

Abstract
Do endocannabinoids (eCBs) participate in long-term synaptic plasticity in the brain? Using pharmacological approaches and genetically altered mice, we show that stimulation of prelimbic cortex afferents at naturally occurring frequencies causes a long-term depression of nucleus accumbens glutamatergic synapses mediated by eCB release and presynaptic CB1 receptors. Translation of glutamate synaptic transmission into eCB retrograde signaling involved metabotropic glutamate receptors and postsynaptic intracellular Ca2+ stores. These findings unveil the role of the eCB system in activity-dependent long-term synaptic plasticity and identify a mechanism by which marijuana can alter synaptic functions in the endogenous brain reward system.
Exogenous cannabinoids, such as the active component of Cannabis sativa L, (−)-transdelta9-tetrahydrocannabinol, as well as endocannabinoids (eCBs; anandamide and 2-arachidonoyl-glycerol) share the same target in the central nervous system: a Gi/Go-coupled receptor named CB1 (1, 2). Activation of CB1 receptors inhibits both inhibitory and excitatory synaptic transmission in the hippocampus (3, 4), substantia nigra pars compacta (5), the cerebellum (6), the prefrontal cortex (7), and the nucleus accumbens (NAc) (8, 9). Both eCBs and CB1 have been involved in a short-lasting form of synaptic regulation: the “depolarization-induced suppression” of both inhibitory (10–15) and excitatory transmission (16). However, the involvement of eCB in long-lasting activity-dependent synaptic plasticity remained to be documented. The mesocorticolimbic dopaminergic system, and particularly the NAc, is essential to the reinforcing properties of addictive drugs (17, 18). Cannabinoids activate mesolimbic dopamine neurons (19) and increase NAc dopamine levels (20), similarly to other drugs of abuse. Our finding that CB1 are localized at the excitatory afferents to the NAc where exogenous cannabimimetics inhibit glutamatergic synaptic transmission (9) raised two questions: how does synaptic activity lead to the production of eCBs in the NAc, and what are the physiological correlates of eCB release on synaptic transmission?
Methods
Slice Preparation and Electrophysiology.
Whole-cell patch–clamp and extracellular field recordings were made from medium spiny neurons in parasagittal slices of mouse NAc. These methods have been described in detail previously (9). In brief, mice (male C57BL6, 4–6 weeks) were anesthetized with isoflurane and decapitated. The brain was sliced (300–400 μm) in the parasagittal plane by using a vibratome and maintained in physiological saline at 4°C. Slices containing the NAc were stored at least 1 h at room temperature before being placed in the recording chamber and superfused (2 ml/min) with artificial cerebrospinal fluid that contained (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, 1.2 NaH2PO4, and 11 glucose, and was equilibrated with 95% O2/5% CO2. All experiments were done at room temperature. The superfusion medium contained picrotoxin (100 μM) to block γ-aminobutyric acid type A receptors. All drugs were added at the final concentration to the superfusion medium.
For field potential recordings, the recording pipette was filled with 3 M NaCl, and both the field excitatory postsynaptic potential (fEPSP) slope (calculated with a least-square method) and fEPSP amplitude were measured (graphs depict amplitudes). For patch–clamp experiments, cells were visualized by using an upright microscope with infrared illumination, and recordings were made with whole-cell electrodes containing the following (mM): CsGluconate 128, NaCl 20, MgCl21, EGTA 1, CaCl2 0.3, Mg-ATP 2, GTP 0.3, cAMP 0.2 buffered with Hepes 10, pH 7.3. Electrode resistance was 4 MΩ, acceptable access resistance was <20 MΩ, and the holding potential was −70 mV. A 5-mV depolarizing step was applied before each evoked excitatory postsynaptic current (eEPSC) to evaluate access resistance. Recordings where access resistance varied by more than 10% were discarded. An Axopatch-1D (Axon Instruments, Foster City, CA) was used to record the data, which were filtered at 1–2 kHz, digitized at 5 kHz on a DigiData 1200 interface (Axon), and collected on a personal computer by using ACQUIS-1 software (Biologic, Grenoble, France). To evoke synaptic currents, stimuli (100–150 μs duration) were delivered at 0.033 Hz through bipolar tungsten electrodes placed at the prefrontal cortex–accumbens border (9, 21). Recordings were made in the rostral–medial dorsal accumbens close to the anterior commissure.
Spontaneous EPSCs (sEPSCs).
sEPSCs were recorded by using Axoscope 1.1.1. sEPSC amplitude and interinterval time were measured by using AXOGRAPH 3.6 as previously described (9, 22).
Generation of CB1−/− Mice.
To construct the targeting vector, we subcloned into pUC19 a 9-kb KpnI-KpnI fragment from a 129SvJ6 mouse genomic library containing the entire coding region of the CB1 gene. We replaced an ApaI-ApaI genomic fragment with a neomycin resistance cassette derived from the plasmid pMC1NeoPolyA (Stratagene). The insertion replaced part of the upstream uncoding region down to the sixth transmembrane region of the receptor, thereby resulting in a nonfunctional receptor (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). The CB1 gene was targeted in embryonic stem cells by using a subclone of the embryonic day 14.1 (129/Ola) line, as previously described (23, 24). Briefly, embryonic stem cells were electroporated with a replacement type of target vector, and G418 (400 μg/ml) was added 24 h later. After 9–10 days, surviving clones were disaggregated and half of the colony screened by nested PCR with oligonucleotides specific for neomycin, CB1, and a reverse inner primer located externally to the short arm of the target vector. Embryonic stem cells were injected into blastocysts, and chimeric mice were generated by standard protocols. DNA of tail biopsies from F1 offspring and subsequent generations was analyzed by PCR (forward: 5-TTCATTGGGCTTGGCACGTTT-3, reverse: 5-TGATAAGCTTGTTCATCTTCCC-3) for the disrupted CB1 allele. CB1 mutant mice were backcrossed to C57BL/6, and heterozygous mice were intercrossed to obtain homozygous CB1-deficient mice.
Extraction of genomic DNA from frozen tail fragments taken from F1 offspring was performed by overnight incubation of the tail fragments in 100 μl of lysis buffer (100 mM Tris⋅HCl/10 mM EDTA/100 mM NaCl/0.2% SDS/0.5 mg/ml of proteinase K) at 56°C with agitation. After proteinase K inactivation (70°C, 10 min) and H2O addition, samples were centrifuged (16,000 × g, 10 min, 4°C). DNA extraction was verified by electrophoresis on 1% agarose gels and ethidium bromide coloration. Homologous recombinants were screened by PCR by using forward primers in the coding sequence: 5′-ATTGCTGTGTTGCCTCTCCT and in the neomycin resistance gene: 5′-CTCGTGCTTTACGGTATCGC and a common reverse oligonucleotide primer, located downstream from the 3′ end of the integration site in the exon: 5′-TCACGGTGGAGTTCAGCAG, giving amplicons of 442 bp for the wild-type allele and of 253 bp for the mutated allele. The two DNA fragments were amplified for 34 cycles of 20, 20, and 90 sec at 94, 56, and 68°C, respectively, by using the Taq polymerase “Expand TM High Fidelity PCR system” (Boehringer). Amplified fragments were visualized by electrophoresis on 2% agarose gels and ethidium bromide coloration.
Total RNA was prepared from homogenized brain by using Trizol (BRL). Random hexamer-primed reverse transcription (RT) was performed with Superscript-RT (BRL) by using 3 μg of total RNA in a 30-μl reaction volume containing 0.04 μl (1.2 ng) random hexamers (Pharmacia), 0.4 mM dNTP (Promega), 50 mM Tris⋅HCl, pH 8.3, 75 mM KCL, 3 mM MgCl2, and 0.5 units RNAsin (Promega). After 90 min of incubation at 37°C, samples were heated at 94°C and then quickly chilled on ice. Samples were diluted 1:10 with H2O to a concentration of 10 ng/μl of cDNA equivalents assuming a 1:1 ratio of reverse transcription. Samples were standardized for the expression of β2-microglobulin by competitive PCR using a mimic control vector essentially as described (25). Standardized cDNA aliquots (4 μl) were amplified by PCR for 35 cycles by using CB1 specific oligonucleotides (forward: 5′-TATAAGAGGATCGTCACCAG-3′ reverse: 5′-TGATAAGCTTGTTCATCTTCCC-3′).
Data Analysis and Materials
All values are given as mean ± SEM. Statistical analyses were done with the Mann–Whitney U test (P < 0.05 was taken as indicating statistical significance) or the Kolmogorov–Smirnof test (P < 0.001 was taken as indicating a statistical difference) by using statview (Abacus Concepts, Berkeley, CA). Drugs used: WIN 55,212,2, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphtalenylmethanone; AM-251, N-piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4methyl-1H-pyrazole-3-carboximide hydrochloride; AM-404, (all-Z)-N-(4-hydroxyphenyl)-5,8,11,14-eicosatetraamide; thapsigargin, 2-methyl-6-(phenylethynyl) pyridine; eGlu, (2S)-α-ethylglutamic acid; LY341495, 2-amino-2-(2-carboxycyclopropan-1-yl)-3-(dibenzopyran-4-yl) propanoic acid; D-AP5, d-amino-5-phosphonovaleric acid; and (S)-DHPG, (S)-3,5-dihydroxyphenylglycine from Tocris Neuramin (Bristol, U.K.); xestospongin C from Calbiochem (France); SCH23390, sulpiride, and ryanodine from RBI (France); BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate acetoxymethyl ester from Alexis (France); BAPTA, kynurenate, N-methyl-d-aspartate, and picrotoxin from Sigma (France); SR 141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride, was a gift from Sanofi (Paris).
Results
In vivo studies have shown the occurrence of low-frequency (up to 10–15 Hz) cell firing in the NAc (26, 27), and that long-term synaptic potentiation can be induced at the cortico-striatal synapses by applying 5-Hz discharge for several minutes (28). We decided to explore the effects of various low-frequency stimulations in a slice preparation containing prelimbic cortex–NAc synapses. It was found that a 10 min stimulation at 13 Hz of the prelimbic cortex afferences to the NAc reliably induced a robust long-term depression (LTD) of evoked excitatory synaptic transmission. Whole-cell recorded eEPSCs clearly expressed the 13-Hz LTD (Figs. 1A and 3B): on average eEPSC was 77.9 ± 13.1% of baseline 30 min after the end of the tetanus. The 13-Hz LTD was also observed measuring fEPSP measured 81.4 ± 3.6% of baseline 60 min after tetanus, n = 39, Fig. 1 B and C). Repetition of the low-frequency tetanus caused saturation of LTD that reached a maximum 40% depression (Fig. 1B).
Figure 1.

eCBs mediate LTD in the NAc. (A) Typical experiment where tetanic stimulation (13 Hz during 10 min) of prelimbic cortical afferents induced a LTD of eEPSC (see Fig. 2B for averaged data). (Inset) Superimposed traces (average of 10 consecutives eEPSCs) taken at time indicated on graph. (Bar = 20 msec, 200 pA.) (B) Typical experiment showing saturation of 13-Hz LTD (as observed in four other experiments). (C) LTD is eliminated in slices perfused with the CB1 antagonist SR141716A (1 μM) (fEPSP was 96.3 ± 3.4% of baseline at 60 min in the presence of antagonist; n = 11, P <0.05 vs. control without antagonist). (D) 13-Hz LTD is eliminated in CB1−/− mice, compared with their wild-type littermates. fEPSP 60 min after tetanus measured 104.2 ± 5.1% of baseline in CB1−/− and 82.7 ± 3.8% in CB1+/+, P < 0.05.
Figure 3.
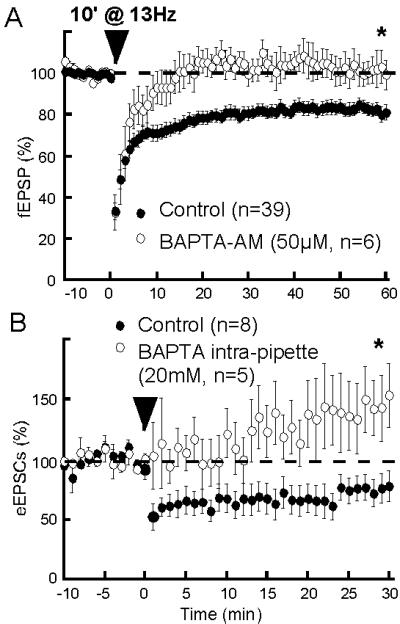
Postsynaptic intracellular Ca2+ rise is necessary to eCB-LTD. (A) The membrane-permeant Ca2+-chelator BAPTA-AM markedly reduced the fEPSP (45.5 ± 7.2% of baseline in BAPTA-AM, n = 4, not shown), suggesting that this concentration effectively buffered intracellular Ca2+. Preincubation of slices with BAPTA-AM (50 μM) prevented eCB-LTD: fEPSP was 99.6 ± 7.0% of baseline at 60 min, n = 6, P < 0.05 in BAPTA-AM-treated slices vs. control without BAPTA-AM. (B) In whole-cell experiment, filling the recording pipettes with BAPTA (20 mM) also suppressed eCB-LTD: eEPSC was 153.4 ± 26.6% of baseline at 30 min in the presence of intracellular BAPTA, n = 5, P < 0.05 vs. control without BAPTA.
Considering the presence of CB1 on glutamatergic afferents to the NAc (9), we reasoned that eCBs might be released by the 13-Hz tetanus and induce LTD. Thus, both pharmacological experiments and CB1-null mutant mice were used to test the role of eCBs in the 13-Hz LTD. First, the effects of selective CB1 antagonists were tested. The selective CB1 antagonist SR141716A (29) completely blocked 13-Hz-LTD induction (fEPSP: see Fig. 1C; eEPSC was 121.4 ± 23.2%, n = 4, of baseline 30 min after tetanus in SR141716A slices, not shown). Identical results were obtained with concentrations as low as 100 nM SR141716A (fEPSP was 100.9 ± 1.9% of baseline at 60 min, n = 6, P < 0.05 vs. control, not shown) and with another CB1 antagonist AM-251 (2 μM, fEPSP was 102.3 ± 8.9% of baseline at 60 min, n = 6, P < 0.05 vs. control, not shown). Noteworthy, SR141716A (1 μM) had no effect when applied once eCB-LTD was already induced (fEPSP was 79.2 ± 5.5% of baseline at 60 min, n = 7, P = 0.7 compared with control without SR141716A, not shown) suggesting that the late phase of 13-Hz LTD is not because of the continuous release of eCBs. Genetically altered mice lacking CB1 were used to unequivocally demonstrate the role of CB1 in 13-Hz LTD. Details on the methods for obtention and characterization of CB1−/− mice are published as supporting information on the PNAS web site (see also Methods). We first verified the absence of CB1-induced fEPSP inhibition in CB1−/− mice: 25 min after 10 μM WIN 55,212,2, fEPSP was 29 ± 9% of baseline in +/+ mice vs. 98 ± 9% in −/− mice (see supporting information). In addition to showing the absence of CB1-mediated inhibition in CB1−/− mice, this result indicates the specificity of high doses of WIN 55,212,2 in this preparation.
As suggested by the pharmacological experiments, we found that, in marked contrast with their wild-type littermates, CB1−/− mice did not display 13-Hz-induced LTD (Fig. 1D). These results confirm that 13-Hz LTD depends on eCB signaling and CB1.
Presynaptic CB1 inhibit glutamate release at the prelimbic cortex–NAc synapses (9), and we reasoned that if CB1 activation and eCB-LTD share common mechanisms to inhibit excitatory synaptic transmission, eCB-LTD must be occluded by exogenous cannabimimetic and endogenously released cannabinoids. Pretreatment with the CB1 agonist WIN 55,212,2 (300 nM) inhibited excitatory synaptic transmission and completely occluded eCB-LTD (Fig. 2A). Blockade of CB1 with SR141716A does not enhance basal fEPSPs in the NAc [not shown (9)], showing that CB1 are not tonically active and indicating that there either is very little basal eCB production or that eCB-transporters totally prevent eCB accumulation. Bath application of 10 μM AM-404, a selective inhibitor of the anandamide/2-arachidonoyl-glycerol transporter (30, 31), caused a small but significant reduction of the fEPSP that was blocked by preincubation with the CB1 antagonist (Fig. 2B Left). This result suggested that in basal conditions, the housekeeping functions of eCB-transporters prevent from the tonic activation of CB1 by circulating eCBs (11). Reminiscent of the effect of the CB1 agonist WIN 55,212,2, pretreatment with AM-404 partially occluded eCB-LTD (Fig. 2B Right, P = 0.06 compared with LTD in the absence of AM-404). Thus, eCB-LTD is occluded by exogenous cannabimimetics and naturally occurring eCBs suggesting common pathways for these CB1-mediated phenomena. On the basis of the previous observation of the presynaptic localization of CB1 at the prelimbic cortex–NAc synapses (9), we decided to verify that eCB-LTD was also expressed at a presynaptic locus. For this purpose, sEPSCs were recorded in control condition and 30 min after eCB-LTD induction. As shown in Fig. 2 C and D, induction of eCB-LTD markedly decreased sEPSC frequency up to 30 min after tetanus (P < 0.0001, Kolmogorov–Smirnof test). In contrast, the distribution of sEPSC amplitude was not affected (P = 0.012, Kolmogorov–Smirnof test). These results show that eCB-LTD is expressed at a presynaptic locus, in agreement with a role of presynaptic CB1.
Figure 2.

eCB-LTD is occluded by CB1 activation and is presynaptic. (A) Typical experiment (time course) and group data (histogram) where the CB1 agonist WIN 55,212,2 (300 nM) occluded eCB-LTD. In the presence of WIN 55,512,2 fEPSP measured 106.7 ± 8.8% of baseline, 60 min after LTD-induction; n = 6, P < 0.05 vs. control without WIN 55,212,2. (B) The eCB transporter blocker AM-404 inhibits fEPSP in a SR141716A-sensitive manner (fEPSP measured 88.4 ± 2.2% of baseline 15 min after AM-404 application, n = 7, and 97.9 ± 4.2% in the presence of SR141716A, n = 5, P < 0.05) and partially occludes eCB-LTD. In the presence of AM-404, fEPSP measured 94.5 ± 3.9% of baseline 60 min after tetanus, n = 7, P < 0.06 vs. control without AM-404. (C) Representative consecutive 1-sec current sweeps from a cell where sEPSCs were recorded in control and 30 min after tetanic stimulation. (Bar = 30 pA, 200 msec.) (D) sEPSC amplitude distribution was unchanged (n = 3, Kolmogorov–Smirnof test, P > 0.001). On the contrary, the distribution of the time intervals between successive sEPSCs in these neurons shows a reduction of sEPSC frequency after tetanus (Kolmogorov–Smirnof test P < 0.0001).
Postsynaptic Ca2+ elevation plays a key role in eCB signaling (32) and eCB-mediated synaptic plasticity in the hippocampus (11) and the cerebellum (10, 16). We decided to evaluate the Ca2+ requirements of eCB-LTD. Fig. 3 demonstrates the importance of the intracellular Ca2+ level increase to eCB-LTD: when slices were preincubated with the membrane permeant Ca2+-chelator BAPTA-AM (50 μM), eCB-LTD was completely prevented. Accordingly, eCB-LTD was eliminated by selectively buffering postsynaptic calcium with intrapipette BAPTA. As shown Fig. 3B, the 13-Hz protocol rather caused synaptic enhancement when BAPTA was perfused into the intracellular compartment.
What are the receptors triggering the release of eCB during the tetanus? Because both glutamatergic and dopaminergic afferents connect to NAc medium spiny neurons (33) and that in the dorsal striatum anandamide signaling is activated by dopamine (34), the involvement of glutamate and dopamine receptors was evaluated. Pretreatment with the nonsubtype-selective ionotropic glutamate receptors kynurenate (1 mM, 10 min before and during tetanus) completely blocked fEPSPs but had no effect on the induction of eCB-LTD, which was normal after kynurenate washout (Fig. 4A), suggesting the lack of involvement of postsynaptic ionotropic glutamate receptors. As summarized in Fig. 4A, similar negative results were obtained with the competitive N-methyl-d-aspartate receptor antagonist D-AP5, a mixture of the selective D1 antagonist SCH23390 and the D2 antagonists sulpiride and by selective blockade of metabotropic glutamate (mGlu2)/3 receptors with a low concentration of LY341495 or (2S)-α-ethylglutamic acid.
Figure 4.

eCB-LTD is mediated by mGlu5 activation and rise of Ca2+ from intracellular store. (A) Antagonists of ionotropic GluR (kynurenate), NMDAR (D-AP5, 50 μM), mGlu2/3 [200 nM 2-amino-2-(2-carboxycyclopropan-1-yl)-3-(dibenzopyran-4-yl) propanoic acid and 200 μM (2S),-α-ethylglutamic acid], and D1 and D2 receptors (SCH23390 and sulpiride, 30 μM) do not affect eCB-LTD: fEPSP measured 81.1 ± 8.8% (n = 6), 84.0 ± 3.5% (n = 5), 73.1 ± 5.0% (n = 3), 86.6 ± 7.9% (n = 8) and 79.5 ± 7.2% (n = 7) of baseline, at 60 min, P = 0.8, 0.2, 0.3, 0.9, and 0.4, respectively, vs. control in the absence of antagonists. (B) The mGlu antagonist LY354740 (50 μM) and the highly specific mGlu5 antagonist 2-methyl-6-(phenylethynyl) pyridine (10 μM) suppressed eCB-LTD: fEPSP measured 109.0 ± 5.2% (n = 6) and 102.1 ± 3.0% (n = 7) of baseline at 60 min, P < 0.05 vs. control without antagonist, respectively. (C) Activation of mGlu5 with (S)-DHPG induced a form of LTD that was absent in CB1−/− mice: 30 min after (S)-DHPG, fEPSP measured 101.9 ± 4.9% of baseline in CB1−/− (n = 4, white dots) and 86.7 ± 5.2% in CB1+/+ (n = 9, black dots), P < 0.05. (D) (S)-DHPG-induced-LTD is occluded after saturation of 13-Hz/eCB-LTD: at 30 min fEPSP measured 88.7 ± 2.1% (n = 13, black dots) and 100.2 ± 3.6% (n = 6, white dots) of baseline, respectively. (E) Blockade of mGlu5/eCB-LTD after depletion of Ca2+ intracellular pools with the Ca2+-ATPase inhibitor thapsigargin. (F) Role of ryanodine-sensitive intracellular Ca2+ stores in eCB-LTD. Ryanodine (20 μM) inhibited eCB-LTD: fEPSP measured 109.7 ± 12.6% (n = 7) of baseline at 60 min after ryanodine (20 μM, 1–2 h) P = 0.03 vs. control without inhibitor.
In the cerebellum, activation of postsynaptic mGlu1 causes retrograde endocannabinoid signaling and inhibition of presynaptic release (14). Medium spiny neurons express a subtype of mGlu receptors coupled to phospholipase C: mGlu5 (21, 35). mGlu5 is predominantly located in postsynaptic elements (35) and because our BAPTA experiments show that eCB-LTD requires postsynaptic Ca2+ elevation, we hypothesized that prolonged stimulation of cortical glutamatergic afferents could activate postsynaptic mGlu5, whose role might be to translate the glutamate signal into eCB signaling. Indeed, Fig. 4B shows that eCB-LTD is completely abolished by the broad-spectrum mGlu antagonist LY341495 and more importantly by the specific mGlu5 antagonist 2-methyl-6-(phenylethynyl) pyridine (MPEP) (36). In accord with a role of mGlu5, it was found that direct stimulation with the specific group 1 mGluR agonist (S)-DHPG (100 μM, 10 min) also causes LTD. Moreover, whereas direct pharmacological stimulation of mGlu5 with (S)-DHPG is sufficient to induce LTD in wild-type mice (Fig. 4C, black dots), the long-term effects of the specific mGlu5 agonist were absent in CB1−/− mice (Fig. 4C, white dots). Accordingly, the CB1 antagonist SR141716A (1 μM) eliminated mGlu5-LTD [30 min after induction of (S)-DHPG-LTD, fEPSP was 100.7 ± 4.2% of baseline, n = 6, not shown]. Occlusion experiments were performed and (S)-DHPG application did not cause LTD in slices where 13-Hz LTD was already induced (Fig. 4D). Together, these results show that stimulation of mGlu5 by both synaptically released glutamate or exogenous agonist is both necessary and sufficient to the expression of LTD at the prelimbic cortex–NAc synapses. mGlu receptors are coupled to the phospholipase C, phospholipase D, and both inositol 1,4,5-triphosphate (IP3)- and ryanodine-sensitive intracellular Ca2+ second messenger pathways (37, 38). In favor of the idea that mGlu5 translate the glutamate signal into eCB signaling via the release of Ca2+ from IP3-sensitive intracellular pools, we found that the Ca2+-ATPase inhibitor thapsigargin blocked eCB-LTD (Fig. 4E).
Because both IP3- and ryanodine-sensitive intracellular Ca2+ stores are depleted by thapsigargin, we next decided to test the effects of xestospongin C (a selective IP3-receptor channel blocker) and of ryanodine. Preincubation for at least 2 h with xestospongin C (2 μM) did not affect eCB-LTD (fEPSP was 86.0 ± 7.2% of baseline at 60 min, n = 6, P = 0.6 compared with control in the absence of xestospongin C, not shown). In contrast, preincubation with ryanodine (1–2 h, 20 μM) blocked eCB-LTD (Fig. 4F), confirming the involvement of intracellular Ca2+ stores in this phenomenon.
Discussion
Here we demonstrate a role of eCB signaling in activity-dependent long-term synaptic plasticity in the brain. We found that tetanic stimulation of prelimbic glutamatergic afferents induced a presynaptic LTD dependent on eCB and CB1. Induction of eCB-LTD required postsynaptic activation of mGlu5 and a rise in postsynaptic Ca2+ mediated by intracellular stores. The retrograde signaling cascade led to postsynaptic eCB release and activation of presynaptic CB1 receptors and LTD.
In addition to LTD in the NAc, eCB retrograde signaling also is important to another form of synaptic regulation in the cerebellum and the hippocampus (depolarization-induced suppression of inhibitory or excitatory transmission) (10–13, 16). In these structures, the brief depolarization of postsynaptic neurons caused eCB release and short-lived presynaptic inhibition: depolarization-induced suppression of inhibitory or excitatory transmission lasts from tens of seconds to 1 min. Thus, retrograde eCB signaling is implicated in both short- and long-term regulation of synaptic transmission. What can explain the difference in the overall duration of eCB-mediated retrograde inhibition? It is likely that the direct stimulation of a large number of prefrontal cortex fibers for 10 min released a larger amount of eCB and activated CB1 over a wider area than the single 1-sec depolarization of a single neuron required to cause depolarization-induced suppression of inhibitory or excitatory transmission. In favor of the idea that the concentration and the duration of the cannabinoid signaling play a key role in triggering eCB-LTD, we have previously reported (ref. 9; Fig. 2) that washout of the inhibitory effects of WIN 512,212,2 by the CB1 antagonist SR 141716A is marked by a reversible and a nonreversible component. The latter component is reminiscent of eCB-LTD, and this observation suggests that prolonged activation of presynaptic CB1 is sufficient to trigger LTD at the prefrontal cortex–NAc synapses. Finally, the signaling cascade responsible for eCB release might be a determinant in shaping the form of eCB-dependent plasticity. In the central nervous system, eCBs are produced by various mechanisms. Depolarization-induced suppression of transmitter release is triggered by a transient postsynaptic depolarization (10–13, 15, 16), and interactions with group I mGlu have recently been proposed (14, 39). In the dorsal striatum, anandamide production involves dopamine and D2 receptors (34), whereas in the cortex, 2-arachidonoyl-glycerol production depends on N-methyl-d-aspartate receptor activation (40). In the NAc, eCB-LTD is independent of these mechanisms. Our study brings to light an additional brain cannabinergic pathway and reveals a potentially important physiological function for mGlu5: translating postsynaptic glutamatergic signaling to presynaptic eCB signaling. Moreover, in agreement with the known dependency of eCB production to intracellular Ca2+ elevation (32) and coupling of group I mGlu to IP3- and ryanodine-sensitive Ca2+ stores (37, 38), our data indicate a role for the ryanodine-sensitive Ca2+ stores mGlu5-mediated eCB production. Thus, in contrast with studies describing modulatory roles of group I mGlu in depolarization-induced suppression of inhibitory transmission (14, 38) or no role of mGlu (ref. 11; see refs. 12, 16), our data strongly support the idea that postsynaptic mGlu5 and intracellular Ca2+ pools are necessary and sufficient to eCB-LTD. Experiments in other brain areas are necessary to determine whether eCB-mediated long-term plasticity always requires group I mGlu.
The fact that eCB-LTD was induced with low-frequency tetanus mimicking naturally occurring frequencies (26–28) is highly indicative of the existence of eCB-LTD in vivo. Retrograde eCB signaling could be engaged in the adaptive responses of many central glutamatergic synapses. Specifically in the NAc, eCB-LTD might be part of a negative feedback loop reducing glutamatergic synapses strength during sustained cortical activity. In all cases, the present findings add to the growing body of evidence pointing toward a major role of the eCB system in short- or long-term synaptic plasticity in brain areas important to the behavioral effects of cannabis use (e.g., the cerebellum, the hippocampus, and the NAc).
A current hypothesis is that addictive drugs participate in and alter normal brain function and cause the remodeling of synaptic functions. To understand how the exogenous cannabinoids affect brain function, one must find out how their endogenous counterparts (eCBs) participate in synaptic transmission and plasticity. In keeping with this idea, our report of a form of LTD induced by eCB and occluded by an exogenous cannabinoid gives insight into how cannabis derivatives such as marijuana alter normal eCB functions in the brain reward system.
Supplementary Material
Acknowledgments
Work in O.J.M.'s laboratory is supported by grants from Mission Interministérielle de Lutte contre la Drogue et la Toxicomanie, Ministère de la Recherche (A.C.I. “Jeunes Chercheurs”), Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, and SANOFI-Synthélabo. We thank Dr. G. Le Fur and David Shire at SANOFI-Synthélabo for the gift of CB1 knockout mice and Dr. P. Chavis for comments on the manuscript.
Abbreviations
- eCBs
endocannabinoids
- NAc
nucleus accumbens
- fEPSP
field excitatory post-synaptic potential
- EPSC
excitatory postsynaptic current
- sEPSC
spontaneous excitatory postsynaptic current
- eEPSC
evoked excitatory postsynaptic current
- LTD
long-term depression
- WIN 55,212,2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphtalenylmethanone
- (S)-DHPG
(S)-3,5-dihydroxyphenylglycine
- SR141716A
N-piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride
- BAPTA
1,2-bis(2-amino-phenoxy)ethane-N,N,N′,N′-tetraacetate
- BAPTA-AM
BAPTA-acetoxymethyl-ester
- mGlu
metabotropic glutamate receptor
- IP3
inositol 1,4,5-triphosphate
- AM-404
(all-Z),-N-(4-hydroxyphenyl)-5,8,11,14-eicosatetraemide
- LY341495
2-amino-2-(2-carboxycyclopropan-1-yl)-3-(dibenzopyran-4-yl) propanoic acid
- D-AP5
d-amino-5-phosphonovaleric acid
- mGlu
metabotropic glutamate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ameri A. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 2.Felder C C, Glass M. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman A F, Lupica C R. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan J M. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 5.Chan P K, Chan S C, Yung W H. NeuroReport. 1998;9:671–675. doi: 10.1097/00001756-199803090-00020. [DOI] [PubMed] [Google Scholar]
- 6.Levenes C, Daniel H, Soubrie P, Crepel F. J Physiol. 1998;510:867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auclair N, Otani S, Soubrie P, Crepel F. J Neurophysiol. 2000;83:3287–3293. doi: 10.1152/jn.2000.83.6.3287. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman A F, Lupica C R. J Neurophysiol. 2001;85:72–83. doi: 10.1152/jn.2001.85.1.72. [DOI] [PubMed] [Google Scholar]
- 9.Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni O J. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohno-Shosaku T, Maejima T, Kano M. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 11.Wilson R I, Nicoll R A. Nature (London) 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 12.Kreitzer A C, Regehr W G. J Neurosci. 2001;21:RC174. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diana M A, Levenes C, Mackie K, Marty A. J Neurosci. 2002;22:200–208. doi: 10.1523/JNEUROSCI.22-01-00200.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- 15.Wilson R I, Kunos G, Nicoll R A. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 16.Kreitzer A C, Regehr W G. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 17.Hyman S E. Science. 1996;273:611–612. doi: 10.1126/science.273.5275.611. [DOI] [PubMed] [Google Scholar]
- 18.Koob G F. Neuron. 1996;16:893–896. doi: 10.1016/s0896-6273(00)80109-9. [DOI] [PubMed] [Google Scholar]
- 19.Gessa G L, Melis M, Muntoni A L, Diana M. Eur J Pharmacol. 1998;341:39–44. doi: 10.1016/s0014-2999(97)01442-8. [DOI] [PubMed] [Google Scholar]
- 20.Tanda G, Pontieri F E, Di Chiara G. Science. 1997;276:2048–2050. doi: 10.1126/science.276.5321.2048. [DOI] [PubMed] [Google Scholar]
- 21.Manzoni O, Michel J M, Bockaert J. Eur J Neurosci. 1997;9:1514–1523. doi: 10.1111/j.1460-9568.1997.tb01506.x. [DOI] [PubMed] [Google Scholar]
- 22.Manzoni O J, Williams J T. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopf M. In: Immunology Methods Manual: The Comprehensive Sourcebook of Techniques. Lefkovits I, editor. San Diego: Academic; 1996. pp. 199–216. [Google Scholar]
- 24.Nitschke L, Kopf M, Lamers M C. Biotechniques. 1993;14:914–916. [PubMed] [Google Scholar]
- 25.Kopf M, Brombacher F, Kohler G, Kienzle G, Widmann K H, Lefrang K, Humborg C, Ledermann B, Solbach W. J Exp Med. 1996;184:1127–1136. doi: 10.1084/jem.184.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang J Y, Janak P H, Woodward D J. Neuroscience. 2000;99:433–443. doi: 10.1016/s0306-4522(00)00218-9. [DOI] [PubMed] [Google Scholar]
- 27.Carelli R M, Ijames S G. Brain Res. 2000;866:44–54. doi: 10.1016/s0006-8993(00)02217-4. [DOI] [PubMed] [Google Scholar]
- 28.Charpier S, Mahon S, Deniau J M. Neuroscience. 1999;91:1209–1222. doi: 10.1016/s0306-4522(98)00719-2. [DOI] [PubMed] [Google Scholar]
- 29.Rinaldi-Carmona M, Barth F, Heaulme M, Alonso R, Shire D, Congy C, Soubrie P, Breliere J C, Le Fur G. Life Sci. 1995;56:1941–1947. doi: 10.1016/0024-3205(95)00174-5. [DOI] [PubMed] [Google Scholar]
- 30.Beltramo M, Piomelli D. NeuroReport. 2000;11:1231–1235. doi: 10.1097/00001756-200004270-00018. [DOI] [PubMed] [Google Scholar]
- 31.Beltramo M, Stella N, Calignano A, Lin S Y, Makriyannis A, Piomelli D. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 32.Mechoulam R, Fride E, Di Marzo V. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 33.Pennartz C M, Groenewegen H J, Lopes da Silva F H. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 34.Giuffrida A, Parsons L H, Kerr T M, Rodriguez de Fonseca F, Navarro M, Piomelli D. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- 35.Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- 36.Pin J P, De Colle C, Bessis A S, Acher F. Eur J Pharmacol. 1999;375:277–294. doi: 10.1016/s0014-2999(99)00258-7. [DOI] [PubMed] [Google Scholar]
- 37.Conn P J, Pin J P. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 38.Fagni L, Chavis P, Ango F, Bockaert J. Trends Neurosci. 2000;23:80–88. doi: 10.1016/s0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- 39.Varma N, Carlson G C, Ledent C, Alger B E. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stella N, Schweitzer P, Piomelli D. Nature (London) 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.