

Abstract
The main malaria parasite, Plasmodium falciparum expresses 6 Hsp70 proteins. Among them, two are Hsp110 homologues localised in the cytosol and endoplasmic reticulum. The cytosolic Hsp110 (PfHsp70-z) has been fairly studied but the ER, PfGrp170 (PfHsp70-y) has not been well characterised. PfGrp170 is essential for parasite survival and is thought to be important for protein folding during exportation of parasite proteins to the host cell cytosol. Therefore, this study optimised the conditions for its overexpression in an E. coli system and investigated the structure-functional features of Grp170 protein. The recombinant full-length version of PfGrp170 was successfully produced under native conditions. The secondary structure analysis showed a predominantly α-helical structural features which exhibit moderate thermal stability with a Tm above 55 °C. The presence of nucleotides, such as ATP perturbed the structural stability of PfGrp170 making it more sensitive to heat stress and proteolytic cleavage. We observed that PfGrp170 exhibits basal ATPase activity and has the capability to suppress the heat induced aggregation of model substrate malate dehydrogenase in a nucleotide independent manner. In addition, using molecular docking and molecular dynamics simulations we show evidence of selective binding to nucleotides and the protein structure is more responsive to ATP binding than ADP. Findings from this study provide the first direct evidence for the independent chaperone function of PfGrp170 and its sensitivity to structural modulation by nucleotides which could be targeted in antimalarial drug development strategies.
Keywords: Malaria, Plasmodium falciparum, PfGrp170/PfHsp70-y, Hsp110, Molecular chaperone
Subject terms: Biochemistry, Biophysical chemistry, Protein folding, Proteins, Enzymes, Protein design, Protein folding, Proteins, Target identification
Introduction
The main malaria agent, Plasmodium falciparum, resides in a terminally differentiated red blood cell for its erythrocytic stage of the life cycle. For survival and development, the parasite continuously remodels the host cell to provide a conducive environment. The parasite exports approximately 400 proteins to the host cell which are processed through its endoplasmic reticulum (ER)1. In P. falciparum, like in other eukaryotes, for efficient ER function there is need for a robust protein folding system to maintain the trafficking cargo in folding competent form2,3. There has been reports implicating the ER stress response in the development of drug resistance against the first line of treatment of malaria, artemisinin monotherapy4–8. The machinery responsible for ER protein quality control system is mainly facilitated by a group of proteins termed glucose regulated proteins (Grp) which are homologs to the cytosolic heat shock proteins (Hsp)9,10.
P. falciparum expresses six Hsp70 proteins; four are canonical Hsp70s and two are Hsp110 proteins. Of the Hsp110 proteins, one paralog resides in the cytosol (PfHsp110c or PfHsp70-z), whilst the other is localized to the ER (PfGrp170 or PfHsp70-y)11. Generally, like members of the Hsp70 protein family, Hsp110s comprise of two domains; an amino-terminal nucleotide-binding domain (NBD), connected by a linker to the C-terminal substrate-binding domain (SBD), which consists of two distinct subdomains, β-sandwich (SBDβ) and α-helical lid (SBDα)12–14. Hsp110 proteins are larger than their canonical Hsp70 counterparts, having an insertion in the SBDβ domain and a C-terminal extension in the SBDα domain15. Hsp70 proteins function primarily as typical ATP-dependent foldase chaperones16,17, whereas Hsp110s have dual functions, with more pronounced holdase activities and as nucleotide exchange factors (NEFs)18,19. As NEFs of the canonical Hsp70s, Hsp110s speeds up the release of ADP in exchange for ATP binding20. In addition, NEFs regulate Hsp70 function, through indirect modulation of the binding and releasing activity of bound substrates by allosteric signals from ATP/ADP binding events of the NBD21. Thus, NEFs dictate substrate dwell time and ensure that adequately folding competent substrates are released, as those that are prematurely released will misfold and are channelled for degradation22. We previously showed that the P. falciparum cytosolic Hsp110 and Hsp70 associate in a nucleotide dependent fashion, with ATP promoting their association suggesting that PfHsp70-z acts as an NEF for PfHsp70-123.
Hsp110 proteins have the capability to function as holdases, where they actively bind and stabilise client proteins in a folding competent form, after which they are passed on to other chaperones for refolding24,25. Using the recombinant form of PfHsp70-z, we previously showed that Hsp110 suppressed heat induced substrate aggregation in a nucleotides independent manner26. This confirmed that nucleotides are dispensable for the open and close states of the parasite Hsp110 SBD, as was described in other organisms27–29. In fact, Hsp110s exhibit better holdase activity than their canonical Hsp70 counterparts23,30. This is through their capability to transiently bind to client peptides, in a rapid and constant cycle of association and dissociation31. The ER-resident parasite Hsp110 protein, PfGrp170 has not been well studied but its expression was shown to be essential for parasite cell survival during febrile episodes at the erythrocytic stages of the parasite life cycle32. Therefore, our study is the first to heterologously express, purify and biochemically characterise the full length essential malarial Grp170 protein. Like other Hsp110 proteins, our results show that PfGrp170 is a thermally resilient molecule that exhibits independent chaperone activity through the capability to suppress the heat induced aggregation of a model substrate. We report the nucleotide independent chaperone function of PfGrp170 and structural perturbation in the presence of nucleotides. These findings show that PfGrp170 has dual independent functions, one dependent on nucleotides possibly as NEF and the other where nucleotides are dispensable for its bona fide chaperone function. This presents PfGrp170 as an attractive drug target whose inhibition may compromise the parasite’s ability to survive the thermal stress it is exposed to in the host during fever episodes.
Materials and methods
Materials
The materials used in this study unless otherwise stated, were purchased from Sigma Aldrich (MA, USA) and Thermo Scientific (MA, USA).
PfGrp170 plasmid construct design
The amino acid sequence of PfGrp170 with PlasmoDB accession number PF3D7_1344200 and UniProt accession number: C0H5H0 residues 20–932 after removal of protein secretion signals (residues 1–19) were submitted to GenScript (NJ, USA) for synthesis. The codon harmonised form of the PfGrp170 was synthesised and inserted in pQE30 vector (Qiagen, USA) that carry an N-terminal 6 × Histidine tag (His-tag) between the 5’ BamHI and 3’ NheI restriction sites. The plasmid construct was confirmed by DNA sequencing and validated by restriction enzyme digestion (Supplementary Figure S1), however, we could not retrieve the full original agarose gel image.
Recombinant protein expression and purification
The expression of the recombinant protein was conducted as previously described33. Briefly, chemically competent E. coli XL1-Blue cells were transformed with pQE30-PfGrp170 and cultured in Luria broth (LB) media supplemented with 100 μg/mL ampicillin and allowed to grow overnight at 37 °C shaking at 250 rpm. The protein over-expression was induced with 0.1 mM Isopropyl ß-D-1-thiogalactopyranoside (IPTG) and cells were harvested after overnight induction. The harvested cells were lysed and clarified to extract the soluble supernatant by centrifugation at 10 000 xg for 60 min at 4 °C. The soluble supernatant with the recombinant PfGrp170 was purified using a HiTrap IMAC FF chromatography column (Cytiva, MA, USA) connected to an automated FPLC system (ÄKTA™ start chromatography system, Cytiva, MA, USA). The chromatography column was prepared and equilibrated with binding buffer (20 mM Tris–HCl, pH 7.5, 500 mM NaCl containing 1 mM PMSF) prior to loading with supernatant of the cell lysate. The eluted PfGrp170-containing fractions were pooled together and further purified using size exclusion chromatography (SEC). The SEC column was equilibrated with SEC buffer (20 mM Tris–HCl, pH 7.4, 200 mM NaCl, 0.1% Complete protease inhibitor) and loaded onto a HiLoad® 16/600 Superdex® 200 pg (Sigma-Aldrich, USA, CA) connected to FPLC system. Subsequently, the pooled fractions from IMAC were then applied to the SEC column and resolved over 20 column volumes with a flow rate of 1 mL/min. The eluted PfGrp170 fractions were buffer exchanged using Amicon® Ultra-15 centrifugal filters, 50 kDa MWCO (Merck, Darmstadt, Germany) and samples were analysed using SDS-PAGE.
Antibody production
To confirm the identity of the purified fractions, we produced polyclonal antibodies against the recombinant PfGrp170 protein by immunising rabbits using a previously described protocol with modifications34. Briefly, trinitrophenylated acid treated–naked Salmonella minnesota R595 (NB) bacterial cells were used to adsorb the recombinantly purified PfGrp170 protein in a ratio of 1: 5 (40 μg PfGrp170 to 200 μg NB) in 0.5 ml PBS (8 mM Na2HPO4, 1.46 mM KH2PO4, 137 mM, NaCl, 2.7 mM KCl, pH 7.2). The mixture was used to immunise female Flemish giant rabbits at 6 months of age obtained from Custom antibody synthesis from the Institutional Faculty of Science Small Animal Research Facility, Stellenbosch University, SA with ethical clearance granted by the Stellenbosch University Animal Ethics Committee (Ethical clearance approval licence number SU-ACU-2020–14892). All experiments were performed in accordance with relevant guidelines and regulations and are reported in accordance with ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines. Immunisation was achieved by injecting the antigen mixture intravenously in the marginal ear vein of the rabbits after EMLA (AstraZeneca, Cambridge, UK) local anaesthetic cream application. On day 42 post immunization, 20 ml of blood was collected from the central auricular artery and allowed to coagulate overnight at 4 °C and subsequently centrifuged at 800 × g for 20 min to separate the cells and serum. After collection of blood the rabbits were all returned to the animal house for breeding. The separated antisera (polyclonal PfGrp170 antibody) were used for western blot to confirm the expression and purification of recombinant PfGrp170. Antibody specificity was investigated by probing another ER based Hsp90 chaperone, PfGrp9435.
Investigation of protein secondary structural features
The secondary structure of the recombinant PfGrp170 protein was analysed using circular dichroism (CD) spectroscopy as previously described36. The far-UV CD spectra of PfGrp170 were recorded between 185 to 260 nm using a ChiraScan™ plus CD instrument (Applied Photophysics, MA, USA). The purified 0.15 μM PfGrp170 protein was suspended in CD buffer (10 mM KH2PO4, pH 7.5, and 100 mM KF) and analysed in a quartz cuvette with a pathlength of 0.01 cm (Hellma Analytics, Müllheim, Germany) at 25 °C. The CD spectra recorded were averaged from 7 scans, from which the baseline spectrum of the buffer without protein was subtracted before analysis. The resultant CD spectra were deconvoluted using the Dichroweb server (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml) using reference set 7 on the CONTIN-LL analysis program37 and validated using AlphaFold predicted PfGrp170 3D structure (accession numbers: AF-C0H5H0-F138).
In a separate experiment, the stability of recombinant PfGrp170 was investigated by thermal and chaotropic denaturation as previously described35,36. Initially, 0.15 μM recombinant PfGrp170 protein suspended in CD buffer was subjected to temperature increase from 25 °C to 90 °C at a rate of 1 °C/min whilst monitoring the CD spectra every 5 min. To monitor refolding capability of the recombinant protein the CD spectra was recorded as the temperature was reduced from 90 °C to 25 °C. This was repeated with different upper limits of the temperature to determine the resilience of the protein to unfold and refold after exposure to different temperatures. In addition, the spectra of PfGrp170 incubated in buffer supplemented with protein exposed to known chaotropic denaturants 0–8 M urea or 0–6 M Guanidine-HCl were conducted to validate its stability. Furthermore, the CD spectra of recombinant PfGrp170 suspended in CD buffer at different pH 2–10 was also used to assess the resilience of the protein to pH changes. The resultant molar residue ellipticity (θ) at 195, 205 and 222 nm were converted to the folded fraction as a ratio of the folded structure at any temperature compared to that at room temperature 25 °C using Eq. 1.
 |
1 |
where θ₅ is the molar residue ellipticity (MRE) of the protein sample at a given temperature, θ95 is the MRE at 95 ºC and θ25 is the MRE at 25 ºC.
Differential scanning fluorimetry
The thermal shift assay of recombinant PfGrp170 was investigated using differential scanning fluorimetry (DSF) as previously described39. Briefly, 4 μM of PfGrp170 recombinant protein was suspended in buffer TBS (20 mM Tris, pH 7.5, and 200 mM NaCl) supplemented with 40 μM (20X) SYPRO™ Orange Protein Gel Stain dye (Thermo Scientific, USA) and incubated for 10 min at 25 °C. To start the assay, the reaction mixture was transferred to a Magnetic Induction Cycler (MicPCR, Bio Molecular Systems, Australia) and temperature was increased from 40 °C to 95 °C at a rate of 0.2 °C/s while monitoring the fluorescence emission signal at 570 nm. To investigate the effect of nucleotides, the experiment was repeated in the presence of 5 mM ADP / ATP and 100 μM of a known ER Hsp90 ATP mimicking inhibitor 5’-N-ethyl-carboxamide-adenosine (NECA) as a control40. The signals recorded from the emission spectra buffer with or without ATP/ ADP/NECA, and SYPRO™ Orange dye were subtracted from the spectra as baseline before analysis. Data was processed by fitting a non-linear regression Boltzmann sigmoidal curve (Eq. 2) to determine the inflexion point (Tm) of the recombinant PfGrp170 protein (Huynh and Partch, 2016). The first derivative of the normalised fluorescence was also calculated using (Eq. 3) as previously described35, and the resulting data were analysed using GraphPad Prism 10.01 (GraphPad Software, MA, USA).
 |
2 |
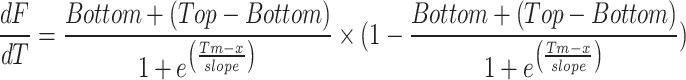 |
3 |
“Bottom” is the lowest value of the melting curve, “Top” is the peak melting curve value, and Tm is the inflexion point (°C).
Limited proteolysis
To further confirm the structural perturbation of recombinant PfGrp170, we conducted a partial proteolysis assay as previously described23. The experiment was initiated by suspending 7 µM PfGrp170 in buffer KNM (20 mM KH2PO4, pH 7.5, 200 mM NaCl, 10 mM MgCl2, 5 mM CaCl2, and 5 mM DTT) and supplemented with 0.072 µM Trypsin from porcine pancreas (Sigma-Aldrich, CA, USA). The reaction mixture was incubated at 25 °C and samples were collected at 5 min, 10 min, and 30 min time points and the proteolytic reaction was stopped using 1 mM phenylmethylsulfonyl fluoride (PMSF) (Roche, Germany). This was followed by the addition of SDS-PAGE loading buffer (100 mM Tris–HCl, pH 6.8, 140 mM SDS, 3 mM bromophenol blue, 30% (v/v) glycerol, and 200 mM β-mercaptoethanol) and boiled at 95 °C for 5 min. To investigate the effect of nucleotides, the assay was repeated in the presence of 2 mM ADP/ ATP incubated for 10 min prior to proteolytic digestion. The collected samples were analysed using SDS-PAGE and the band intensities were quantified using Image J (https://imagej.net/ij/index.html).
Basal ATPase activity
The basal ATPase activity of PfGrp170 and PfGrp94 as control35 was measured using the ATPase/GTPase Activity Assay Kit (Sigma-Aldrich, USA) following the manufacturer’s protocol. Optical density was measured at 620 nm using a Multiskan Sky Microplate reader (ThermoScientific, MA, USA). The sample OD values were normalized by subtracting the no enzyme control OD values. The standard curve of inorganic phosphate was used to determine free phosphate concentration ([Pi], µM), and ATPase activity was calculated using Eq. 4.
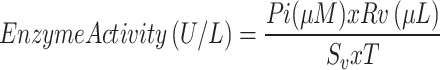 |
4 |
where T is the reaction time in minutes, Sv is the sample volume added in the well in µL and Rv is the total reaction volume.
Determining dissociation constants
The dissociation constants of ATP and recombinant PfGrp170 proteins were determined with varying ATP concentrations range of 0-2.4 µM using Nuclear Magnetic Resonance (NMR) as previously described41 with minor modifications. The recombinant proteins were dialyzed using SnakeSkin™ 10 000 MWCO dialysis membrane (Thermo-Fisher Scientific, USA, MA) into buffer MES (25 mM MES, 5 mM MgCl2, pH 6.5) according to the manufacturer’s instructions (Thermo-Fisher Scientific, MA, USA). Next, 0.135 µM protein and 10% (v/v) Deuterium oxide (D20) (Thermo-Fisher Scientific, MA, USA) and the desired ATP concentration were suspended in Wilmad NMR tubes (Sigma-Aldrich, CA, USA). NMR experiments were conducted using a Bunker NMR Ascend™ 600 MHz spectrometer (Bruker, Faellanden, Switzerland). The temperature of the probe was set at 25 °C and the WATERGATE pulse sequence was used to supress the water signal. The 1H NMR spectra of ATP were recorded after every 20 min for 2 h for the different ATP concentrations. To determine the dissociation constant (Kd) of PfGrp170 with ATP the chemical shift changes of the H2 (8.25 ppm) and H8 (7.88 ppm) protons of ATP were monitored as the ATP concentration was gradually increased. These particular proton signals were chosen because they exhibited significant chemical shift changes in response to varying ATP concentrations, allowing for accurate determination of the Kd value. The NMR data were processed using MestReNova software v15.0.1–35,756 (https://metrelab.com/download) and a non-linear fitting method was used to determine the Kd using the Eq. 5.
 |
5 |
where: Lo represents the molar concentration of PfGrp170, Po represents the molar concentration of ATP, Dmax is the maximum chemical shift change at saturation and Dobs is the observed chemical shift change.
Investigation of chaperone function
The chaperone activity of PfGrp170 was investigated by analysing its capability to suppress heat-induced aggregation of a model substrate, Malate dehydrogenase (MDH) as previously described23,36. First, 1.0 μM MDH from pig heart (Roche Diagnostics, Mannheim, Germany) together with varying concentrations of 0.125-2 μM PfGrp170 were suspended in buffer TBM (20 mM Tris–HCl, pH 7.5, 200 mM NaCl, and 5 mM MgCl2) and incubated at 48 °C for 150 min. The protein aggregation was monitored by recording light scattering at 360 nm every 10 min using a Multiskan Sky Microplate Spectrophotometer (Thermo-Fisher Scientific, MA, USA). The experiment was repeated in the presence of 5 mM ATP/ADP or 100 μM NECA. The data was analysed after baseline subtraction of absorbance reading in the absence of protein. The absorbance reading of MDH with or without a control protein, BSA, at 150 min was set as 100% and used to normalise all the other readings to generate the relative aggregation proportion on GraphPad Prism 10.0.1 (GraphPad Software, MA, USA).
Molecular docking
To elucidate the nucleotide binding mode of PfGrp170 NBD (AF-C0H5H0-F1), molecular docking was conducted using Schrödinger’s Maestro program 2022–1 as previously described42. In brief, the standard protocol was used to prepare the protein and ligand for generating up to 20 poses for the receptor and the box was set to the centre of specific residues of select regions with potential ligand-binding sites (ATP binding pocket). Furthermore, the box size was set to dock ligands with a length of 46 Å or less. The constraints settings were left as default and for the ligand settings, the sampling ring conformations were set to an energy window of 2.5 kcal/mol. The default Glide docking receptor scaling parameters were set as follows: Receptor van der Waals (0.50), Ligand van der Waals (0.50), maximum number of poses (20). The default prime refinement setting was set to refine residues within 5.0 Å of ligand poses and to optimize side chains. For glide redocking, the settings were set to redock into structures within 30.0 kcal/mol of the best structure and within the top 20 structures. The precision was set to standard protocol with the jobs set to a glide and prime CPUs of 5. The nucleotides ATP/ADP and control NECA were then analysed using MD simulations.
Molecular dynamics simulation
Molecular dynamics (MD) simulation to predict the time-dependent motion of atoms, simulating the dynamic behaviour of PfGrp170 in the presence and absence of nucleotides as a molecular system was conducted as previously described42. The initial step of the molecular dynamics simulations was to use the system builder function on the Desmond module of Schrodinger suite 2022–1 to solvate the energy minimized docked. The TIP3P solvent parameter was used, and boundary conditions were described as a cubic box with dimension distances of 90 Å and angles of 90° minimized to a volume of 10 Å. The force field was set to OPLS_2005, neutralized by adding Cl- or Na+ ions to salt concentration at 0.15 M. The MD simulations of the 3D structure of the PfGrp170 NBD were conducted using the Schrödinger Maestro enabled Schrodinger Desmond version. The ensemble class was set to NPT with bar pressure of 1 bar and a temperature of 300 K in all runs. The model system was set to relax before simulation using OPLS_2005 force field. The prepared molecular dynamics simulation jobs were written in Linux under the gpu_1 (36.1) host, and the files were submitted to the Lengau Centre for High Performance Computing (CHPC, CSIR, SA) cluster. The simulations were repeated as three independent experiments for 250 ns each to a total of 750 ns, and the post-dynamic analysis of the behaviour and the type of bonds for the interaction between the protein and ligand was recorded. The MD simulation stability was monitored by the protein–ligand Root mean square deviation (RMSD), protein–ligand Root mean square fluctuation (RMSF), Radius gyration, protein–ligand properties, and trajectories of ligand torsion profile.
Results
Overexpression and purification of recombinant forms of PfGrp170
The recombinant form of full length PfGrp170 protein was successfully expressed in E. coli XL1-Blue cells and purified using affinity chromatography (Fig. 1A). The SDS-PAGE analysis of the size exclusion chromatography of the recombinant protein was observed as a species at approximately 108 kDa which was confirmed by western blot analysis using the polyclonal α-PfGrp170 antibodies we developed (Fig. 1B). We validated the specificity of our in-house produced rabbit raised antibodies using another parasite ER localised PfGrp94 protein and the antibodies were specific for PfGrp17035. Furthermore, pre-immunisation sera from the same rabbit did not detect the recombinant proteins.
Fig. 1.

The production of recombinant PfGrp170 and antibodies. (A) The SDS-PAGE analysis of the PfGrp170 expression in E. coli XL1 Blue cells and (B) the purification using affinity chromatography followed by size exclusion chromatography. The lower panels are western blot confirmation of the expression and purification using rabbit raised polyclonal α-PfGrp170 as indicated. Lane M-protein molecular weight marker (kDa), lane C-total cell lysate of cells transformed with pQE30 plasmid, lane 0–12, total cell lysate of cells transformed with PfGrp170 at 0—12 h and lane O/N overnight post induction with IPTG, lane S- is the total cell lysate soluble fraction, lane F – is the flow through of unbound protein and lane W, are the washes to remove unbound protein, lanes E1-E3, elution at 150–500 mM imidazole. The full SDS-PAGE gels and Western blots are available as supporting data.
Secondary structure analysis of PfGrp170
The secondary structure of recombinant PfGrp170 was analysed using circular dichroism (CD) spectroscopy. CD measurements were recorded after optimising the buffer conditions (Supplementary Figure S1). We retrieved the predicted 3D model of PfGrp170 from AlphaFold to use as control in the secondary structure comparisons (Fig. 2A). The CD spectra of PfGrp170 observed a maximum (positive peak) at 192 nm, and deep minima (troughs) from 207–220 nm (Fig. 2B). The spectra deconvolution predicted 38% α-helices, 20% β-sheets and 42% unfolded/unordered regions. These deconvoluted experimental CD data were in agreement with the predicted AlphaFold secondary structure element distribution with 36% α-helices, 20% β-sheets and 44% unfolded/turns (Supplementary Table S1).
Fig. 2.

Circular dichroism spectroscopy of PfGrp170. (A) A 3D schematic showing the domain organization of PfGrp170 (PlasmoDB ID: PF3D7_1344200). The nucleotide binding domain (NBD) is shown in red, β substrate binding domain (βSBD) in green and the α substrate binding domain (αSBD) in grey. (B) The CD spectra of PfGrp170 had maximum at 192 nm and minima between 207 and 222 nm. The folded fraction of the PfGrp170 as a ratio of native protein compared to protein subjected to (C) different buffer pH, (D) chaotropic agents—urea and guanidine HCl. (E) The folded fraction of PfGrp170 as the protein was exposed to heat induced unfolding in the presence and absence of nucleotides 100 μM ATP/ADP, monitored as CD spectra when the temperature was increased from 25 °C, and (F) the refolding capability of the secondary structure of PfGrp170 monitored at 195, 205 and 222 nm as a ratio of the protein spectra at 25 °C when the temperature was reduced. The PfGrp170 structure was obtained from the AlphaFold server (https://alphafold.ebi.ac.uk/) with accession number AF-C0H5H0-F1-v4 and further refined using Schrödinger software (Maestro, Schrödinger Release 2022–1; https://www.schrodinger.com/platform/products/maestro/). Error bars represent standard error of the mean (n = 3).
To evaluate the structural stability of PfGrp170, the effect of pH on the mean residue ellipticity of the protein was monitored at 195, 205 and 222 nm. We observed that PfGrp170 is most stable at pH 7.5 (Fig. 2C). Interestingly, the observed signal at pH 6, pH 8 and pH 10 were lower than the signal at pH 7.5. This observation suggests that PfGrp170 undergoes a two-step unfolding from pH 2 to pH 6 then reaches a more stable state at pH 7.4 and second unfold step from pH 8 to pH 10. Considering that the pH ranges in the ER lumen is 7.2 ± 0.3 and that of the Golgi is at 6.7 ± 0.2 43,44. This suggests that PfGrp170 is possibly stable both in the ER lumen and Golgi’s if it gets exported from the ER before its return during protein secretion processes based on its ER retention signal KSDEL. These findings further support that PfGrp170 as an ER resident chaperone may facilitate the parasite to thrive under different pHs in weakly buffered solutions 45. Taken together, this suggest that PfGrp170 is structurally resilient to pH changes within a limited range around pH 7, which may assist the parasite to resist deleterious effects of pH changes as it traverses the different environments during its lifecycle in mosquito vector and human host.
To further validate the stability of the recombinant PfGrp170, we exposed the protein to chaotropic agents, urea and guanidine-HCl (GdHCl) which are known to denature proteins. The increase in concentration of either urea or GdHCl resulted in PfGrp170 losing its secondary structure fold (Fig. 2D). We observed that PfGrp170 unfolded in a two-step manner with the first unfolding from 0–2 M for urea, and a second unfolding with a steeper gradient from 4–6 M (Fig. 2D). On the other hand, PfGrp170 exposed to GdHCl had a more constant and more steeper denaturation profile from 0–2 M which became less steep towards 6 M. Taken together, this suggest that at lower concentrations, GdHCl is a more effective denaturant for PfGrp170 than urea. It is interesting to note that exposure to chemical denaturant, PfGrp170 follows a similar two step structural change as observed when exposed to different pHs. These observations validated that we purified a folded PfGrp170 which is stable as its structure was perturbed by exposure to denaturants.
After these observations, we further investigated the stability of PfGrp170 under heat stress. We exposed the protein to increasing temperatures and observed that as temperature increased the protein lost its relative folded structure (Fig. 2E). The protein became more unfolded with an increase in temperature in a 2-step fashion. The first rapid step was observed from 50 °C to 60 °C and the second step was more gradual after 65 °C to 75 °C, with a resultant Tm of 55 ± 1.8 °C. It was previously shown that the cytosolic parasite Hsp110s structure was perturbed by presence of nucleotides23, here we investigated the effect of nucleotides on the structural stability of the recombinant PfGrp170 protein. The presence of nucleotides affected the secondary structure stability of the protein (Fig. 2E). The presence of ATP/ADP were not significantly different, as presence of ATP resulted in a Tm of 52 ± 2.4 °C and ADP with a Tm of 55 ± 1.6 °C (Table 1). The presence of nucleotides resulted in a more rapid loss of structure up to 55 °C than in the apo state. After which the protein in the presence or absence of nucleotides followed a similar constant loss of structure up to 70 °C and thereafter a more gradual unfolding was observed. To further interrogate the effects of these folded fraction events on the refolding capability of the protein, we allowed the denatured protein to refold as the temperature was lowered from different maximum temperatures (Fig. 2F). The protein refolding capability was inversely proportional to the maximum temperature it was exposed to. Thus, protein exposed to a maximum temperature of 55 °C managed to refold up to 78% of its original structure. However, the protein exposed to 90 °C only managed to regain to 30% of its original folded structure. This suggest that PfGrp170 enters into a heat induced unfolded state at high temperatures and remains in a folding competent form that refolds when temperatures drop. Taken together this suggest that PfGrp170 partially unfolds under extreme stress and has the capability to refold when stress is removed.
Table 1.
Melting temperatures of PfGrp170 and its docking scores with nucleotides.
| Method | PfGrp170 Tm (°C) | |||
|---|---|---|---|---|
| Apo | ATP | ADP | NECA | |
| DSF | 58.8 ± 1.4 | 57.8 ± 0.9 | 59.9 ± 2.3* | 58.3 ± 1.6 |
| CD | 53.0 ± 1.8 | 52.0 ± 1.4 | 55.6 ± 1.6* | ND |
Analysis of PfGrp170 structure stability
The thermal stability analysis of PfGrp170 structure was conducted using differential scanning fluorimetry (DSF). We observed that the recombinant PfGrp170 unfolded as the temperature was raised and lost its secondary and tertiary structure fold by exposing hydrophobic patches which were bound by the dye resulting in increased fluorescence (Fig. 3A). The melting point Tm from the thermal shift assay for PfGrp170 of 58.8 ± 1.4 °C (Table 1), was marginally higher than for CD spectrum (Tm 54 ± 1.8 °C), which suggest that both the secondary and tertiary structure of PfGrp170 are thermally resilient. To further investigate the effect of nucleotides on the conformational changes during unfolding events, we repeated the experiment in the presence of nucleotides. There were changes observed in the presence of ATP/ ADP to the melting temperature of PfGrp170. Thus, the presence of ATP shifted the melting curve to the left reducing the melting temperature to 57.8 ± 0.9 °C, whilst, presence of ADP promoted resilience increasing the melting temperature to 59.9 ± 2.3°C (Table 1). We used a known ATP mimicking inhibitor, NECA, to determine its effects on the melting temperature of PfGrp170. Based on our observations, NECA failed to shift the meting temperature of PfGrp170, although as an ATP mimicking compound, it is specific for the ER Hsp90 proteins and not Hsp110s40. Taken together, this suggest that PfGrp170 was responsive to nucleotides with ATP promoting sensitivity to heat stress while ADP promoted resilience. Our current findings are in line with previous observation on the parasite cytosolic version PfHsp70-z whose structure was perturbed by the presence of nucleotides23,26.
Fig. 3.

Tertiary structure analysis of PfGrp170. (A) DSF melting curve fit onto sigmoidal curves used to derive the inflexion points of the melting temperatures indicated. (B) The analysis of change in fluorescence over time. (C) SDS-PAGE analysis of PfGrp170 subjected to proteolytic digestion by trypsin and (D) the densitometric analysis of the protein fractions after proteolytic digestion in the presence or absence of 2 mM ATP/ADP at 5 min, 15 min and 30 min intervals. Lane MW: molecular weight ladder (kDa), lane 0: Apo-PfGrp170 without proteolysis, lanes 5, 15 and 30: PfGrp170 digested with trypsin after 5 min, 15 min and 30 min, respectively in the absence or presence of 2 mM ATP/ADP. The error bars indicate the standard error of the mean from three independent assays (n = 3). The full SDS-PAGE gels are available as supporting data. Melting temperatures are reported as the mean of three independent assays and the standard error of the mean is shown. ND – not determined. Statistical analysis was conducted using one way ANOVA, significant differences from the apo state is shown by * at p < 0.05 (n = 3).
Investigation of protein conformation and stability using limited proteolysis
The tertiary structure conformational stability of PfGrp170 was further investigated using partial proteolysis by Trypsin. The SDS-PAGE analysis of the remaining protein after partial digestion were quantified using densitometric analysis. Based on the band intensities, PfGrp170 showed digestion under all conditions tested (Fig. 3C), based on the intensity of the band at ~ 100 kDa. Under all conditions, the protein degraded as time increased from 0 to 30 min. The presence of ATP seems to expose the protein to more proteolytic digestion compared to ADP. We repeated this using proteinase K and observed a similar proteolytic profile (Supplementary Figure S3). This possibly confirms the observations made using the DSF and CD thermal shift assays that PfGrp170 structure perturbation is responsive to the presence of ADP, but ATP modulate its sensitivity to thermal stress.
Investigation of PfGrp170 chaperone activity
The chaperone function of PfGrp170 was investigated by monitoring its capability to suppress the heat-induced aggregation of model substrate malate dehydrogenase (MDH). First, MDH suspended in buffer and subjected to heat stress at 48 ºC showed a marked increase in absorbance depicting high light scattering. The light scattering was monitored at 360 nm and was set as 100% aggregation (Fig. 4). Thereafter the experiment was repeated in the presence of protein control, an equimolar BSA, and a similar aggregation profile was observed. The recombinant PfGrp170 when exposed to these conditions did not aggregate in line with CD and DSF results (Fig. 2 & 3). As such, we then mixed varying concentrations of PfGrp170 with the MDH which showed a reduced aggregation to 30% at an equimolar ratio of 1:1 chaperone: substrate (Fig. 4A). We further interrogated the effect of nucleotides on the chaperone function of PfGrp170. The presence of ATP did not significantly affect the chaperone activity of PfGrp170 (Fig. 4B). Similarly, the presence of ADP did not significantly improve the aggregation suppression capability of PfGrp170. This was in line with our previous finding of its cytosolic homology PfHsp11023. This suggest that the chaperone function of both versions of parasite Hsp110 (cytosolic and ER localised) are nucleotide independent. However, higher concentrations of NECA abrogated the chaperone function of PfGrp170 which suggest that the protein may be amenable to inhibition by ATP mimicking inhibitors despite its nucleotide independent chaperone function.
Fig. 4.

Chaperone suppression of heat-induced aggregation of malate dehydrogenase and basal ATPase activity. (A) Suppression of MDH aggregation by the varying amounts of PfGrp170. (B) The aggregation suppression activity of 1 μM PfGrp170 in the absence of nucleotides or the presence of 5 mM ATP/ADP or 100 μM NECA nucleotides. (C) The enzyme activity of PfGrp170 and PfGrp94 as control. (D) The structure of ATP highlighting the protons adenine H2 and H8. (E) Representative plot change in chemical signal of protons versus molar ratio of ATP and PfGrp170. The basal ATPase activity and dissociation constants are indicated. Error bars represent the standard error for the mean of three independent repeats and one way ANOVA was used for the experimental conditions compared to the control (MDH), ns: no significant difference (p ≤ 0.1: *, p ≤ 0.01: **, p ≤ 0.001: ***, p ≤ 0.0001: ****).
Basal ATPase activity
The basal ATPase activity of PfGrp170 was determined through a colorimetric assay, which involved measuring the rate of inorganic phosphate release during ATP hydrolysis. PfGrp170 exhibited a basal ATP hydrolysis rate of 2.0 ± 0.5 μU/L (Fig. 4C). We then investigated the dissociation constant (Kd) which represents the rate at which the ATP dissociates from the PfGrp170 using Nuclear Magnetic Resonance (NMR) titration by monitoring the adenine H2 and H8 protons over time (Fig. 4D). There were noticeable chemical shift changes in the H2 and H8 proton signals of ATP at each titration point (Supplementary Figure S4). The analysis of the chemical shift data reveal that PfGrp170 exhibits a slower dissociation rate (Kd) of 143.0 ± 15.6 nM (Fig. 4E). This was within the range of ATP dissociation constants for Hsp110 proteins from the yeast, S. cerevisiae, Sse1p which were reported between 95.0 nM to 153.0 nM28. However, this was lower than for the C. albicans of 40.6 nM25. This indicates that PfGrp170 has the capability to bind and hydrolyse ATP as its homologues but with species specific dissociation rate. These findings provide significant insights into unique potential ATP-binding and hydrolysis properties of this chaperone.
Molecular docking
After we observed that PfGrp170 had subtle structural changes induced by nucleotides or the control inhibitor NECA, we next investigated the localised effects of these nucleotides on the nucleotide binding domain. Using the PfGrp170 NBD truncated 3D model from AlphaFold, we docked ATP/ADP/NECA onto the prepared protein structure (Fig. 5). We observed that the protein bound with compound NECA with the highest docking scores of -9.31 kcal/mol followed by ADP of -8.83 kcal/mol and -8.30 kcal/mol for ATP (Table 2). The docking results indicated that these nucleotides and compound interacted with the protein through specific interactions. ATP formed hydrogen bonds with, Glu34, Tyr35, Lys58, Asp256, Ser259 (Fig. 5A). ADP interacted with PfGrp170 residues Asp30, Asn33, Asn92, Lys211, Lys260, Asn261, and Ser404 (Fig. 5B). Similarly, NECA formed hydrogen bonds with residues Asp30, Asn33, Asn263, Ser404 and Asp431 with an additional, pi-pi stacking interaction with Phe31 (Fig. 5C). Additional interactions occurred through Pi-cation interactions for ADP and NECA with Lys37. Hydrophobic interactions with several other residues within 4 Å stabilized the docked complexes. These findings further support the chaperone function profile of the nucleotides where there were minor differences between ATP/ADP but drastic effects in the presence of NECA, which may be due to these binding events.
Fig. 5.

The 3D and 2D analysis of molecular docking of nucleotides with PfGrp170. The 3D active site pocket surface bound with (A) ATP, (B) ADP and (C) NECA and its orientation. The middle panel show the 3D structure of the binding complex which are shown in 2D on the third panel respectively. The docking predictions were generated using Schrödinger Maestro 2022 (https://www.schrodinger.com/platform/products/maestro/).
Table 2.
The molecular docking and dynamics simulation summary data. The standard deviation of the mean is shown.
| PfGrp170 in complex with | Docking scores /kcal/mol | Rg (mean ± SD) Å | RMSD (mean ± SD) Å | RMSF (mean ± SD) Å | Ligand RMSD (mean ± SD) Å |
|---|---|---|---|---|---|
| Apo | - | 23.56 ± 0.13 | 3.26 ± 0.38 | 1.87 ± 1.21 | - |
| ATP | -8.30 | 23.43 ± 0.13 | 2.86 ± 0.47 | 1.95 ± 1.50 | 2.11 ± 0.53 |
| ADP | -8.83 | 23.33 ± 0.11 | 2.90 ± 0.38 | 1.92 ± 1.17 | 1.38 ± 0.38 |
| NECA | -9.31 | 23.09 ± 0.11 | 2.66 ± 0.30 | 1.63 ± 0.86 | 2.46 ± 0.36 |
Molecular dynamics simulations
To further validate the molecular docking findings, MD simulations were conducted to investigate the binding stability and dynamics of selected compounds with PfGrp170. Here, the dynamic behaviour of PfGrp170 protein and its nucleotide ligand complexes was investigated by comparing the Rg, RMSF, RMSD values and the type of protein–ligand intermolecular interactions for 250 ns. The mean Rg (± SD) values for Cα atoms were 23.56 ± 0.13 Å for PfGrp170-Apo, 23.43 ± 0.13 Å for PfGrp170-ATP, 23.33 ± 0.11 Å for PfGrp170-ADP, and 23.09 ± 0.11 Å for NECA. Comparative Rg profile analysis revealed ATP and ADP induced significant conformational changes, resulting in more compact structure similar to the apo state (Fig. 6A). The binding PfGrp170 to NECA, even though had the lowest Rg profile did not result in dramatic conformational changes to the protein. These finds suggest that the nucleotides bind to the protein and stabilise it without inducing major protein conformational changes.
Fig. 6.

The molecular dynamics simulations stability of PfGrp170 and its complexes with nucleotides. (A) The Rg of Cα atoms illustrating the compactness of the structure, (B) The RMSD plot of PfGrp170 Cα atoms illustrating the stability of the protein backbone, (C) the flexibility of PfGrp170-NBD in its apo form and in complex with nucleotides, (D) the ligand RMSD graph showing the stability of nucleotides, within the PfGrp170-binding site over 250 ns. These graphs were generated using GraphPad Prism v10.0.2 (GraphPad Software, MA, USA) and are summarised in Table 2.
RMSD of the protein’s Cα atoms was monitored over 250 ns to understand the conformational dynamics of the simulated systems (Fig. 6B). The PfGrp170 complex with ATP displayed the low conformational change with an RMSD value of 2.86 ± 0.47 Å, followed by ADP, 2.90 ± 0.38 Å, and in the apo state had the highest conformational change at 3.26 ± 0.38 Å. The lowest conformational changes were recorded for NECA with RMSD value of 2.66 ± 0.30 Å. NECA had a similar but unique binding profile compared to nucleotides. The restricted dynamics of PfGrp170 in complex with ATP may suggest structural changes to facilitate its hydrolytic activity compared to ADP and then goes into a relaxation in the absence of nucleotides with more conformational changes. This further validates the structural stability profile perturbations observed in the presence of ATP from the CD and DSF analysis.
To investigate residue flexibility, RMSF analysis was performed on the side chains of PfGrp170 NBD during MD simulations (Supplementary Figure S5). The RMSF profiles of the apo-protein and with nucleotide complexes, overlap with the apo-protein profile (Fig. 6C). The mean RMSF values for side chains of PfGrp170 with nucleotides ATP and ADP or without nucleotides were within the same fluctuation range 1.95 ± 1.50 Å, 1.92 ± 1.17 Å, 1.87 ± 1.21 Å respectively. However, PfGrp170 in the presence of NECA had the least residue fluctuations with RMSF of 1.63 ± 0.86 Å. These results suggest that nucleotides do not cause dramatic structural changes to PfGrp170 but alter the protein structure by binding to the least fluctuating regions of the protein. Interestingly, NECA could potentially cause structural changes to Grp170 as it bound to unique residues than the nucleotides. The stability of the nucleotides within the PfGrp170-binding pocket was investigated through ligand RMSD. The mean RMSD (± SD) values for side chains were highest for PfGrp170-NECA with 2.46 ± 0.36 followed by PfGrp170-ATP at 2.11 ± 0.53 Å and PfGrp170-ADP had the lowest at 1.38 ± 0.38 Å. The results show that ADP exhibited a more stable behaviour compared to ATP and NECA (Fig. 6D). It should be noted that, ATP, ADP and NECA all remained stable and bound to the same active site throughout the simulation (Supplementary Figure S5).
The molecular basis of ligand binding, protein–ligand intermolecular interactions were analysed throughout the MD simulation for PfGrp170 and its ligands (Fig. 7; Supplementary Table S2). The results show that ADP exhibited a higher interaction fraction (3.4) compared to ATP (2.9) and NECA (1.75), with dominant hydrogen bond interactions and water bridges, indicating stronger and more frequent interactions between ADP and PfGrp170 residues (Supplementary Figure S6). Specifically, hydrogen bond interaction fraction of ADP was significantly higher than for ATP and NECA, forming strong and stabilized protein–ligand complexes (Fig. 7A). Additionally, water-bridged interactions between ADP were also dominant, with a higher interaction fraction compared to ATP and NECA. As a nonspecific inhibitor, NECA formed less stronger bonds but with more effects on the structure of PfGrp170 NBD. These findings suggest that ADP form more stable complexes with PfGrp170 than ATP, due to stronger and more frequent intermolecular interactions. Taken together, these results in part confirm the potential catalytic activity of PfGrp170 to destabilize the structure of ATP resulting in its hydrolysis to more stable interactions with ADP and release of inorganic phosphate as confirmed by the basal ATPase enzymatic assay.
Fig. 7.

The interaction patterns between PfGrp170 with nucleotides during a 250 ns simulation. The frequency and type of bond contacts between PfGrp170 with nucleotides (A) ATP, (B) ADP and (C) NECA. The 2D rendering of specific atom-level interactions between ATP /ADP/NECA with residues within the protein active site, for more than 80 ns. The key binding interactions that stabilize the complex observed were generated using Maestro v13.1.8's simulation interaction diagram algorithm.(https://www.schrodinger.com/platform/products/maestro/).
Analysis of residues interacting with ligands within 4 Å for over 30% of the simulation time revealed the stability of the formed structures (Fig. 7). Specifically, the O- and O atoms on the last phosphate group of ATP formed stabilizing hydrogen bonds with Lys37, Lys58 and Ser404 for more than 35% of the simulation period (Fig. 7A; Supplementary Table S3). The O- and O atoms on the second phosphate group of ATP formed stabilizing hydrogen bonds and water bridges with Thr35, Glu34 and Asn33 respectively for more than 40%. Lastly, the O- and O atoms on the first phosphate group of ATP formed stabilizing hydrogen bonds and water bridges with Asn33, Ser259, Lys260 and Asn261 respectively for more than 50% of the simulation period. Asp256 and Asn261 formed a hydrogen bond and water bridge with the hydroxyl group (89%) and oxygen group (36%) on the ribose sugar. Asp30 and Ser202 interacted with the amine group on the 5-carbon ring of adenine for 39% and 93% of the simulation period respectively. The last phosphate group of ADP formed numerous hydrogen bonds and water bridges with Asn33, Asn92, Asn93, Lys211, Lys260, Asn261, Glu291 and Asn292 for more than 47% of the simulation period (Fig. 7B). The first phosphate of ADP also formed hydrogen bonds and water bridges with Asn33, Ser259, Lys260 and Asn261 for more than 46% of the simulation period. These nucleotide binding poses were similar to the predictions made using the human Grp17046. Further, analysis of the residues implicated in PfGrp170 binding to NECA were predominantly hydrogen bonds with Phe31, Asp256, Ser259, Asp431, Ser404 and formed water bridges with Asp30 and Lys37 (Fig. 7C). Taken together, these findings suggest that PfGrp170 form unique interactions with ADP, ATP and NECA, however, the differences in residues and bond types potentially cause conformational changes to the PfGrp170 structure influencing its thermal stability profiles. This further validates our findings that PfGrp170 is responsive to nucleotides to structural perturbation, which may not be enough to cause allosteric changes required for chaperone function modulation. However, the unique binding profiles of NECA may have resulted in PfGrp170 undergoing restricted conformational dynamics and its failure to hydrolyse NECA may have caused the inhibition of its chaperone function.
Discussion
The ER localised PfGrp170 is an essential protein during the schizont development at the asexual stages of the parasite32. PfGrp170 is thought to function as a chaperone that facilitates the folding of parasite proteins that are destined for secretion and export through the ER (Chakafana et al., 2019). Furthermore, PfGrp170 has been shown to associate with the ortholog PfGrp78 and its deletion resulted in activation of eIF2α through the protein kinase PK4 pathway32. This is an important signalling pathway for ER stress response to activate the unfolded protein response (UPR)47. Therefore, this implicates PfGrp170 as an important player in the parasite cell ER stress response which may explain its essential role in parasite survival. However, its function has not been well established. Here, we present the first direct evidence for the overexpression, purification and in vitro biochemical and biophysical characterization of PfGrp170. There are reports that in part, artemisinin mode of action is through downstream protein alkylation leading to protein degradation by the ER stress response systems which activate the parasite UPR4,48 through a yet to be validated mechanism. As it has been proposed that, parasites under artemisinin drug pressure that elicit a robust ER stress response through the UPR, develop resilience to the drug action resulting in parasite resistance49. It is tempting to speculate that PfGrp170 function in the ER may be an antagonist to the effects of artemisinin treatment thereby masking the effects of the drug by facilitating a robust ER stress response. Therefore, in light of the broad functions of PfGrp170 and its possible implication in drug resistance development its characterization is important.
Firstly, we sought to optimise the production of recombinant PfGrp170, and using a combination of immobilized metal affinity chromatography and size exclusion chromatography we achieved more than 90% purity of the recombinant protein (Fig. 1). We established that PfGrp170 secondary structure is comprised of predominantly α-helical, β-sheets regions and turns (Fig. 2), these findings are in line with our previous findings of the predominantly α-helical structure of the cytosolic parasite homologue PfHsp70-z26. Considering that Hsp110s are larger distant family members of the Hsp70 super family, they are characterised by insertions on the NBD and SBD lid50, this justifies the presence of a large portion of unordered/turns on the structure of PfGrp170 (Fig. 2A). PfGrp170 also exhibited resilience to thermal stress to temperatures of up to 70 °C as it was able to gain its secondary structure fold upon removal of the heat stress (Fig. 2F). These findings further confirm its potential essential role in parasite survival when the temperature spikes during host fever episodes where body temperatures can rise up to 42 °C51. This heat stress resilience of PfGrp170 is consistent with previous findings made with the parasite cytosolic PfHsp70-z26 and with its function as a molecular chaperone against the development of parasite cells under heat stress32. Moreover, analysis of the structure stability confirmed that the protein is thermally resilient, and the presence ADP promoted its stability more than ATP (Fig. 2E; 3A, B). Furthermore, PfGrp170 exhibited an ATPase activity of 2.0 ± 0.5 μU/L which was further confirmed with NMR spectroscopy (Fig. 4C,E). This may suggest that as the protein binds and hydrolyse ATP and during this enzymatic reaction the nucleotide induced conformational changes are potentially important for its nucleotide exchange function on the ER localised Hsp70, PfGrp78. This may partly explain the reduction in its thermal stability in the presence of ATP. Similarly, this conformational change in the presence of ATP predisposed the protein to proteolytic cleavage by proteases (Fig. 3C; Supplementary Figure S3). On the other hand, the presence of ADP seems to have a protective role to thermal stress and proteolytic cleavage by trypsin (Fig. 3C,D) and proteinase K (Supplementary Figure S3). Our findings are in contrast with the yeast cytosolic Hsp110, which was shown to be sensitive to ATP structure stabilization52. This may be due to differences between these two proteins as they have low sequence identities (19%) between PfGrp170 when compared with its yeast equivalent similar to our previous observations26. In addition, this highlight species specific differences between the parasite and the yeast homologues, as was observed between yeast and human Hsp110s, to show that Hsp110s are species specific25,28.
Hsp110s bind and reduce unproductive protein folding under thermal stress as holdases50,18. They eventually pass their substrates to foldases such as Hsp70 that can actively facilitate the refolding of the bound substrates53,54). Our results showed that PfGrp170 suppressed the heat induced aggregation of MDH in equimolar ratio (Fig. 4A). The presence of ADP/ADP did not affect the capability of PfGrp170 to suppress MDH aggregation when compared to the apo state (Fig. 4B). This shows that PfGrp170 binds substrates tightly in the presence or absence of nucleotides, which is in contrast to the nucleotide induced structural perturbation observed in the CD, DSF and limited proteolysis structural analysis (Fig. 2E, 3A,B,C). However, this is in line with the findings from the human Grp170 homologue, whose chaperone function was shown to be unresponsive to ATP presence in contrast to the canonical Hsp70, Bip16. To further validate the modulation of PfGrp170 function by small molecule inhibitors we used an ATP mimicking inhibitor, NECA, at low concentration, it had similar effect on the chaperone activity of PfGrp170 as was observed with ADP/ATP albeit with marginal loss of activity (Fig. 4B). However, the presence of higher concentration of NECA abrogated the chaperone function of PfGrp170, which offers promise to further development of ATP mimicking inhibitors targeting the parasite Grp170 ATP binding pocket as it is different from the GHKL family of Bergerat fold of Hsp90s55, only sharing about 42% identity46. It should be noted that NECA was effective to abrogate the chaperone function of PfGrp170 in micro molar range whilst ATP/ADP at milli molar range could not (Fig. 4B). To further explore this, since NECA has an adenine moiety similar to nucleotides, we conducted in silico studies using the PfGrp170 NBD 3D structure (Fig. 5). The molecular docking and molecular dynamics simulation studies showed the presence of unique interaction residues between ATP/ADP (Fig. 5A,B, Fig. 7A,B) and with NECA (Fig. 5C,Fig. 7C). These differences in binding and stabilization of the protein structural conformation (Fig. 6) further support the nucleotide induced changes to possibly prime the protein for its NEF function. However, these structural changes due to nucleotide binding are potentially dispensable for chaperone function as the PfGrp170 protein was not responsive to nucleotides as previously shown in yeast cytosolic and ER Hsp110 homologues50,52. However, the presence of an ATP mimicking inhibitor caused both structural changes and abrogated the chaperone function of PfGrp170 (Fig. 3A,B, 4B). This suggests that similarly to the nucleotide induced structural changes on PfGrp170, small molecules could abrogate its chaperone function which may result in parasite death.
Our current study findings support that PfGrp170 exhibit bona fide nucleotide independent holdase function to suppress the aggregation of model substrate MDH. In addition, we showed that nucleotides were capable of modulating its structure which may be enough to promote its NEF function on PfGrp78 in the ER. Furthermore, we showed that the chaperone function of the PfGrp170 is modulated by small molecules suggesting an avenue for malaria parasite specific inhibition as the protein is essential for parasite survival. These observations pave a way for further biophysical analysis to elucidate the role of PfGrp170 in malaria parasites development as a potential drug target.
Limitations to the study
While we overexpressed and purified the protein under non denaturing conditions, we could not validate native fold of the protein. Therefore, the recombinantly produced protein structure and function should be interpreted based on the experimental setup and not in the parasite under physiological conditions.
Supplementary Information
Acknowledgements
The authors would like to acknowledge the South African Council for Scientific and Industrial Research (CSIR) Centre for high performance computing (CHPC) for computational platform on Schrodinger software and GPU usage. TZ is a recipient of the Georg Foster fellowship from the Alexander von Humboldt Foundation and the South African Department of Science and Technology Future Professor’s fellowship. Finally, the authors would like to acknowledge the Stellenbosch University Sub-Committee B for financial support to TZ
Author contributions
FLM wrote the manuscript, conducted some experiments, and performed data interpretation. MLS: conducted some experiments, interpreted the data. WM: conducted some experiments and interpreted the data, ES: co-supervised the study. TZ: conceptualized the study, interpreted data, wrote and edited the manuscript and implemented the study, supervised the study.
Funding
National Research Foundation,148045,617193, South African Medical Research Council
Data availability
Data is provided within the manuscript or supplementary information files. The sequence datasets generated and/or analyzed during the current study are available in the European Nucleotide Archive (ENA) repository, Accession number: ERA31197427.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-98317-0.
References
- 1.Jonsdottir, T. K., Gabriela, M., Crabb, B. S., de Koning-Ward, T. F. & Gilson, P. R. Defining the essential exportome of the malaria parasite. Trends Parasitol.37(7), 664–675 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Braakman, I. & Hebert, D. N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol.5(5), a013201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Florentin, A., Cobb, D. W., Kudyba, H. M. & Muralidharan, V. Directing traffic: Chaperone-mediated protein transport in malaria parasites. Cell. Microbiol.22(7), e13215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharjee, S. et al. Remodeling of the malaria parasite and host human red cell by vesicle amplification that induces artemisinin resistance. Blood J. Am. Soc. Hematol.131(11), 1234–1247 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandit, K., Surolia, N., Bhattacharjee, S. & Karmodiya, K. The many paths to artemisinin resistance in Plasmodium falciparum. Trends Parasitol.39(12), 1060–1073 (2023). [DOI] [PubMed] [Google Scholar]
- 6.Peng, M., Chen, F., Wu, Z. & Shen, J. Endoplasmic reticulum stress, a target for drug design and drug resistance in parasitosis. Front. Microbiol.12, 670874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rocamora, F. et al. Oxidative stress and protein damage responses mediate artemisinin resistance in malaria parasites. PLoS Pathog.14(3), e1006930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ray, A., Mathur, M., Choubey, D., Karmodiya, K. & Surolia, N. Autophagy underlies the proteostasis mechanisms of artemisinin resistance in P. falciparum malaria. MBio13(3), 00630–00722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haas, I. G. BiP (GRP78), an essential hsp70 resident protein in the endoplasmic reticulum. Experientia50(11), 1012–1020 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Kozutsumi, Y., Segal, M., Normington, K., Gething, M. J. & Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature332(6163), 462–464 (1988). [DOI] [PubMed] [Google Scholar]
- 11.Shonhai, A., Boshoff, A. & Blatch, G. L. The structural and functional diversity of Hsp70 proteins from Plasmodium falciparum. Protein Sci.16(9), 1803–1818 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li, H. et al. An unexpected second binding site for polypeptide substrates is essential for Hsp70 chaperone activity. J. Biol. Chem.295(2), 584–596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu, Q. & Hendrickson, W. A. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell131(1), 106–120 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuermann, J. P. et al. Structure of the Hsp110: Hsc70 nucleotide exchange machine. Mol. cell.31(2), 232–243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Easton, D. P., Kaneko, Y. & Subjeck, J. R. The Hsp110 and Grp170 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones5(4), 276 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behnke, J. & Hendershot, L. M. The large Hsp70 Grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional Hsp70s. J. Biol. Chem.289(5), 2899–2907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekhar, A., Rosenzweig, R., Bouvignies, G. & Kay, L. E. Hsp70 biases the folding pathways of client proteins. Proc. Natl. Acad. Sci.113(20), E2794–E2801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andréasson, C., Rampelt, H., Fiaux, J., Druffel-Augustin, S. & Bukau, B. The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J. Biol. Chem.285(16), 12445–12453 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mattoo, R. U., Sharma, S. K., Priya, S., Finka, A. & Goloubinoff, P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem.288(29), 21399–21411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dragovic, Z., Broadley, S. A., Shomura, Y., Bracher, A. & Hartl, F. U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J.25(11), 2519–2528 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer, M. P. & Gierasch, L. M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem.294(6), 2085–2097 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandal, A. K. et al. Hsp110 chaperones control client fate determination in the Hsp70–Hsp90 chaperone system. Mol. Biol. Cell21(9), 1439–1448 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zininga, T. et al. Plasmodium falciparum Hsp70-z, an Hsp110 homologue, exhibits independent chaperone activity and interacts with Hsp70-1 in a nucleotide-dependent fashion. Cell Stress Chaperones21(3), 499–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakafana, G. & Shonhai, A. The role of non-canonical Hsp70s (Hsp110/Grp170) in cancer. Cells10(2), 254 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, Y. et al. Purification and biochemical characterization of Msi3, an essential Hsp110 molecular chaperone in Candida albicans. Cell Stress Chaperones26(4), 695–704 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zininga, T. et al. Overexpression, purification and characterisation of the Plasmodium falciparum Hsp70-z (PfHsp70-z) protein. PLoS ONE10(6), e0129445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia, V. M., Nillegoda, N. B., Bukau, B. & Morano, K. A. Substrate binding by the yeast Hsp110 nucleotide exchange factor and molecular chaperone Sse1 is not obligate for its biological activities. Mol. Biol. Cell28(15), 2066–2075 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raviol, H., Bukau, B. & Mayer, M. P. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett.580(1), 168–174 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Shaner, L., Sousa, R. & Morano, K. A. Characterization of Hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry45(50), 15075–15084 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu, X. et al. Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. J. Biol. Chem.287(8), 5661–5672 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang, J. et al. Conformation transitions of the polypeptide-binding pocket support an active substrate release from Hsp70s. Nat. commun.8(1), 1201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kudyba, H. M. et al. The endoplasmic reticulum chaperone PfGRP170 is essential for asexual development and is linked to stress response in malaria parasites. Cell. Microbiol.21(9), e13042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakafana, G., Mudau, P. T., Zininga, T. & Shonhai, A. Characterisation of a unique linker segment of the Plasmodium falciparum cytosol localised Hsp110 chaperone. Int. J. Biol. Macromol.180, 272–285 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Bellstedt, D. U., Van der Merwe, K. J. & Galanos, C. Immune carrier properties of acid-treated Salmonella minnesota R595 bacteria The immune response to TNP-bacterial conjugates in rabbits and mice. J. Immunol. Methods108(1–2), 245–254 (1988). [DOI] [PubMed] [Google Scholar]
- 35.Muzenda, F. L. et al. Characterization and Inhibition of the Chaperone function of Plasmodium falciparum Glucose-regulated protein 94 kDa (Pf Grp94). Proteins: Struct., Funct., Bioinf.10.1002/prot.26779 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makumire, S. et al. Mutation of GGMP repeat segments of Plasmodium falciparum Hsp70–1 compromises chaperone function and Hop co-chaperone binding. Int. J. Mol. Sci.22(4), 2226 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miles, A. J., Janes, R. W. & Wallace, B. A. Tools and methods for circular dichroism spectroscopy of proteins: a tutorial review. Chem. Soc. Rev.50(15), 8400–8413 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature596(7873), 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maake, R. & Achilonu, I. Expression, purification and biophysical characterisation of Klebsiella Pneumoniae Protein Adenylyltransferase: a systematic integration of empirical and computational modelling approaches. Protein J.43, 751–770. 10.1007/s10930-024-10210-3 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huck, J. D. et al. NECA derivatives exploit the paralog-specific properties of the site 3 side pocket of Grp94, the endoplasmic reticulum Hsp90. J. Biol. Chem.294(44), 16010–16019 (2019). [DOI] [PMC free article] [PubMed]
- 41.Yamamoto, S. et al. ATPase activity and ATP-dependent conformational change in the co-chaperone HSP70/HSP90-organizing protein (HOP). J. Biol. Chem..289(14), 9880–9886 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stofberg, M. L., Muzenda, F. L., Achilonu, I., Strauss, E. & Zininga, T. In silico screening of selective ATP mimicking inhibitors targeting the Plasmodium falciparum Grp94. J. Biomol. Structure Dynamics10.1080/07391102.2024.2329304 (2024). [DOI] [PubMed] [Google Scholar]
- 43.Klonis, N. et al. Evaluation of pH during cytostomal endocytosis and vacuolar catabolism of haemoglobin in Plasmodium falciparum. Biochem. J.407(3), 343–354. 10.1042/BJ20070934 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohring, F. et al. Determination of glutathione redox potential and pH value in subcellular compartments of malaria parasites. Free Radical Biol. Med.104, 104–117 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Saliba, K. J. & Kirk, K. pH regulation in the intracellular malaria parasite, Plasmodium falciparum: H+ extrusion via a V-type H+-ATPase. J. Biol. Chem.274(47), 33213–33219 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Pagare, P. P., Wang, H., Wang, X. Y. & Zhang, Y. Understanding the role of glucose regulated protein 170 (GRP170) as a nucleotide exchange factor through molecular simulations. J. Mol. Graph. Model.85, 160–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galluzzi, L., Diotallevi, A. & Magnani, M. Endoplasmic reticulum stress and unfolded protein response in infection by intracellular parasites. Fut. Sci. OA.3(3), FSO198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haldar, K., Bhattacharjee, S. & Safeukui, I. Drug resistance in Plasmodium. Nat. Rev. Microbiol.16(3), 156–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang, M. et al. Inhibiting the plasmodium eIF2α kinase PK4 prevents artemisinin-induced latency. Cell Host Microbe.22(6), 766–776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oh, H. J., Easton, D., Murawski, M., Kaneko, Y. & Subjeck, J. R. The chaperoning activity of Hsp 110: Identification of functional domains by use of targeted deletions. J. Biol. Chem.274(22), 15712–15718 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Miller, A., Koeneman, S., Suneja, M., Cavanaugh, J. & Polgreen, P. Diurnal temperature variation and the implications for diagnosis and infectious disease screening: a population-based study. Diagnosis11(1), 54–62. 10.1515/dx-2023-0074 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andreasson, C., Fiaux, J., Rampelt, H., Mayer, M. P. & Bukau, B. Hsp110 is a nucleotide-activated exchange factor for Hsp70. J. Biol. Chem.283(14), 8877–8884 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Rampelt, H. et al. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J.31(21), 4221–4235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sztangierska, W. et al. Early steps of protein disaggregation by Hsp70 chaperone and class B J-domain proteins are shaped by Hsp110. Elife13, RP94795 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergerat, A. et al. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature386(6623), 414–417 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is provided within the manuscript or supplementary information files. The sequence datasets generated and/or analyzed during the current study are available in the European Nucleotide Archive (ENA) repository, Accession number: ERA31197427.