

Abstract
Molecular evolutionary studies of eukaryotes have relied on a sparse collection of gene sequences that do not represent the full range of eukaryotic diversity in nature. Anaerobic microbes, particularly, have had little representation in phylogenetic studies. Such organisms are the least known of eukaryotes and probably are the most phylogenetically diverse. To provide fresh perspective on the natural diversity of eukaryotes in anoxic environments and also to discover novel sequences for evolutionary studies, we conducted a cultivation-independent, molecular phylogenetic survey of three anoxic sediments, including both freshwater and marine samples. Many previously unrecognized eukaryotes were identified, including representatives of seven lineages that are not specifically related to any known organisms at the kingdom-level and branch below the eukaryotic “crown” radiation of animals, plants, fungi, stramenopiles, etc. The survey additionally identified new sequences characteristic of known ecologically important eukaryotic groups with anaerobic members. Phylogenetic analyses with the new sequences enhance our understanding of the diversity and pattern of eukaryotic evolution.
Reconstruction of eukaryotic phylogeny, and thus history, is hindered by incomplete knowledge of extant microbial eukaryotic diversity. Current models of deep eukaryotic evolution have relied on comparisons among only a few microbes, mainly cultivated model organisms and pathogens (1). It is well established, however, that sparse taxon representation can dramatically influence the accuracy of phylogenetic reconstructions (2–4). Yet, expanding eukaryotic taxonomic representation by discovery of novel organisms has provoked relatively little interest from researchers. In the cases of the phylogenetic domains Bacteria and Archaea, cultivation-independent identification of environmental organisms by rRNA gene sequences has revealed a diverse microbial world that is not represented by cultured organisms (5, 6). It would be surprising were this not also the case for eukaryotes. Indeed, recent rRNA gene-based surveys of eukaryotes in oxic planktonic environments have discovered novel rRNA gene sequences. Organisms detected thus far by sequence are affiliated phylogenetically with recognized major eukaryotic groupings, however, and do not branch deeply in phylogenetic trees (7, 8).
New rRNA sequences from primitively divergent phylogenetic lines of descent would be particularly useful for resolving the ancient patterns of eukaryotic evolution. The most deeply divergent of known eukaryotic lineages in phylogenetic trees are represented by anaerobic or aerotolerant organisms, the inhabitants of anoxic environments. Anoxic environments have occurred continuously throughout the history of Earth, and cultivation and descriptive microscopic studies have shown that such environments harbor a diverse assemblage of eukaryotic microbes (9, 10). The cultivation of most microbes is seldom successful, however, and microscopic descriptions tend to be biased toward morphologically conspicuous organisms. Consequently, organisms detected by cultivation or morphology do not necessarily represent the abundance and diversity of naturally occurring ones. To describe more fully the eukaryotic microbial diversity in anoxic habitats and potentially to discover new sequences useful for phylogenetic studies, we surveyed the constituents of three such habitats by using culture-independent, rRNA gene-based methods that target eukaryotes.
Materials and Methods
Sample Collection and DNA Extraction.
Samples for analysis were collected from three anoxic sediments (>1 cm below surface of black, reducing sediments) and stored at −70°C until processing. Two of the sediments collected for analysis were marine [24°C, pH 7.8, 1–3 cm depth at low tide (Berkeley Aquatic Park, Berkeley, CA) and 30°C, pH 7.8, 1–3 cm depth at low tide (Bolinas Tidal Flat, Bolinas, CA)] and one sediment was freshwater [28°C, pH 7.5, 3–5 cm depth in loose sediment (Lake Lemon, Bloomington, IN)]. Total community DNA was prepared as reported with a combined physical, chemical, and enzymatic method of cell lysis (11), and high molecular weight DNA was purified further (12) to remove substances that coprecipitate with DNA and can inhibit PCR reactions. Environmental DNA greater than 5 Kb (as determined by comparison to molecular weight standards) was eluted from agarose gel slices, and the purified environmental DNA was subsequently used as a template for eukaryote-specific PCR reactions.
PCR Amplification of Eukaryotic rDNA.
Degenerate, eukaryote-specific small-subunit (SSU) rRNA forward primers were designed and used together with modified “universal” reverse primers to selectively amplify eukaryotic SSU rDNA genes from the total community DNA pool. Two sets of degenerate oligonucleotide primers were used in the PCR reactions. Primer combination A used the forward primer 82FE 5′-GAADCTGYGAAYGGCTC-3′ [numbers based on the rRNA sequence of Gracilariopsis sp. isolate England-1 (GenBank accession no. M33639)] and the universal reverse primer 1391RE 5′-GGGCGGTGTGTACAARGRG-3′. Primer combination D used the forward primer 360FE 5′-CGGAGARGGMGCMTGAGA-3′ with the universal reverse primer 1492R 5′-ACCTTGTTACGRCTT-3′.
Molecular identification of rare sequences, as well as the quantification of microbes by environmental DNA analysis, can be biased because of various PCR reaction parameters, including template concentration, primer design, and reaction (13). To maximize our coverage in PCR amplifications, we used a temperature gradient PCR approach to provide a range of conditions to amplify eukaryotic rDNA genes. Each 50-μl eukaryote-specific PCR reaction contained 30 mM Tris⋅HCl (pH 8.4), 50 mM KCl, 1.5 mM MgCl2, 0.05% BSA (Sigma), 0.2 mM of each dNTP, 0.5 ng of each oligonucleotide primer, 0.5 units of AmpliTaq Gold (Perkin–Elmer), and 50–200 ng of the environmental DNA template. Twelve duplicate reaction mixtures were incubated by using an Eppendorf Gradient Thermocycler at 94°C for 12 min, followed by 35 cycles of 94°C for 1 min, an annealing temperature gradient of 45–65°C for 1 min, and 72°C extension for 2 min followed by a final 72°C extension for 10 min.
Cloning and Sequence Analysis.
PCR reactions positive for inserts of the expected size were pooled and cloned with the TOPO TA Cloning kit (Invitrogen) in accordance with the manufacturer's instructions. Four eukaryotic SSU rDNA clone libraries were made with one or both PCR primer combinations: “LEMD,” with Lake Lemon DNA and the D primer set; “BAQA” and “BAQD,” with both primer sets with the Berkeley Aquatic Park DNA; and “BOLA,” with the A primer set with the Bolinas Lagoon DNA. Plasmid DNA from recombinant clones was purified with a described 96-well plate method for subsequent restriction fragment-length polymorphism (RFLP) analysis and automated sequencing (11). Approximately 1,000 rDNA clones from each of the three environments (500 each from the two Berkeley Aquatic Park rDNA clone libraries) were screened by RFLP to identify unique sequence types or “phylotypes.” Representative clones were then sequenced entirely and analyzed by several phylogenetic inference methods (see Figs. 1–4 and Figs. 6–8, which are published as supporting information on the PNAS web site, www.pnas.org). The 125 sequences of representative rDNA clones have been deposited in GenBank under the accession nos. AF372704–AF372828.
Figure 1.

Summary of unique eukaryotic phylotypes identified in these anoxic environments. A is a summary of the abundance of phylotypes from each of the three anoxic environments surveyed. LEM, freshwater Lake Lemon sediment in Bloomington, IN; BAQ, brackish sediment in Berkeley Aquatic Park, Berkeley, CA; and BOL, marine intertidal sediment from the Bolinas Lagoon, Bolinas, CA, grouped by kingdom-level affiliation. (FUNGI, all fungi, including chytrids; CERCZN, Cercozoa; ACANTH, acanthamoebids; CHOANO, choanoflagellates; NNCRWN, novel, noncrown kingdom-level groups; NCROWN, novel, crown kingdom-level groups; ALVEO, alveolates; STRAM, stramenopiles; ANIMAL, all metazoans). B is a summary of the total number of phylotypes that was identified and grouped by kingdom-level affiliation. All 125 novel sequences were used in these summaries.
Figure 4.

Molecular phylogeny of novel kingdom-level lineages in Eucarya. Consensus phylogenetic tree of representative eucaryal rRNA sequences including novel lineages from these environmental surveys. The tree is a summary of 100 multiple bootstrapped replicates with four phylogenetic methods [maximum parsimony (MP), ME, ML, and Bayesian inference (MB)] to infer the tree topologies. The bootstrap values, determined as percentages of 100 trees inferred by each type of analysis, are given for branches with greater than 50% support, and presented in the order of MP/ME/ML/MB. Bootstrap values for each of the major kingdom-level cultivated and environmental lineages (excluding red algae and Acanthamoebae) also showed greater than 75% bootstrap support with each tree inference method (not shown). Fifty-four representative rRNA sequences incorporating 789 unambiguously homologous nucleotide positions were used in the phylogenetic analyses. The scale bar indicates 0.10 changes per site. Analysis of alternative branching orders of the novel lineages is presented in Table 1.
Environmental rDNA sequences initially were compared with a current database of genetic sequences (GenBank) by using gapped blast analysis (14) to determine their approximate phylogenetic affiliation, then sequences were aligned to an updated database of more than 5,000 aligned eukaryotic SSU rRNAs in the arb software package (15) and in the Ribosomal Database Project (RDP) (16). Secondary-structure predictions and the check_chimera software program provided by the RDP identified only three chimeric sequences (16). DNA sequences were aligned within arb according to conserved primary and secondary structural elements; these conserved positions were used in subsequent phylogenetic analyses. The final selection of taxa for phylogenetic analysis was based on the results of preliminary phylogenetic analyses from within the aligned ARB database.
Nonparametric Bootstrapped Phylogenetic Analyses.
Unique phylotypes were classified initially by their inclusion into or exclusion from major evolutionary lineages within the Eucarya by using four phylogenetic inference methods: maximum parsimony, ME, and maximum likelihood (ML) in the paup* software package (17), and Bayesian analyses with MR. BAYES 2.0 (18). All heuristic searches were unrooted and performed with random, stepwise addition of taxa with the tree bisection/reconnection branch-swapping algorithm. To assess the reliability of individual branches (both peripheral and internal nodes) in the phylogenetic analyses, we first used nonparametric bootstrapped analyses with resampling. Several optimality criteria and estimated models of character change (when applicable) were evaluated with the paup* 4.0 software package: maximum parsimony; minimum evolution under the HKY85 substitution model; maximum likelihood with a six-category, estimated general time-reversible (GTR) substitution model alone; and maximum likelihood with an estimated six-category, GTR substitution model, with among-site rate heterogeneity, an estimated shape parameter (G), and estimated invariant sites (I). Individual base frequencies were determined empirically with paup*.
Bayesian Maximum Posterior Probability (MB) Analysis.
Nonparametric bootstrap analysis can give underestimates of accuracy at high bootstrap values and overestimates at low values depending on the data set (19). As an alternative to nonparametric bootstraps, we used Bayesian analysis to evaluate uncertainties in the evolutionary relationships of Cercozoa and kingdom-level lineages. Bayesian phylogenetic inferences confer an advantage over nonparametric bootstrapped analysis in the consideration and weighting of all potential trees according to the posterior probability that each is correct (20). Posterior probabilities of trees were approximated with MR. BAYES 2.0 (18), with four-chain Metropolis-coupled Markov chain Monte Carlo (MCMCMC) analysis. In these analyses, base frequencies were empirically determined, the substitution rate matrix was estimated, and the gamma distribution was estimated with invariant sites. Based on the comparisons of the mean, variance, and credible interval for all generations, the first 35,000 generations were discarded as “burn-in,” based on the assumption that these generations reflect when the chain was not stationary. The chains were sampled every 1,000 generations, and inferences from each run were based on a total of 100,000 sampled trees.
Evaluation of Alternative Topologies of Novel Lineages by Using Kishino–Hasegawa (KH) Tests.
To evaluate alternative hypotheses of evolutionary relationships among the novel kingdom-level lineages, we constrained the branching position of each novel lineage to one of six positions (A–F) within the most likely ML global eukaryotic SSU rRNA tree and evaluated these hypotheses for each novel lineage with a two-tailed KH test, with 1,000 bootstrapped replicates and an estimated RELL distribution in paup*. The alternative branching orders of the novel “kingdom-level” lineages were constrained within the macclade software package (21) and imported as user-defined constraint trees into paup* for these calculations. Both the log likelihoods (ln −L) and the probabilities (P) of excluding the null hypothesis (no significant difference among the best or lowest log likelihood and the five other alternatives) were evaluated for each of the proposed novel uncultivated eukaryotic lineages (summarized in Table 1 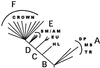 ).
).
Table 1.
Evaluation of six alternative topologies for each novel kingdom-level lineage by using KH tests
| BAQ1
|
BOL1
|
BOL2
|
BOL3
|
BOL4
|
LEM1
|
LEM2
|
LEM3
|
|
|---|---|---|---|---|---|---|---|---|
| BAQA065 | BOLA048 | BOLA212 | BOLA267 | BOLA187 | LEM267 | LEM119 | LEM052 | |
| A | 18404.016 | 18428.304 | 18438.566 | 18425.017 | 18452.513 | 18439.902 | 18441.679 | 18440.541 |
| P = 0.61 | P = 0.10 | P = 0.01 | P = 0.02 | P = 0.01 | P = 0.03 | P = 0.01 | P = 0.04 | |
| B | 18402.244 | 18423.811 | 18423.811 | 18416.137 | 18430.551 | 18428.015 | 18427.857 | 18427.768 |
| BEST | BEST | P = 0.12 | P = 0.13 | P = 0.05 | P = 0.12 | P = 0.12 | P = 0.17 | |
| C | 18407.759 | 18428.523 | 18419.906 | 18407.103 | 18420.614 | 18419.906 | 18422.224 | 18421.552 |
| P = 0.43 | P = 0.093 | BEST | BEST | P = 0.16 | BEST | P = 0.30 | P = 0.39 | |
| D | 18416.195 | 18432.910 | 18421.194 | 18413.109 | 18412.789 | 18419.906 | 18419.906 | 18420.932 |
| P = 0.04 | P = 0.037 | P = 0.35 | P = 0.36 | P = 0.31 | P = 0.48 | BEST | P = 0.24 | |
| E | 18427.866 | 18433.351 | 18420.949 | 18414.508 | 18409.160 | 18420.135 | 18420.932 | 18419.906 |
| P = 0.00 | P = 0.037 | P = 0.41 | P = 0.30 | P = 0.43 | P = 0.44 | P = 0.24 | BEST | |
| F | 18431.571 | 18438.732 | 18430.644 | 18411.504 | 18407.103 | 18430.762 | 18434.013 | 18425.181 |
| P = 0.00 | P = 0.01 | P = 0.05 | P = 0.66 | BEST | P = 0.04 | P = 0.05 | P = 0.21 |
Alternative branching orders of the novel kingdom-level lineages were constrained to one of six tree regions (A–F, diagram), and the alternative topologies were evaluated by using the KH log likelihood tests from the paup* package with empirically defined general time-reversible substitution rate matrix, estimated invariant sites (I), and empirical rate correction or gamma (G). Potential branch positions of the novel lineages are compared by log likelihood (ln − L) differences with the alternatives and the probabilities (P) of excluding the null hypothesis of no significant difference between the best (lowest log likelihood) and each alternative topology. CROWN, crown group; SM/A, slime molds/amoebae; EU, euglenozoa; HL, heterolobosea; DP, diplomonads; MS, microsporidians; TR, trichomonads.
Relative Rate Tests.
To compare the branch length between neighboring basal or “noncrown” lineages and the novel environmental lineages, relative rate computations were performed with the most likely (best) ML global eukaryotic SSU rRNA tree within the rrtree software program (22).
Results
Novel Eukaryotic Diversity in Anoxic Environments.
Environmental DNAs were purified from two marine and one freshwater anoxic sediments and used as templates for amplification of rRNA genes by PCR, with degenerate primers selective for eukaryotic rDNA (Materials and Methods). PCR products were cloned and screened by RFLP to identify the unique types, which were sequenced. In total, more than 3,000 clones were screened by RFLP, and 125 unique eukaryotic SSU rDNA sequences from the three anoxic sediment environments were determined. Sequences were aligned with a database of ≈5,000 other eucaryal rDNA sequences, and phylogenetic analyses were conducted to compare the sequences with those of known organisms.
The frequencies with which the unique phylotypes occurred in the environmental rDNA libraries, as assessed in the RFLP screens, provide a rough census of the dominant eucaryal groups that comprise the communities sampled, which are summarized in Fig. 1. The number of rRNA genes per cell is highly variable, however, therefore the relative abundance of rRNA genes in the clone libraries does not necessarily directly reflect relative numbers of cells. Additionally, technical variabilities such as differential extraction or different efficiencies of PCR of different rRNA genes can influence the relative recoveries of particular clones (13, 23). Nonetheless, the clone libraries probably identify the most abundant eukaryotes in the samples. Each of the environmental rDNA libraries was dominated by relatively few rRNA sequence types or phylotypes. Many of the sequences represent ecologically important eukaryotic groups that are known to have anaerobic members such as stramenopiles (18% of the total), alveolates (31%), Cercozoa (10%), and fungi (26%). Sequences representative of the animal (ANIMAL), choanoflagellate (CHOANO), and Acanthamoebid (ACANTH) lineages were less abundant among our rDNA isolates. In general, the sequences detected are not closely related to any previously known sequence. Only about one-fifth of the environmental sequences are related as closely as the “genus level” (>95% sequence identity) to known rDNA sequences. (For comparison, Tetrahymena thermophila and Tetrahymena pyriformis SSU rRNA sequences are about 95% identical.)
Some of the novel sequences indicate unsuspected breadth of diversity and organization in known eucaryal groups. For example, the rRNA relatedness group represented by BAQA072, BAQD018, and BAQD220 identifies a deeply divergent clade in the phylogenetic kingdom of stramenopiles (Fig. 2). The most closely related characterized stramenopiles are all heterotrophs, possibly indicating that the organisms detected by the sequences also are heterotrophic. Another example of novel diversity in known kingdoms is indicated by sequences affiliated with Cercozoa, a relatedness group that includes cercozoan flagellates, testate amoebae, and chlorarachniophytes (Fig. 3). The environmental sequences detected in this study more than double the known deep branchings in this group. Novel and diverse clades additionally were indicated for alveolates (Figs. 6 and 7) and acanthamoebids (Fig. 8), other main relatedness groups of eukaryotes. Fungal sequences were related to known ascomycetes, basidiomycetes, and chytrids. Animal sequences were most closely related to flatworms (data not shown) and other invertebrates.
Figure 2.

Phylogenetic tree of Stramenopiles. This figure depicts a consensus phylogenetic tree of the evolutionary relationships of cultivated and uncultivated stramenopiles and summarizes 100 multiple bootstrapped replicates with two phylogenetic methods (ME and ML; ME, minimum evolution) to infer the tree topologies. Fifty-three representative rRNA sequences incorporating 1,064 unambiguously homologous nucleotide positions were used to infer the phylogenetic trees. The bootstrap values, determined as percentages of 100 trees inferred by each type of analysis, are given for branches with greater than 50% support (ME values shown above lines and ML values shown below). The scale bar indicates 0.10 changes per site. Phototrophic stramenopiles form a phylogenetic group to the exclusion of several deeply branching lineages of heterotrophic stramenopiles.
Figure 3.

Phylogenetic tree of Cercozoa. Consensus bootstrapped phylogenetic tree showing evolutionary relationships of uncultivated members of Cercozoa, comprised of cercomonads, testate amoebae, thaumatomonads, and chlorarchaniophytes. Taxa with the designation “LKM” are from a recent molecular survey of a detritus-fed, continuous flow bioreactor (29). Forty-one representative rRNA sequences comprised of 954 unambiguously homologous nucleotide positions were used to create the tree. The bootstrap values, determined as percentages of 100 trees inferred by each type of analysis, are given for branches having greater than 50% support (ME values shown above lines and ML shown values below). The scale bar indicates 0.10 changes per site.
Novel Kingdom-Level Diversity.
Eight independent clades identified in the three environments in this study are not specifically affiliated with known, kingdom-level eukaryotic lineages. As shown in Fig. 4, one typical phylogenetic tree that includes a broad representation of eucaryal rRNA sequences, seven of these novel lineages (represented by sequences BAQA065, BOLA048, BOLA212/458, BOLA267, LEMD003/LEMD145/LEMD119, and LEMD267) consistently branch outside the eukaryote “crown” radiation of animals, plants, fungi, etc. (24). (We discuss below statistical tests of the branching topology.) In addition, the BOL4 environmental lineage (represented by sequences BOLA187 and BOLA366 in Fig. 4) branches within the crown radiation but independently of any other crown-group lineage. In some analyses (data not shown) these latter sequences affiliate loosely with the crown lineage that contains the amoeboid pelobiont Mastigamoeba invertans.¶
Several of the new kingdom-level lineages branch at intermediate levels in the overall eukaryotic radiation. The new lines punctuate the long lines that previously separated the basal radiation from more peripheral branches in most phylogenetic analyses (Fig. 4). Specifically, the inclusion of BAQA065 and BOLA048 in phylogenetic calculations interrupts the long branch between the heterolobosea and the unresolved radiation of amitochondriate lines (diplomonads, etc.) located at the base of the eukaryotic global phylogeny. [The BAQ1 lineage is affiliated with the microsporidian lineage in some phylogenetic analyses but not in analyses that exclude bacterial and archaeal outgroups (data not shown)]. Additionally, divergences represented by BOLA212/458, LEMD267, and BOLA267 intersect the long branch between euglenozoa and the heterolobosea. Finally, the lineage comprised of LEMD003, LEMD145, and LEMD119 interrupts the long branch between the crown radiation and the euglenozoa lineage.
Analysis of Rates and Alternative Tree Topologies.
Our nonparametric bootstrap analyses highlight particular difficulties in establishing precise branching order and evolutionary relationships among diverse eukaryotic groups in deep eukaryotic phylogenies. For example, high rates of evolution of some of the deeply branching eukaryotic lineages, manifest by long branch lengths in phylogenetic trees, potentially cause artifacts in molecular phylogenetic reconstructions (1). To evaluate the evolutionary rates of the novel kingdom-level lineages relative to others, we compared branch lengths by maximum likelihood and Bayesian inferences and used relative rate tests (25). These methods concurred in the conclusion that several of the deeply divergent environmental sequences (e.g., BOLA048, BOLA458/212, BOLA267, and BAQA065) have evolved at significantly lower rates than those that represent neighbor branches in the eucaryal tree (data not shown). In general, the deeply divergent novel lineages have evolved at roughly half the rate of the known basal-derived lineages represented by diplomonads or trichomonads. Furthermore, the BAQA065 lineage has evolved at roughly half the rate of the neighboring microsporidian line.
The specific topologies of phylogenetic trees, even based on the same data set, depend on methods and parameters used in calculations. To evaluate the optimal calculated branching orders for the sequences that represent the novel kingdom-level lineages, we initially used nonparametric bootstrap analysis by three standard phylogenetic inference methods: maximum parsimony, evolutionary distance, and maximum likelihood (Methods and Materials). Results with the different methods concur generally in overall topology, but bootstrap support for the specific location of most internal nodes is low (Fig. 4). In some cases, low bootstrap values for the positions can be explained by local branch-swapping. For example, the sequence BOLA048 had 35% support for the position shown in Fig. 4, 35% support for branching in the vicinity of Heterolobosea, and 30% support for branching near the novel lineage represented by BAQA065. Because of the generally low bootstrap support for specific internal nodes in the tree, we tested six possible tree topologies for each novel lineage with the KH log likelihood test and additionally used an alternative tree inference method, Bayesian maximum posterior probability analysis, to help evaluate the variance in deep branching orders.
Methods to estimate the SE and confidence intervals for particular phylogenetic topologies generally evaluate differences in log-likelihoods of alternative tree topologies. Because we had no preconceived hypotheses of branch placement, we used the KH log-likelihood ratio test to estimate a “best” tree and the probability (P) of branching of each of the novel lineages at alternative positions in the global eukaryotic tree. Results are summarized in Table 1. The specific branching orders of the new lineages are not resolved by this analysis and the available sequence data set. Nonetheless, five of the eight novel lineages (BAQ1, BOL1, BOL2, LEM1, and LEM2) are excluded (P ≤ 0.05) from the crown group (region F in Table 1), consistent with their deep branching in other analyses. The BOL1 lineage is likely associated with region B in Table 1. Except for BAQ 1, all of the new lines are essentially excluded from the basal radiation, region A. Lineage BOL4 can be excluded only from the deeper regions of the tree, but in most analyses (e.g., Fig. 4) this group is associated with the crown radiation.
To address potential artifacts in phylogenetic reconstructions with standard nonparametric methods with bootstrap analysis (20), we also used an alternative phylogenetic inference method that evaluates posterior likelihood probabilities of clades with Bayesian analysis (Materials and Methods). With this method, the posterior probabilities of deep nodes were moderately higher than bootstrap values obtained with nonparametric methods (Fig. 4). Specifically, the nodes leading to the novel lineages BAQ1, BOL1, BOL2, BOL3, LEM1, and LEM2 showed greater statistical support (0.5–0.70 of posterior probabilities) than that obtained with nonparametric bootstrapping analyses. The stronger support for internal branching order with Bayesian analysis suggests that nonparametric bootstrapping estimates of deep nodes could be too conservative.
Discussion
These new environmental sequences significantly expand the known extent of eucaryal rRNA diversity. It is possible that some of the organisms that we detect by sequence analysis have been identified in previous culture or other studies, but are not represented by rRNA sequences. At this time only a few thousand rRNA sequences of eucaryal microbes are known, and it is estimated that more than 200,000 “morphospecies” have been described by morphology or other property (26). This disparity between the number of identifying gene sequences and the number of classically described organisms highlights the need for further molecular surveys, both in the environment and in culture collections, if we are to understand the diversity and evolution of eukaryotes.
The new sequences collectively represent a broad span of rRNA phylogeny. There is no convention for the taxonomic organization of sequence-based relatedness groups of eukaryotes. Based on various traditional or molecular classification schemes, eukaryotes have been categorized into from 3 (27) to over 70 major taxonomic kingdoms (28). Eucaryal rRNA sequences available in the databases fall into about 30 independent relatedness clusters, which we take to represent the kingdom-level taxa. Most of the environmental sequences that we detect, about 90%, are affiliated with recognized and molecularly defined kingdoms. None of the sequences is identical to a known sequence, however. In some cases, the environmental sequences indicate deeply branching, and thus diverse groups within documented phylogenetic kingdoms (Figs. 2, 3, and 6–8).
Several of the new sequences, by multiple analytical criteria, are not specifically affiliated with any molecularly described taxonomic group and therefore indicate novel, kingdom-level relatedness groups. The new sequences contribute substantial variation to the sequence collection, which increases the accuracy of the phylogenetic calculations. The general results of these and other phylogenetic studies based on rRNA sequence comparisons are interpreted in Fig. 5. The overall topology of the eucaryal rRNA tree was seen as a basal radiation of lines of descent, only one of which gave rise successively to other kingdom-level lines and culminated in the unresolved crown radiation. The specific positions of intermediate branches in the rRNA tree are only approximate, but the general branching order is indicated by all of the analyses. The accuracy with which the kingdom-level lines can be resolved will improve as the sequence collection available for analysis grows. As indicated in Fig. 5 by the areas of the wedges that represent the phylogenetic kingdoms, the currently available collection of rRNA sequences is heavily biased toward only a few of those groups. Consequently, larger sets of more diverse sequences than are currently available will be particularly informative for the further resolution of the eucaryal tree. As we show here, the natural environment is one source of readily available sequence diversity.
Figure 5.

Schematic diagram of the evolution of Eucarya. Schematic summary tree of global SSU rRNA phylogeny including the novel lineages from the three environments surveyed in this study. The areas of the wedges reflect the number of SSU rRNA sequences of these groups in GenBank. The DRIP lineage is a recently defined protistan clade near the animal–fungal divergences (30). This figure is based on nonparametric bootstrap analyses of MP, ME, and ML trees, Bayesian inferences, and the analysis of alternative likelihood topologies of the novel lineages with the KH likelihood test in the PAUP* package (see Table 1).
The view of successive branching in the eucaryal phylogenetic tree contrasts with the results of some comparisons of protein-encoding genes, with limited phylogenetic representation. Those results have been interpreted to indicate that there is no particular branching order, and that the contemporary kingdom-level lines of descent derived from a single expansive radiation (1). Proponents of this latter view have argued that extensive sequence differences between basal-derived and crown-group rRNA genes do not reflect great evolutionary distance of the basal lines from the crown radiation, but rather are a consequence of relatively rapid evolution in the basal lines. However, seven of the candidate kingdoms identified in this study branch more deeply in the tree than the crown radiation, and thereby punctuate the long lines of previously identified, deeply branching relatedness groups. Moreover, as judged by branch lengths and relative rate tests, several of the deeply divergent environmental lineages apparently have evolved at significantly slower rates than neighboring branches in the eucaryal tree. The identification of multiple novel lineages of deeply divergent eukaryotes with relatively slow rates of evolution indicates that the high evolutionary rates previously ascribed to the basal divergences in rRNA trees are not the norm. Thus, deeper branches than the crown radiation are not a systematic error because of rate effects.
Phylogenetic trees based on a single gene, that of SSU rRNA in this case, do not reflect the genealogies of all genes that specify the organism. Genomic and other studies have provided ample evidence that many genes have undergone more or less extensive lateral transfers during their evolution. In contrast, there is no indication that rRNA genes have done so. The phylogenies of genes that specify most other components of the cellular nucleic acid-based information-processing system are congruent with the rRNA phylogeny, therefore the rRNA tree seems to reflect the evolutionary path of the core genetic machinery. Genes with phylogenies that are incongruent with the rRNA tree possibly have undergone lateral transfer in their evolution.
Many of the organisms identified here by sequences are phylogenetically highly diverse from any known ones and consequently may have novel properties. The potential novelty and apparent abundance of the organisms justify their further study. Study of the properties of environmental organisms generally has required the development of cultured models, however, and most microbes are not readily cultivated. The availability of the rRNA sequences opens new avenues to the study of these organisms, even if they prove intractable to culture. The sequences are incisive identifiers of the organisms in any place or form and also are the basis of molecular tools, fluorescent hybridization probes, and others that can be used to visualize the organisms and study them in their natural habitats.
Supplementary Material
Acknowledgments
We acknowledge Elizabeth Pine, Fred Harbinski, and Rachel Whitaker for their contributions to these and other molecular surveys; and Carl Woese, Ford Doolittle, Mitchell Sogin, David J. Patterson, John R. Spear, Allen G. Collins, and other colleagues for insightful comments on the manuscript. This research was supported by grants from the National Science Foundation and the National Aeronautics and Space Administration Astrobiology Institute.
Abbreviations
- SSU
small subunit
- RFLP
restriction fragment-length polymorphism
- KH
Kishino–Hasegawa
- ML
maximum likelihood
- ME
minimum evolution
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF372704–AF372828).
Edgcomb, V. P., Simpson, A. G. B., Zettler, L. A., Walker, G., Nerad, T., Patterson, D. J. & Sogin, M. L. (2000) Abstr. Gen. Meet. Am. Soc. Microbiol. 100, 680 (abstr.).
References
- 1.Philippe H, Lopez P, Brinkmann H, Budin K, Germot A, Laurent J, Moreira D, Muller M, Le Guyader H. Proc R Soc London Ser B Biol Sci. 2000;267:1213–1221. doi: 10.1098/rspb.2000.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hillis D M. Syst Biol. 1998;47:3–8. doi: 10.1080/106351598260987. [DOI] [PubMed] [Google Scholar]
- 3.Poe S. Syst Biol. 1998;47:18–31. doi: 10.1080/106351598261003. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler W. In: Extinction and Phylogeny. Novacek M J, Wheler Q D, editors. New York: Columbia Univ. Press; 1992. pp. 205–215. [Google Scholar]
- 5.Hugenholtz P, Goebel B M, Pace N R. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barns S M, Delwiche C F, Palmer J D, Pace N R. Proc Natl Acad Sci USA. 1996;93:9188–9193. doi: 10.1073/pnas.93.17.9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moon-van der Staay S Y, De Wachter R, Vaulot D. Nature (London) 2001;409:607–610. doi: 10.1038/35054541. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Garcia P, Rodriguez-Valera F, Pedros-Alio C, Moreira D. Nature (London) 2001;409:603–607. doi: 10.1038/35054537. [DOI] [PubMed] [Google Scholar]
- 9.Bernard C, Simpson A G B, Patterson D J. Ophelia. 2000;52:113–142. [Google Scholar]
- 10.Fenchel T, Finlay B J. Ecology and Evolution in Anoxic Worlds. Oxford: Oxford Univ. Press; 1995. [Google Scholar]
- 11.Dojka M A, Hugenholtz P, Haack S K, Pace N R. Appl Environ Microbiol. 1998;64:3869–3877. doi: 10.1128/aem.64.10.3869-3877.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barns S M, Fundyga R E, Jeffries M W, Pace N R. Proc Natl Acad Sci USA. 1994;91:1609–1613. doi: 10.1073/pnas.91.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki M T, Giovannoni S J. Appl Environ Microbiol. 1996;62:625–630. doi: 10.1128/aem.62.2.625-630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strunk O, Gross O, Reichel B, May M, Hermann S, Stuckmann N, Nonhoff B, Lenke M, Ginhart A, Vilbig A, et al. PAUP* 4.0. Munich, Germany: Technische Universität München; 1998. [Google Scholar]
- 16.Maidak B L, Cole J R, Lilburn T G, Parker C T, Saxman P R, Farris R J, Garrity G M, Olsen G J, Schmidt T M, Tiedje J M. Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swofford D L. MR. BAYES 2.0. Sunderland, MA: Sinauer; 2000. [Google Scholar]
- 18.Huelsenbeck J P, Ronquist F. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 19.Hillis D M, Moritz C. Molecular Systematics. Sunderland, MA: Sinauer; 1990. [Google Scholar]
- 20.Huelsenbeck J P, Ronquist F, Nielsen R, Bollback J P. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- 21.Maddison W. J Gen Physiol. 1993;102:9A–10A. [Google Scholar]
- 22.Robinson-Rechavi M, Huchon D. Bioinformatics. 2000;16:296–297. doi: 10.1093/bioinformatics/16.3.296. [DOI] [PubMed] [Google Scholar]
- 23.Burgmann H, Pesaro M, Widmer F, Zeyer J. J Microbiol Methods. 2001;45:7–20. doi: 10.1016/s0167-7012(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 24.Knoll A H. Science. 1992;256:622–627. doi: 10.1126/science.1585174. [DOI] [PubMed] [Google Scholar]
- 25.Li P, Bousquet J. Mol Biol Evol. 1992;9:1185–1189. doi: 10.1093/oxfordjournals.molbev.a040779. [DOI] [PubMed] [Google Scholar]
- 26.Levine N D, Corliss J O, Cox F E, Deroux G, Grain J, Honigberg B M, Leedale G F, Loeblich A R, 3rd, Lom J, Lynn D, et al. J Protozool. 1980;27:37–58. doi: 10.1111/j.1550-7408.1980.tb04228.x. [DOI] [PubMed] [Google Scholar]
- 27.Whittaker R H, Margulis L. Biosystems. 1978;10:3–18. doi: 10.1016/0303-2647(78)90023-0. [DOI] [PubMed] [Google Scholar]
- 28.Patterson D J. Am Nat. 1999;154:S96–S124. doi: 10.1086/303287. [DOI] [PubMed] [Google Scholar]
- 29.van Hannen E J, Mooij W, van Agterveld M P, Gons H J, Laanbroek H J. Appl Environ Microbiol. 1999;65:2478–2484. doi: 10.1128/aem.65.6.2478-2484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ragan M A, Goggin C L, Cawthorn R J, Cerenius L, Jamieson A V C, Plourde S M, Rand T G, Söderhäll K, Gutell R R. Proc Natl Acad Sci USA. 1996;93:11907–11912. doi: 10.1073/pnas.93.21.11907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.