Binder A & Brody JD In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 9, 1966–1981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson DB, Puzanov I & Kelley MC Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 7, 611–619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang TY et al. Immunogenicity assessment of AAV-based gene therapies: an IQ consortium industry white paper. Mol. Ther. Methods Clin. Dev. 26, 471–494 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anselmo AC & Mitragotri S Nanoparticles in the clinic: an update. Bioeng. Transl. Med. 4, e10143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shirley JL, de Jong YP, Terhorst C & Herzog RW Immune responses to viral gene therapy vectors. Mol. Ther. 28, 709–722 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen R et al. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 41, 262–272 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bloom K, van den Berg F & Arbuthnot P Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 28, 117–129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hou X, Zaks T, Langer R & Dong Y Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 6, 1078–1094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huayamares SG et al. High-throughput screens identify a lipid nanoparticle that preferentially delivers mRNA to human tumors in vivo. J. Control. Release 357, 394–403 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson LA et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 7, 275ra222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwartz LH et al. RECIST 1.1—update and clarification: from the RECIST committee. Eur. J. Cancer 62, 132–137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw IVIS images for Extended Data Fig. 8 are available from the Georgia Institute of Technology Research Repository (repository@library.gatech.edu) upon publication of this paper. The remaining data are available within the article and Supplementary Information or upon reasonable request to the corresponding author. Source data are provided with this paper.