

Abstract
G protein-coupled inward rectifiers (GIRKs) are activated directly by G protein βγ subunits, whereas classical inward rectifiers (IRKs) are constitutively active. We found that a glutamate residue of GIRK2 (E315), located on a hydrophobic domain of the C terminus, is crucial for the channel activation. This glutamate (or aspartate) residue is conserved in all members of the Kir family. Substitution of alanine for the glutamate on GIRK1, GIRK2, and IRK2, expressed in HEK293 cells, greatly reduced the whole-cell currents. The whole-cell current of GIRK channels with a constitutively active gate, GIRK2(V188A), [Yi, B. A., Lin, Y. F., Jan, Y. N. & Jan, L. Y. (2001) Neuron 29, 657–667] was also reduced by the same glutamate mutation. Mean open time and conductance of single channels in GIRK2 and IRK2 were not affected by the mutation, indicating that the reduced whole-cell current resulted from a lowered probability of channel activation. The mutated GIRK and IRK showed normal trafficking to the cell membrane. The mutated GIRK2 retained the ability to interact with G protein βγ subunits, and it showed almost the same inwardly rectifying property as the wild type. The mutated GIRK1 and GIRK2 retained ion selectivity to K+ ions. This glutamate residue corresponds to one of the residues causing Andersen's syndrome [Plaster, N. M., Tawil, R., Tristani-Firouzi, M., Canun, S., Bendahhou, S., Tsunoda, A., Donaldson, M. R., Iannaccone, S. T., Brunt, E., Barohn, R., et al. (2001) Cell 105, 511–519]. Our interpretation is that this region of the glutamate residue is crucial in relaying the activating message from the ligand sensor region to the gate.
Inward rectifier K+ (Kir) channels are a family of K+ channels with a simple molecular structure. Dysfunctions of Kir channels are linked to human diseases such as persistent hyperinsulinemic hypoglycemia of infancy (1), Bartter's syndrome (2), and Andersen's syndrome (3). The Kir family includes several subfamilies, such as classical inward rectifier K+ channels (IRKs) (4) and G protein-coupled inward rectifier K+ channels (GIRKs) (5, 6). IRKs are constitutively active, and they function to maintain the resting potential and conserve cellular K+ ions. By contrast, the activity of GIRK channels is regulated by transmitters and hormones. This regulation is transduced by G protein βγ subunits (Gβγ), which, by interacting with the N and C termini of GIRK, enhance GIRK activity (7, 8). The interaction of Gβγ with GIRK causes openings of the channel gate located in the transmembrane helices (M1 and M2) (9, 10).
Given this sequence of activation, an important problem is to determine how the interaction between GIRK and Gβγ translates into the openings of the channel gate. Here, we report that a mutation of a glutamate residue located on the C terminus of Kirs greatly reduces the whole-cell currents generated by both GIRKs and an IRK. This glutamate residue is located on a conserved hydrophobic region. This current reduction is caused by a decrease of the opening probability of the channel without altering other attributes of the K+ channels. Our results suggest that this region of the C terminus of Kir is one of the determinants for controlling gate opening for both GIRKs and IRKs. Coincidentally, a mutation of the corresponding glutamate residue in IRK1 has recently been reported to be one of the causes for Andersen's syndrome (3).
Materials and Methods
Molecular Biology.
cDNAs for GIRK1, GIRK2, and IRK2 were isolated from a rat brain cDNA library by PCR or by colony hybridization (11). Point mutations were introduced into the cDNAs by using the QuickChange Site-directed Mutagenesis kit (Stratagene). To test the membrane expression of Kirs, the influenza virus hemagglutinin (HA) epitope was inserted into the extracellular loops (12, 13) between I126 and E127 of GIRK2, and between A115 and E116 of IRK2 by using an ExSite PCR-based site-directed mutagenesis kit (Stratagene). cDNA for HA-tagged Kir6.2 was kindly supplied by Lily Jan and Blanche Schwappach. For immunoprecipitation experiments, N-terminal-tagged HA-GIRK1 and Myc-GIRK2 were constructed with the PCR method. Plasmids of Gβ1 and Gγ2 were constructed as described before (14). All PCR-manipulated DNAs were verified by sequencing.
Cell Culture and Transfection.
Human embryonic kidney (HEK) 293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. One day before transfection, HEK 293 cells were cultured in 3.5-cm dishes coated with rat tail collagen (Boehringer Mannheim). Transfection was carried out with TransIT-LT1 reagent (PanVera, Madison, WI). For whole-cell recordings, 0.5 μg per dish of each kind of cDNA was used for transfection. Green fluorescent protein (GFP) plasmid (0.1 μg) was cotransfected to visualize the transfected cells. For single-channel recordings of the wild-type (wt) GIRK and IRK channels, the amount of each kind of cDNA was reduced to 0.04 μg; otherwise, records contained the activity of too many channels. For single-channel recording of the mutated GIRK2, the amount of each kind of cDNA was increased to 0.06 μg per dish. For the mutated IRK2 the amount of cDNA was increased to 0.2–0.8 μg.
Electrophysiolology.
Electrophysiological experiments were performed between 24 and 36 h after transfection. Some of the single-channel recordings of mutated IRK2 channels were performed 48 h after transfection. All recordings were made on isolated cells that demonstrated GFP fluorescence. For whole-cell recordings, the bath solution was composed of 146 mM sodium gluconate, 10 mM potassium gluconate, 2.4 mM CaCl2, 1.3 mM MgCl2, 5 mM Hepes–NaOH, and 0.5 μM tetrodotoxin (pH 7.4). The pipette solution contained 151 mM potassium gluconate, 5 mM Hepes–KOH, 0.1 mM CaCl2, 0.5 mM EGTA–KOH, 4 mM MgCl2, 3 mM Na2ATP, and 0.2 mM GDP (pH 7.2).
For inside-out single-channel recordings, the bath solution contained 141 mM potassium gluconate, 8.7 mM NaCl, 5 mM Hepes–KOH, 5 mM EGTA–KOH, 1 mM MgCl2, 2 mM Na2ATP, and 0.1 mM GDP (pH 7.2). The pipette solution contained 156 mM KCl, 2.4 mM CaCl2, 1.3 mM MgCl2, 5 mM Hepes–NaOH, and 0.5 μM tetrodotoxin (pH 7.4). Recordings were made with a frequency response of 2 kHz and were analyzed at a sampling frequency of 20 kHz. Recordings were performed at room temperature, and statistical results are expressed as mean ± SEM.
Immunocytochemistry.
HEK 293 cells grown on Aclar plastics coated with rat-tail collagen were transfected with HA-tagged channel cDNAs by using TransIT-LT1 reagent. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde for 20 min, blocked in either 5% donkey serum in PBS (nonpermeabilized cells) or 5% donkey serum with 0.1% Triton X-100 in PBS (permeabilized cells) for 45 min, and were then labeled with rat monoclonal anti-HA antibody (3F-10, Roche, 0.75 μg/ml) for 2 h. After washing thoroughly, cells were incubated with Cy-3-conjugated donkey anti-rat IgG secondary antibody (Jackson ImmunoResearch, 1:500) for 1 h. Extensive rinsing was carried out between each step.
Confocal Image Analysis.
Immunofluorescence images of Cy-3-stained cells were acquired with a Zeiss 40×, 0.8 numerical aperture water-immersion objective on an Axioskop 2FS with a Prairie Technologies laser scanning Confocal microscope. Twelve-bit gray-scale tiff images were analyzed by using Scion IMAGE BETA 4.02 (based on NIH image for Macintosh; Scion, Frederick, MD). Because the thickness of the cells was less than 5 μm after fixation, a pinhole of 150 μm was chosen to produce an optical section of 8.5 μm, so that fluorescence along the optical axis including both the upper and lower plasmalemma as well as the cell interior would be included in an optical volume.
Immunocoprecipitation.
Transfected cells on 10-cm dishes were harvested and solubilized in 550 μl of lysis buffer (20 mM Hepes–NaOH, pH 7.5/150 mM NaCl/5 mM MgCl2/1 mM EDTA/1 mM EGTA/1 mM Na3VO4/10 mM NaF/25 mM β-glycerophosphate/0.5% Triton X-100/10 μg/ml pepstatin A/10 μg/ml leupeptin/2 μg/ml aprotinin) on ice for 20 min. The samples were centrifuged at 15,000 × g for 20 min at 4°C, and the supernatants were subjected to immunoprecipitation. The cell lysates (150 μl) were incubated for 1 h at 4°C with antibody and staphylococcal protein A-Sepharose. Immune complexes with protein A-Sepharose were washed three times with a washing buffer (20 mM Hepes–NaOH, pH 7.5/150 mM NaCl/5 mM MgCl2/0.5% Triton X-100). Immunoprecipitated proteins were boiled in sample buffer (50 mM Tris⋅HCl, pH 6.8/1% SDS/1% 2-mercaptoethanol/0.1% bromophenol blue/8% glycerol) and resolved by SDS/PAGE. Rabbit polyclonal anti-Gβ antibody (T-20) was purchased from Santa Cruz Biotechnology. Mouse monoclonal antibody 9E10 against c-Myc epitope was from Berkeley Antibody (Richmond, CA).
Results
Mutation of GIRK2 E315 to Alanine and the Corresponding Mutations of GIRK1 and IRK2 Reduce Inward Rectifier K+ Current.
We fortuitously encountered a triple mutant of GIRK2 (R216Q, K247Q, E315G) caused by PCR errors. Interestingly, unlike the wild-type (wt) GIRK2, this mutant was incapable of generating large inwardly rectifying currents when expressed together with wt GIRK1 in HEK 293 cells. A further study indicated that substitution of glycine for glutamate at 315 on GIRK2 was responsible for this current reduction. In fact, either glycine substitution or alanine substitution produced greatly reduced currents. The glutamate residue in question (E315 of GIRK2) is well conserved among all Kirs. The residue is located on a C-terminal hydrophobic region that was designated as C4 region by Taglialatela et al. (15) (Fig. 1A). When cells were transfected with GIRK2(wt)/GIRK1(wt), we observed large inward rectifying currents (Fig. 1 B, D, and E). These currents would approximately correspond to transmitter-activated currents, because cells were also transfected with Gβ1 and Gγ2, which activated GIRK channels almost maximally. Previously, we observed (11) that the current caused by the basal activity of GIRK2(wt)/GIRK1(wt) (without expression of exogenous Gβ1γ2) is only about 1/5 of the current amplitude in the presence of the exogenous Gβ1γ2 (11).
Figure 1.

Alanine substitution for E315 of GIRK2 and for the corresponding residues of GIRK1 and IRK2 reduced the whole-cell currents. (A) Alignment of the C-terminal sequence of GIRK1, GIRK2, and IRK2 around the glutamate residue in question. (B) Samples of current traces produced by voltage steps from cells transfected with wild-type GIRK2 or mutated GIRK2, together with GIRK1(wt), Gβ1, and Gγ2. Voltage pulses were applied from −127 mV to −27 mV at 10-mV increments. Holding potential was −77 mV. (C) Sample records from cells transfected with IRK2(wt) and IRK2(E304A). (D) Whole-cell currents (at −117 mV) records from cells transfected with GIRK2(wt) or GIRK2(E315A), both together with GIRK1(wt), Gβ1, and Gγ2. (E) Cells transfected with GIRK1(wt) or GIRK1(E304A), together with GIRK2(wt), Gβ1, and Gγ2. (F) Cells transfected with IRK2(wt) or IRK2(E304A). The * represents P < 0.05, and ** represents P < 0.001 (t test). The rectification factor (see text) in GIRK2 experiment (D) was evaluated as follows: In separate experiments we evaluated endogenous currents, and this endogenous conductance was subtracted from the I–V relationship of each record to obtain the “net” current ascribed to transfection of the Kir channels. In 2 of 10 mutated cells, this rectification factor could not be determined, because the recorded current in mutated GIRK2 was smaller than the average endogenous current.
Substitutions of alanine for the glutamate in GIRK2(E315A), GIRK1(E304A), and IRK2 (E304A) all resulted in much smaller inwardly rectifying currents (Fig. 1 B–F). [GIRK channels are heterotetramers (16–18); therefore, when a mutant GIRK1 is tested, GIRK2(wt) was also transfected.] Compared with the large inward rectifying currents generated by GIRK2(wt)/GIRK1(wt) channels, currents generated by the mutants, GIRK2(E315A)/GIRK1(wt) and GIRK1(E304A)/GIRK2(wt), were reduced to 12% and 6%, respectively (Fig. 1 D and E). IRK2(E304A) produced a small current, only 2% of the IRK2(wt) (Fig. 1 C and F).
The Mutant GIRK2 (E315A)/GIRK1(wt) Shows Inward Rectification and Is Selectively Permeable to K+ Ions.
In some cells that expressed the mutant Kirs, we saw a clear indication of inward rectification (Fig. 1 B and C). However, this was not the case for all cells. We, therefore, evaluated the inwardly rectifying properties quantitatively. The data showed that the rectification factor [the ratio between the depolarizing conductance (from −67 to −57 mV) and the hyperpolarizing conductance (from −117 to −67 mV)] was 0.162 ± 0.028 (n = 8) for wt GIRK2 and 0.208 ± 0.059 for the mutant GIRK2 (P ≈ 0.5) (see Fig. 1 legend). The result suggests that the mutation did not significantly alter the rectification of the GIRK2(E315A)/GIRK1(wt) channel. In the GIRK1 and the IRK2 mutants, the current amplitudes approached the amplitude of the endogenous current (see Fig. 1 legend) and were too small to make quantitative evaluations.
Another question was whether the mutations altered the ion selectivity of Kir channels. In these experiments we used 20 mM K+ Krebs buffer for the external solution. In the mutant GIRK2(E315A)/GIRK1(wt), the inward current with the 20 mM [K]o external solution at −116 mV was 32.5 ± 10.7 pA/pF (n = 8), whereas in K-free solution (K replaced by Na), the current was only 1.8 ± 0.8 pA/pF (n = 6) (these values include currents endogenous to HEK293 cells). In the GIRK1 mutant [GIRK1(E304A)/GIRK2(wt)], the inward current with 20 mM [K]o external solution at −116 mV was 27.0 ± 8.1 pA/pF (n = 6), and in K-free solution the current was reduced to 2.9 ± 0.6 pA/pF (n = 7). Thus, the inward current with the K-free external solution was negligibly small compared with that in elevated [K]o, indicating that the K+ selectivity is maintained in the mutant GIRK1 and mutant GIRK2.
Single-Channel Properties.
We explored the possible mechanism of the current reduction by recording single-channel properties (Fig. 2). The data, summarized in Table 1, indicate that the mutant GIRK2(E315A)/GIRK1(wt) shows practically same open duration and elementary conductance as those of the wt counterpart.
Figure 2.
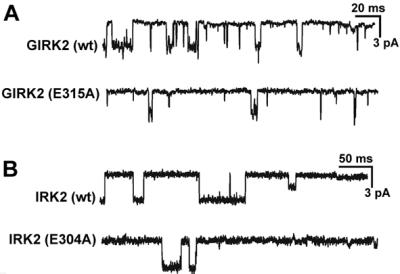
(A) Samples of single-channel records from cells transfected with GIRK2(wt) or GIRK2(E315A). Both cells were transfected together with Gβ1, Gγ2, and GIRK1(wt). (B) Samples of single-channel activity recorded from cells transfected with IRK2(wt) or IRK2(E304A). Potential was −100 mV.
Table 1.
Single-channel properties
| Channel | af | τf, ms | τs, ms | To, ms | γ, pS | n |
|---|---|---|---|---|---|---|
| G2(wt)/G1(wt) | 0.49 ± 0.04 | 0.21 ± 0.02 | 2.0 ± 0.71 | 1.00 ± 0.21 | 30.5 ± 0.63 | 13 |
| G2(E315A)/G1(wt) | 0.43 ± 0.03 | 0.26 ± 0.04 | 1.4 ± 0.37 | 0.95 ± 0.27 | 29.6 ± 1.1 | 5 |
| G2(V188A)/G1(wt) | 0.41 ± 0.05 | 0.22 ± 0.01 | 1.5 ± 0.14 | 1.02 ± 0.12 | 29.9 ± 1.1 | 8 |
| G2(V188A, E315A)/G1(wt) | 0.45 ± 0.07 | 0.23 ± 0.03 | 2.2 ± 0.42 | 1.28 ± 0.27 | 29.9 ± 1.3 | 6 |
| IRK2(wt) | 0.26 ± 0.04 | 1.43 ± 0.18 | 43.7 ± 5.4 | 32.8 ± 4.2 | 31.6 ± 0.71 | 6 |
| IRK2(E304A) | 0.38 ± 0.06 | 1.40 ± 0.28 | 32.4 ± 5.4 | 21.0 ± 4.3 | 29.9 ± 1.83 | 5 |
G2 = GIRK2; G1 = GIRK1. The number of the control patches [G2(wt)/G1(wt)] was 6 for the G2(E315A)/G1(wt) experiment and 7 for the G2(V188A)/G1(wt) experiment. The open-time histogram was fit by the sum of two exponentials. af is the relative area of the fast component. τf and τs are, respectively, the fast and slow time constants of the double exponentials. To = mean open time; namely, af × τf + (1 − af) × τs. γ is the chord conductance. Holding potential = −100 mV. [K]o = 156 mM. The following data on Npo were derived from all analyzable single-channel records, including those with no channel activity and with very vigorous channel activity. Npo for the control [G2(wt)/G1(wt)] during the experiment of G2(E315A)/G1(wt) was 0.66 ± 0.37 (n = 9), and Npo for G2(E315A)/G1(wt) was <0.13 ± 0.088 (n = 18). Npo for IRK2(wt) was 0.64 ± 0.24 (n = 11), and Npo for IRK2(E304A) was <0.021 ± 0.0093 (n = 36). The less than sign (<) indicates that we did not account for the difference in the amount of cDNAs between the mutant experiment and the control experiment.
However, the frequency of the channel activation in the mutant was low even with a 50% increase of both cDNAs used for transfection. The mean value of the number of channels multiplied by the open probability (Npo) of the mutant [GIRK2(E315A)/GIRK1(wt)] was less than ≈20% of the wt (Table 1 legend). Similarly, for IRK2 channels the mutant and the wt showed essentially the same mean open time and the unitary conductance. Again the chance of obtaining active channels in the mutant was very low despite the fact that we increased the amount of cDNAs. The Npo for the mutant IRK2 was less than ≈3% of that for IRK2(wt) (Table 1, legend).
Interestingly, IRK2 channels, as well as GIRK2/GIRK1 channels (not shown), sometimes show small conductance channels (Fig. 2B), as described by Picones et al. (19).
Mutation on the M2 Segment (V188A) Does Not Affect the Influence of the C-Terminus Mutant, E315A, on Channel Activity.
A valine residue located within the M2 segment (V188 in GIRK2) plays an important role in channel gating. When this residue is replaced with almost any amino acid except for a nonpolar one, GIRK channels become constitutively active (9). We used this property to investigate possible relations of the C4 glutamate residue (E315) and the M2 segment. Cells transfected with GIRK2(wt)/GIRK1(wt) (without Gβγ) showed a small inward rectifier K+ current, which probably represents GIRK activation by endogenous Gβγ (Fig. 3A). Cells transfected with the mutant GIRK2(V188A)/GIRK1(wt) showed a large increase in current as compared with the wt. The double mutant—an additional mutation E315A to the constitutively active GIRK2(V188A)—resulted in drastic current reduction (Fig. 3A).
Figure 3.

(A) Mutation of the glutamate residue reduces the currents by constitutively active GIRK2(V188A). Without exogenous Gβ1 and Gγ2, current generated by GIRK2(wt)/GIRK1(wt) was small. Current generated by the constitutively active GIRK2(V188A)/GIRK1, in the absence of Gβ1 and Gγ2, was substantial. Double mutant GIRK2(V188A, E315A)/GIRK1(wt) resulted in a drastic reduction of the current. The * represents P < 0.05 in reference to GIRK2(wt), and † means P < 0.05 in reference to GIRK2(V188A) (nonparametric ANOVA with the Bonferroni test). (B) Single-channel recordings. Holding potential was −100 mV.
The single-channel properties of these mutants were studied. To increase the chance of observing single-channel activity from the double mutant GIRK2(V188A, E315A), cDNAs for Gβ1 and Gγ2 were cotransfected, and the amount of cDNAs was increased. The data showed that single-channel properties of the channels formed by GIRK2(V188A) or by GIRK2(V188A, E315A) were almost the same as those of the GIRK2(wt) channels (Table 1).
The Glutamate Mutation Does Not Affect the Channel Trafficking.
Membrane expression of Kir channels depends partly on structural elements of the C terminus (12). We therefore examined the possibility that the glutamate residue might be important for channel subunit trafficking to the surface membrane. To test this possibility, we inserted an HA epitope into the extracellular loop of the GIRK2 and IRK2 (12, 13). Cells were stained, 24 h after the transfection, by using anti-HA antibody. When we applied the primary antibody after permeabilization, most of the staining was located in a perinuclear compartment (Fig. 4A). When the primary antibody was applied without permeabilization, strong staining was mostly confined to the cellular contour without staining of intracellular organelles (Fig. 4A). The same pattern was observed with the expression of all four HA-tagged wt and mutant GIRK2 and IRK2 tested. As a control (Fig. 4A), cells were transfected with the cDNA for Kir6.2 without the sulfonylurea receptor. Without permeabilization no staining on the cell surface was observed, consistent with the known failure of Kir6.2 (without the sulfonylurea receptor) to be inserted into the plasmalemma (13).
Figure 4.

Mutation of the glutamate residue did not affect the expression level on the cell membrane. The expression was measured by confocal microscopy using Kirs in which the HA epitope was inserted into the extracellular loops (Materials and Methods). Cells transfected with HA-tagged Kir6.2 cDNA without sulfonylurea receptor were used as a control. (A) (Upper) Cells stained with anti-HA antibodies after permeabilization. (Lower) Cells stained with anti-HA antibodies without permeabilization. (B) Degree of membrane expression was quantified by fluorescence intensity (a.u., arbitrary unit) from single nonpermeabilized cells by measuring the mean intensity per voxel located within the cell contour (the contour was determined by Nomarski optics). The fluorescence intensity is linear up to 4,095 a.u.
To quantify the level of surface expression, the images of single nonpermeabilized cells were analyzed by measuring the mean fluorescence intensity within the cell contour. Only cells isolated completely from others were used. No statistically significant differences were found in the expression level between the wt and the mutant in either GIRK2 or IRK2 subunit, indicating that the mutation does not interfere with the subunits trafficking to the cell membrane (Fig. 4B).
Mutant GIRK2(E315A) Interacts with Gβγ.
Immunoprecipitation was performed to test whether GIRK2(E315A) interacts with Gβγ. Myc-GIRK2, which has Myc epitope at the N terminus of GIRK2, with or without E315A mutation, was coexpressed with Gβ1 and FLAG-Gγ2 in HEK 293 cells. Aliquots of the cell lysates were immunoblotted with anti-Myc and anti-Gβ antibodies. Expression levels of the wt and of the E315A mutant of Myc-GIRK2 were similar (Fig. 5 Left). Endogenous Gβ of HEK 293 cells was detected (left lane). Other aliquots were used for immunoprecipitation with anti-Myc antibody. As shown in Fig. 5 Right, Gβ1 coprecipitated with Myc-GIRK2(E315A) as well as Myc-GIRK2(wt), suggesting that reduction of GIRK currents by the E315A mutation was not caused by impairment of Gβγ binding to GIRK2.
Figure 5.

Both GIRK2(wt) and GIRK2(E315A) coimmunoprecipitate with Gβγ. Myc-GIRK2(wt) (WT) or Myc-GIRK2(E315A) (EA) was coexpressed with Gβ1 and FLAG-Gγ2 in HEK 293 cells. Aliquots of the cell lysates were used for immunoprecipitation (IP) with anti-Myc antibody and immunoblotted (IB) with anti-Myc and anti-Gβ antibodies. Other aliquots were immunoblotted with the same antibodies (Left).
The Mutant GIRK2 Interacts with GIRK1.
We performed a competition assay to test whether the glutamate mutant of GIRK2 can assemble with GIRK1 (Fig. 6). To avoid the interference from the Gβγ, we used the constitutively active mutant GIRK2(V188A). Cotransfection of GIRK2(V188A) and GIRK1(wt) formed functional channels as shown by the large inward currents produced (left bar). Cotransfection of GIRK1(wt) with the same amount of double mutant GIRK2(V188A, E315A), and GIRK2(V188A) cDNAs (middle bar) considerably reduced the whole-cell currents compared with those generated by GIRK2(V188A)/GIRK1(wt). One of the explanations for this result is that the double mutant GIRK2(V188A, E315A) competes with GIRK2(V188A) for GIRK1(wt) with the result that less GIRK1(wt) is available for GIRK2(V188A) to form functional channels. The result agrees with the idea that the ability of the mutant GIRK2(E315A) to assemble with GIRK1(wt) remains intact.
Figure 6.
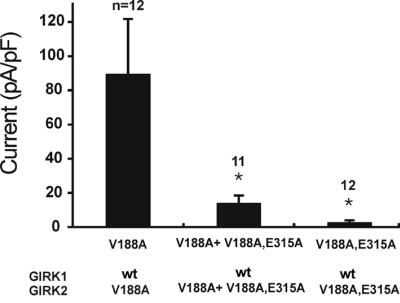
GIRK2(E315A) can assemble with GIRK1. Double mutant GIRK2(V188A, E315A) was cotransfected together with GIRK2(V188A) to test whether the double mutant GIRK2(V188A, E315A) competes with GIRK2(V188A) for GIRK1. The current produced by GIRK2(V188A)/GIRK1 was large (left bar). The current produced by GIRK2(V188A, E315A)/GIRK1 was very small (right bar). Transfection of GIRK2(V188A) plus GIRK2(V188A, E315A) together with GIRK1 produced an intermediate size of current, suggesting that GIRK2(V188A) and GIRK2(V188A, E315A) compete with each other for GIRK1. *, P < 0.05 in reference to V188A (nonparametric ANOVA with the Bonferroni post test).
One could think of an alternative explanation for the results of Figs. 1, 3, 6. Suppose that the C-terminus mutation renders GIRK2 more prone to form homomultimers than heterotetramers. If homomultimers are less efficient in producing channel activity, the result of Figs. 1 and 3 could be explained. This alternative explanation does not agree with the constancy of the single-channel properties among various mutants (Table 1). Homomultimers of GIRK2 produce short-duration “spiky” channels (16–18). Table 1 indicates the proportion of the short-duration component (af) was not altered by any of the C-terminus mutations of GIRKs. A similar argument can be made for the result of Fig. 6.
Furthermore, the IRK2 mutation also showed a reduced whole-cell current, while the single-channel property was unaltered. In this case, we can conclude that the channel activity of the mutant IRK2 homomultimers is suppressed because of the C-terminus mutation, independent of the effect of homo- vs. heteromultimer formation. Taking together the data in Figs. 1, 3, and 6 and Table 1, and the IRK2 result, the simplest explanation is that the mutant GIRK can interact and form a heteromultimer with GIRK1 as readily as the wt GIRK does, and that the channel activity of the heteromultimers of GIRK2 and GIRK1(wt) is drastically reduced if the C-terminal glutamate residue is replaced by alanine.
Discussion
The Role of the C4 Region.
The present experiments have demonstrated that alanine substitution for the glutamate residue on the C4 region of the C terminus of GIRK2, GIRK1, and IRK2 causes a sharp decrease in the whole-cell currents. This glutamate (or aspartate) residue is conserved among all Kir channels, suggesting that corresponding mutations could yield a similar phenotype in all Kir channels. Furthermore, the C4 region as a whole is highly conserved among all Kir channels. We have preliminary results showing that other amino acid residues in this region play a role similar to the glutamate residue, suggesting that maintaining a correct conformation of this region is crucial for normal Kir channel functions. Interestingly, the region corresponding to the C4 in Kir6.2 is one of several places that interact with the N terminus (20).
The mechanism underlying the whole-cell current reduction in the mutated channels is a reduction of opening probability of the channels. The open duration and elementary conductance of single channels were unaltered in the mutants. The number of expressed channels was also unaffected by the mutations. Therefore, the whole-cell currents would be in direct proportion to the open probability of the single channel. Further, single-channel recordings showed that the overall average of Npo was much reduced in the mutant channel as compared with the wt for both GIRK1/2 and IRK2 experiments (Table 1 legend).
The relationship between the glutamate residue and the GIRK channel gate was further elucidated by the introduction of a V188A mutation to GIRK2. The pore-facing V188 located on the M2 segment is considered to be one of the component residues of the mechanical gate of the GIRK channel (9). Mutation of the valine tends to lock the channel in the open conformation (9). The present study shows that the introduction of the additional mutation, E315A, to the GIRK2(V188A) molecule again resulted in a drastic decrease in GIRK currents, indicating that the glutamate residue has essentially the same effect on the modified gate. The single-channel property of the channels formed by GIRK2(V188A/E315A)/GIRK1 remains the same as that of the channels formed by GIRK2(V188A)/GIRK1, indicating that the physical gate itself is not impaired by the mutation, but that only the instruction coming from the C terminus is changed.
Our experiments have excluded the possibility that the mutation of the glutamate residue of GIRK affects channel expression in the membrane and the subunit assembly. Furthermore, our coimmunoprecipitation experiments suggested that the mutant molecule retains the ability to bind to Gβγ. Thus, the glutamate residue may not be a major binding site for Gβγ. Membranous level of phosphatidylinositol 4,5-bisphosphate (PIP2) is known to influence the channel functions in Kirs (21, 22). The glutamate residue, however, is negatively charged, and would not bind PIP2, which is also negatively charged. No positively charged residues are found in its vicinity. Deficiency of PIP2 alters single-channel properties of the Kir channel: open duration is shortened (23) or a subconductance state emerges (24). In contrast, the present results showed that the single-channel characteristics remain unaltered in the mutated Kir channels. Finally, Soom et al. (25) recently showed that the C4 region [corresponding to C5 in Soom et al. (25)] is not a PIP2 binding site.
Similarity of the Gating Mechanisms in GIRK and IRK.
An important aspect of our results is that the glutamate mutation causes the same effect on both IRK and GIRK channels. The C and N termini of GIRKs may contain a ligand sensor (or receptor region) that receives information from Gβγ. We hypothesize that IRK channels could be constitutively active because the sensor region is in the correct conformation for channel activity in the absence of interaction with Gβγ. The C4 glutamate mutation did not affect the functions of the channel pore or polyamine blockage sites (normal selectivity in GIRK1 and GIRK2, and normal inward rectification in GIRK2). The unitary conductance and open duration of the mutant channels were normal (in GIRK2, IRK2, and the constitutively active GIRK2), suggesting that the mutation did not impair the movement of the gate, nor did it alter the inner pore (9, 10, 26, 27). Possibly, the role of the C4 region could be a relay station transmitting the information from the sensor region to the gate. The fact that the mutation affects both GIRK and IRK in the same way suggests that the basic mechanism of the ‘mechanical gates’ of GIRK and IRK could be the same. The mechanical gate of the GIRK channel resides in the M2 segment (9, 10), which rotates relative to the M1 segment (9). Similarly, the IRK channel could possess an M2 mechanical gate, which is more prone to be in the open position. Yet the IRK gate is also regulated by the C4 region, and the activity is turned off as shown in the present experiments.
Implication for Andersen's Syndrome.
A recent publication (3) has revealed that mutations of IRK1 channels have been linked to Andersen's syndrome: one of the clinical manifestations is abnormal cell excitability, such as cardiac arrhythmias and periodic paralysis. An IRK1 mutation (E303K) is one of the causes of this syndrome, and this residue corresponds to the glutamate residues in our present study. Because of the similarity between IRK and GIRK discussed above, it is likely that the molecular mechanism of channel functional loss of Kirs elucidated by the present study may well apply to IRK1. Interestingly, the mutation IRK1(E303K), which is specifically responsible for one of Andersen's mutations, produces hardly any current (unpublished data), suggesting that it would be difficult to investigate the ionic mechanism of this mutation itself. A possible way to investigate the ionic mechanism of this syndrome is to take an indirect method like the one used in this investigation.
Acknowledgments
We thank Lily Jan and Blanche Schwappach for the supply of cDNA for HA-tagged Kir6.2 and Dusica Bajic for invaluable help. This work was supported by U.S. National Institutes of Health Grant MH57837 and a grant from the Japanese Ministry of Education, Science, Sport, and Culture.
Abbreviations
- Kir
inward rectifier K+ channels
- GIRK
G protein-coupled inward rectifier K+ channels
- HA
hemagglutinin
- wt
wild type
- Npo
number of channels multiplied by the open probability
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Thomas P, Ye Y, Lightner E. Hum Mol Genet. 1996;5:1809–1812. doi: 10.1093/hmg/5.11.1809. [DOI] [PubMed] [Google Scholar]
- 2.Simon D B, Karet F E, Rodriguez-Soriano J, Hamdan J H, DiPietro A, Trachtman H, Sanjad S A, Lifton R P. Nat Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 3.Plaster N M, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson M R, Iannaccone S T, Brunt E, Barohn R, et al. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 4.Kubo Y, Baldwin T J, Jan Y N, Jan L Y. Nature (London) 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- 5.Dascal N, Schreibmmayer W, Lim N F, Wang W, Chavkin C, DiMagno L, Labarca C, Kieffer B L, Gaveriaux-Ruff C, Trollinger D, et al. Proc Natl Acad USA. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubo Y, Reuveny E, Slesinger P A, Jan Y N, Jan L Y. Nature (London) 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- 7.Logothetis D E, Kurachi Y, Galper J, Neer E J, Clapham D E. Nature (London) 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 8.Huang C L, Slesinger P A, Casey P J, Jan Y N, Jan L Y. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 9.Yi B A, Lin Y F, Jan Y N, Jan L Y. Neuron. 2001;29:657–667. doi: 10.1016/s0896-6273(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 10.Sadja R, Smadja K, Alagem N, Reuveny E. Neuron. 2001;29:669–680. doi: 10.1016/s0896-6273(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 11.Kawano T, Chen L, Watanabe S Y, Yamauchi J, Kaziro Y, Nakajima Y, Nakajima S, Itoh H. FEBS Lett. 1999;463:355–359. doi: 10.1016/s0014-5793(99)01656-7. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy M E, Nemec J, Corey S, Wickman K, Clapham D E. J Biol Chem. 1999;274:2571–2582. doi: 10.1074/jbc.274.4.2571. [DOI] [PubMed] [Google Scholar]
- 13.Zerangue N, Schwappach B, Jan Y N, Jan L Y. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 14.Yamauchi J, Kaziro Y, Itoh H. J Biol Chem. 1997;272:7602–7607. doi: 10.1074/jbc.272.12.7602. [DOI] [PubMed] [Google Scholar]
- 15.Taglialatela M, Ficker E, Wible B A, Brown A M. EMBO J. 1995;14:5532–5541. doi: 10.1002/j.1460-2075.1995.tb00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krapivinsky G, Gordon E A, Wickman B, Velimirovic B, Krapivinsky L, Clapham D E. Nature (London) 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 17.Kofuji P, Davidson N, Lester H A. Proc Natl Acad Sci USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Velimirovic B M, Gordon E A, Lim N F, Navarro B, Clapham D E. FEBS Lett. 1996;379:31–37. doi: 10.1016/0014-5793(95)01465-9. [DOI] [PubMed] [Google Scholar]
- 19.Picones A, Keung E, Timpe L C. Biophys J. 2001;81:2035–2049. doi: 10.1016/S0006-3495(01)75853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones P A, Tucker S J, Ashcroft F M. FEBS Lett. 2001;508:85–89. doi: 10.1016/s0014-5793(01)03023-x. [DOI] [PubMed] [Google Scholar]
- 21.Huang C L, Fen S, Hilgemann D W. Nature (London) 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, He C, Yan X, Mirshahi T, Logothetis D E. Nat Cell Biol. 1999;1:183–188. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]
- 23.Sui J L, Petit-Jacques J, Logothetis D E. Proc Natl Acad Sci USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leung Y M, Zeng W Z, Liou H H, Solaro C R, Huang C L. J Biol Chem. 2000;275:10182–10189. doi: 10.1074/jbc.275.14.10182. [DOI] [PubMed] [Google Scholar]
- 25.Soom M, Schonherr R, Kubo Y, Kirsch C, Klinger R, Heinemann S H. FEBS Lett. 2001;490:49–53. doi: 10.1016/s0014-5793(01)02136-6. [DOI] [PubMed] [Google Scholar]
- 26.Lu T, Nguyen B, Zhang X, Yang J. Neuron. 1999;22:571–580. doi: 10.1016/s0896-6273(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 27.Doyle D A, Cabral J M, Pfuetzner R A, Kuo A, Gublis J M, Cohen S L, Chait B T, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]