

Abstract
Establishment of mucosal and/or luminal colonization is the first step in the pathogenesis of many gastrointestinal bacterial pathogens. The pathogen must be able to establish itself in the face of competition from the complex microbial community that is already in place. We used culture-independent methods to monitor the colonization of the cecal mucosa of Helicobacter-free mice following experimental infection with the pathogen Helicobacter hepaticus. Two days after infection, H. hepaticus comprised a minor component of the mucosa-associated microbiota, but within 14 days, it became the dominant member of the community. Colonization of the mucosa by H. hepaticus was associated with a decrease in the overall diversity of the microbial community, in large part due to changes in evenness resulting from the relative dominance of H. hepaticus as a member of the community. Our results demonstrate that invasion of the complex gastrointestinal microbial community by a pathogenic microorganism causes reproducible and significant disturbances in the community structure. The use of non-culture-based methods to monitor these changes should lead to a greater understanding of the ecological principles that govern pathogen invasion and may lead to novel methods for the prevention and control of gastrointestinal pathogens.
The gastrointestinal (GI) tract of mammals is inhabited by a complex microbial community that plays a crucial role in maintaining GI tract homeostasis (4, 32). The GI microbiota can perform a variety of beneficial metabolic functions including the catabolism of complex carbohydrates to yield short-chain fatty acids such as butyrate (35). The gut microbiota also directly interacts with the intestinal mucosa, aiding in the development of the mucosal epithelium and maturation of the mucosal immune system (27).
Another beneficial function of the indigenous GI microbiota is to provide resistance to colonization by pathogenic microorganisms, a defense mechanism commonly referred to as “colonization resistance” (6, 17, 45). Although there is wide variation in the specific composition of the climax microbial community of the GI tract between individuals, within an individual, the climax community appears to be relatively stable over time (26, 51). This stability is reflected in the development of colonization resistance. In spite of colonization resistance, certain pathogenic bacteria are able to establish residence in the gastrointestinal tract despite the presence of the indigenous microbiota. It is not known if the introduction of an “invasive species” causes detectable changes in the overall ecologic structure of the intestinal microbial community.
The examination of complex consortia of bacteria has been transformed by the development of culture-independent methods to determine the composition and structure of the community (34). In large part, this has been accomplished through the retrieval of small subunit (SSU) rRNA gene sequences, which provide a phylogenetic context in which to describe the diversity of the community. Several methods have been developed to examine the SSU rRNA-encoding gene (15). One method is to directly amplify DNA extracted from a community using primers that target the conserved regions of the SSU rRNA-encoding gene. These amplicons are then cloned, and the sequences of a number of these clones were determined.
While rRNA-encoding gene sequencing provides unambiguous phylogenetic identification, the richness of many microbial communities and the laboriousness of the technique make this approach unwieldy when applied to a large number of communities. Hence, other approaches, such as terminal restriction fragment length polymorphism (T-RFLP), were developed as rapid methodologies with high throughput that are more suitable for this type of analysis (31). T-RFLP targeting SSU rRNA-encoding genes (2, 3) has been used to profile complex microbial communities (8, 9, 30). Although phylogenetic identification of specific members of a community is difficult, T-RFLP can rapidly provide information regarding the richness and evenness of a complex community.
In this study, we monitored the community structure of the mucosa-associated microbiota of the murine cecum during the establishment of colonization by the murine pathogen Helicobacter hepaticus (43). We used T-RFLP analysis and 16S rRNA-encoding gene clone library construction to provide the first detailed examination of the microbial ecology of the GI tract during invasion by a bacterial pathogen.
(Parts of the information presented here were presented at the 104th General Meeting of the American Society for Microbiology, 23 to 27 May 2004, New Orleans, LA.)
MATERIALS AND METHODS
Animals and housing.
The initial infection studies were performed with C57BL/6 animals purchased from Jackson Laboratories (Bar Harbor, ME). For subsequent experiments, a breeding colony of wild-type C57BL/6 mice was established using breeding stock purchased from Jackson Laboratories. All animal protocols were reviewed and approved by the Michigan State University All University Committee on Animal Use and Care. Mice were housed with autoclaved food, bedding, and water. Cage changes were performed in a laminar flow hood. Animals were housed in groups of up to four animals per microisolator cage, and animals experienced a cycle of 12 h of light and 12 h of darkness.
Helicobacter hepaticus and growth conditions.
The type strain of H. hepaticus, strain 3B1 (ATCC 51449), was obtained from the American Type Culture Collection (ATCC), Manassas, VA, and was cultured on trypticase soy agar plates containing 5% sheep blood. A microoxic environment was maintained in vented GasPak jars without catalyst which were evacuated to −20 mm Hg and then equilibrated with a gas mixture consisting of 80% N2, 10% H2, and 10% CO2. An incubation temperature of 37°C was used for growth. H. hepaticus suspensions for animal challenge were prepared by harvesting organisms from culture plates into trypticase soy broth (TSB).
Experimental mouse infection.
Four- to six-week-old C57BL/6 wild-type mice were challenged orally with 1 optical density at 600 nm unit (approximately 1 × 108 CFU) of a suspension of H. hepaticus strain 3B1 in TSB. Control animals were given 300 μl of sterile TSB. The H. hepaticus suspension and the TSB control were administered directly into the stomach using a 24-gauge ball-tipped gavage needle. Infected and control groups of mice were kept in separate cages.
Monitoring of colonization.
The colonization status of H. hepaticus-infected animals was monitored weekly by culture and PCR analysis of fecal pellets taken from three mice in each experimental group as described previously (49).
Necropsy and microbiological culture.
Mice were euthanized by CO2 asphyxiation. The tip of the cecum of each mouse was removed, quartered, and washed in phosphate-buffered saline to remove luminal contents. Three sections were snap-frozen in dry ice/ethanol, and one section was cultured on CVA selective agar plates (20 mg/ml cefoperazone, 10 mg/ml vancomycin, 2 mg/ml amphotericin B, 5% sheep blood, 1.5% trypticase soy agar).
DNA extraction.
Total DNA was extracted from the cecal samples using a commercial kit (DNeasy tissue kit; QIAGEN, Germantown, MD), as recommended by the manufacturer, except that the cecal samples were digested in the supplied ATL buffer overnight prior to continuing with the extraction procedure.
T-RFLP analysis.
T-RFLP was performed as outlined previously (5). PCR amplification employing primers targeting bacterial 16S rRNA genes (8F and 1492R [38]) was performed on each DNA sample. The 8F primer was linked to the fluorescent dye 6-FAM (Integrated DNA Technologies, Coralville, IA), and the 1492R primer was unlabeled. Each 25-μl PCR mixture contained 20 pmol of each primer, 200 μM of each deoxynucleoside triphosphate, and 1.5 U of Taq DNA polymerase in a final concentration of 10 mM Tris-HCl-50 mM KCl-1.5 mM MgCl2 (Ready To Go PCR beads; Amersham Pharmacia Biotech, Piscataway, NJ). PCR was performed under the following cycle conditions: an initial denaturation step at 94°C for 2 min and 30 cycles of denaturation at 94°C for 30s, annealing at 58°C for 45s, and extension at 72°C for 90s. A final extension step at 72°C for 5 min was performed. The PCR product was purified using GFX purification columns (Amersham Pharmacia Biotech). Two hundred nanograms of purified PCR amplicon was cut individually with the restriction enzymes HhaI and MspI (New England Biolabs, Beverly, MA) for 1 to 2 h at 37°C (30). The DNA fragments were separated on an ABI 3100 Genetic Analyzer automated sequence analyzer (Applied Biosystems Instruments, Foster City, CA) in GeneScan mode at Michigan State University's sequencing facility. The 5′-terminal restriction fragments (TRFs) were detected by excitation of the 6-FAM molecule attached to the forward primer. The sizes and abundance of the fragments were calculated using GeneScan 3.7. The PCR conditions and the restriction digest conditions were chosen to allow maximal reproducibility.
Analysis of T-RFLP profiles.
Profiles were analyzed as follows using Microsoft Excel and the JMP statistical package. Calculations were performed on profiles generated by digestion of fluorescently labeled PCR amplicons with both HhaI and MspI. To standardize each profile for the quantity of labeled DNA present in each sample, the sum of TRF peak heights in each profile being compared was calculated. The sum of peak heights generally varied less than twofold over all of the profiles. Each sum of TRF peak heights was normalized to the lowest sum of peak heights of the comparison samples. This yielded a correction factor that was applied to each peak in a given profile. The resultant peak heights were filtered to eliminate peaks with a height below the noise threshold (set at a relative fluorescence value of 50).
Calculation of diversity indices.
For each normalized T-RFLP profile, the number and height of peaks in each profile were considered to represent the number and relative abundance of different phylotypes present in the sample. Phylotype richness (S) was calculated as the total number of distinct TRF peaks in each normalized profile. The Shannon-Weiner diversity index was calculated as follows: 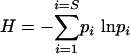 , where pi is the proportion of the ith peak relative to the sum of all peak heights. Evenness was calculated as H/Hmax, where Hmax = ln(S) (the case when all pi's are equal; therefore, pi = 1/S). An additional calculation was preformed for T-RFLP profiles from H. hepaticus-infected animals. In this case, the H. hepaticus-specific TRF was removed from each profile prior to the normalization procedure and the calculation of diversity indices.
, where pi is the proportion of the ith peak relative to the sum of all peak heights. Evenness was calculated as H/Hmax, where Hmax = ln(S) (the case when all pi's are equal; therefore, pi = 1/S). An additional calculation was preformed for T-RFLP profiles from H. hepaticus-infected animals. In this case, the H. hepaticus-specific TRF was removed from each profile prior to the normalization procedure and the calculation of diversity indices.
Clone libraries.
The community structure of infected and uninfected mice was also analyzed by the construction of 16S clone libraries. Unlabeled 8F and 1492R primers were used to amplify DNA samples using the same conditions as those for T-RFLP analysis. Following purification, the PCR products were ligated into a T-tailed plasmid vector (pCR 2.1; Invitrogen, Carlsbad, CA). DNA sequence and sequence analysis were performed as detailed previously (50). Briefly, each clone was sequenced with a single primer (519R) that typically yielded ∼500 bases of readable sequence. Sequences were analyzed for the formation of chimeras using the Chimera Check program from the Ribosomal Database Project (10). Potential chimeric sequences were excluded from additional analysis. Sequences were aligned to one another using the ARB suite of programs (available through http://www.arb-home.de). Regions of ambiguous alignment (primarily stem structures of variable length) were excluded from the final comparison of sequences such that 342 positions were used in the final phylogenetic analyses. Phylogenetic trees were calculated using the ARB neighbor-joining algorithm.
Grouping of 16S clone library sequences and rarefaction analysis (23) were performed using the FastGroup program (39) (available at http://phage.sdsu.edu/research/projects/fastgroup/). Coverage estimations were calculated by the method described previously by Good (21).
Statistical differences in the compositions of clone libraries from infected and uninfected samples were determined using LIBSHUFF (version 1.2) (40). As was done for the T-RFLP profiles, an additional analysis using LIBSHUFF was performed, this time removing rRNA-encoding gene sequences corresponding to H. hepaticus prior to statistical analysis.
In silico terminal restriction fragment length polymorphism analysis.
Terminal restriction fragments using the restriction enzymes HhaI and MspI were calculated for each of the 16S rRNA-encoding gene clones using a software tool designed to be integrated into the ARB program suite (36). This in silico T-RFLP analysis tool (TRF-CUT) predicts TRFs based on the ARB-aligned sequences. After deleting sequences homologous to H. hepaticus, the predicted TRFs were plotted as a histogram using the DeltaGraph software program (Red Rock Software, Salt Lake City, Utah). A uniform bin size of 5 base pairs was used in generating the histograms.
Nucleotide sequence accession numbers.
The partial 16S rRNA-encoding gene sequences were deposited in GenBank under the accession numbers AY914179 to AY914315.
RESULTS
Mouse infections with H. hepaticus.
H. hepaticus strain 3B1 (the type strain) was administered via oral gavage to four wild-type C57BL/6 mice. An equal number of control animals received sterile culture broth. The animals were monitored for colonization with H. hepaticus by culture and H. hepaticus-specific PCR performed on freshly voided fecal pellets. All of the animals infected with H. hepaticus remained colonized with H. hepaticus for the 30-day duration of this experiment (data not shown).
Thirty days after infection with H. hepaticus, the animals underwent gastrointestinal necropsy. The cecal tip was harvested, and the luminal contents were removed. The tissue was longitudinally divided into four sections and frozen for subsequent DNA extraction. None of the animals were found to have gross lesions at necropsy, and histologic examination of the cecum and colon did not reveal any typhlocolitis in either the control or infected animals (data not shown).
T-RFLP analysis of cecal tissue 30 days after H. hepaticus infection.
DNA was extracted from frozen cecal tissue and analyzed by T-RFLP using the restriction enzymes HhaI and MspI. To determine the expected location for the H. hepaticus-specific TRF within a T-RFLP profile, purified genomic DNA from H. hepaticus strain 3B1 was also subjected to T-RFLP analysis.
Visual inspection of the T-RFLP traces reveals that infected animals possessed a prominent TRF corresponding to an H. hepaticus TRF (Fig. 1), whereas profiles from uninfected animals lacked this TRF. To quantify changes in the community structure of the mucosa-associated microbiota associated with colonization by H. hepaticus, traditional indices of ecologic diversity were calculated for both the HhaI and MspI T-RFLP profiles. As seen in Fig. 2, colonization with H. hepaticus was associated with a significant decrease in both overall diversity (as measured by the Shannon-Weiner diversity index, H) and evenness (H/Hmax). There was no significant difference in the number of TRFs encountered in the T-RFLP profiles from infected animals compared to uninfected animals (data not shown).
FIG. 1.

Representative T-RFLP traces comparing the mucosa-associated microbiota in the cecae of an uninfected C57BL/6 mouse and two mice infected with H. hepaticus 30 days previously. The relative fluorescence of each peak is plotted against the size of the peak. In the traces from the two infected animals, a TRF corresponding to an H. hepaticus-specific TRF is clearly seen and is the dominant TRF.
FIG. 2.

Changes in diversity measures of the mucosa-associated microbiota of the murine cecum 30 days after experimental infection with H. hepaticus. The Shannon diversity index (H) and Shannon evenness (H/Hmax) were calculated for each mouse based on normalized T-RFLP profiles obtained with HhaI and MspI digestion. Compared to uninfected animals (“Uninfected” category), animals experimentally infected with H. hepaticus (“Infected” category) had significant decreases in both diversity and evenness. When the analysis was repeated, this time suppressing the H. hepaticus terminal restriction fragment before normalization of the profiles (“Infected [suppressed]” category), the change in diversity and evenness in infected animals was no longer apparent. Comparisons for all pairs of time points was performed by analysis of variance using Tukey-Kramer honestly significant difference. Categories within each plot not connected by the same roman numeral are significantly different with an alpha level set to 0.05.
To determine to what extent the changes in diversity and evenness were due to the dominance that H. hepaticus assumed within the community, these indices were recalculated. This time, the H. hepaticus-specific TRF was removed from each profile obtained from an infected animal before calculation of the diversity and evenness. When the H. hepaticus-specific TRF was suppressed, the differences between the infected animals and uninfected animals were no longer statistically significant (Fig. 2).
Clone library analysis.
T-RFLP analysis provides a relatively inexpensive and high-throughput method for performing microbial community analyses. However, although an overall “fingerprint” of the community is obtained by T-RLFP, it is difficult to identify specific members of the community, unless their presence is suspected beforehand (as is the case with H. hepaticus in experimentally infected animals).
To provide insight into the specific bacterial species present in the tissue from H. hepaticus-infected and uninfected mice, 16S clone libraries were constructed. SSU rRNA-encoding genes were amplified from cecal tissue DNA from an H. hepaticus-infected mouse and an uninfected control using the same bacterial primers used for T-RFLP analysis. The resultant amplicons were cloned into a T-tailed plasmid vector, and the DNA sequence was determined for a set of randomly selected clones from each library.
Examination of a phylogenetic tree that combines the 16S sequences obtained from an uninfected animal and an infected animal reveals that H. hepaticus 16S sequences were present only in the library from the infected animal (Fig. 3). LIBSHUFF analysis (40) indicated that the composition of the two libraries was significantly different when the uninfected library was compared to the infected library, suggesting that the underlying communities being sampled were different. However, if 16S sequences homologous to H. hepaticus are deleted from the library constructed from the infected animal prior to LIBSHUFF analysis, this difference is no longer significant (Fig. 3).
FIG.3.

Phylogenetic tree showing the distribution of 16S rRNA-encoding gene sequences from clone libraries constructed from cecal DNA samples obtained from an uninfected mouse (green circles) or a mouse 30 days after infection with H. hepaticus (red squares). Brackets outline major clusters of organisms. The scale bar represents evolutionary distance (5 substitutions per 100 nucleotides). The tree was constructed by neighbor-joining analysis using the MEGA program. Analysis was performed on a multiple-sequence alignment generated using the ARB suite of programs. Clones representing H. hepaticus were found only in the clone library constructed from DNA from the infected animal. The graph shows rarefaction curves for each library along with Good's coverage estimate in parentheses. The results of LIBSHUFF analysis (40) of the two libraries, with and without inclusion of H. hepaticus 16S rRNA-encoding gene sequences, are shown in the inset.
To compare the community profile obtained by T-RFLP analysis with that provided by clone library analysis, an in silico analysis of the partial 16S rRNA-encoding gene sequences was performed. HhaI and MspI TRFs were predicted for each 16S rRNA-encoding gene clone, with the exception of clones homologous to H. hepaticus, using an ARB software-integrated tool (36). The in silico-generated TRFs were plotted in histogram format to reflect the number of times a specific in silico TRF was encountered, and these plots were compared to actual T-RFLP traces from uninfected mice (Fig. 4). The plots of the in silico-generated TRFs were quite similar to the actual T-RFLP traces.
FIG. 4.

Comparison between T-RFLP analysis and in silico terminal restriction fragment length polymorphism analysis. HhaI and MspI TRFs were predicted for each 16S rRNA-encoding gene clone depicted in Fig. 3, with the exception of clones corresponding to H. hepaticus. The in silico-generated TRFs are plotted in histogram format below a corresponding actual T-RFLP trace from an uninfected animal for each enzyme. The gray areas of the histograms above 470 base pairs represent the areas where we would not expect to see predicted TRFs, given that the maximal length of any given partial 16S sequence was 470 base pairs (see Materials and Methods).
Time course of infection with H. hepaticus.
The above-described results indicate that H. hepaticus becomes a dominant member of the cecal mucosa-associated microbiota 1 month after experimental infection and that H. hepaticus colonization results in a decrease in the diversity of the microbiota. In order to follow the dynamics of colonization of the cecal mucosa by H. hepaticus, a time course experiment was performed. Groups of three C57BL/6 mice were infected with H. hepaticus via oral gavage. An equal number of control animals received sterile culture broth. At 2, 8, 14, and 42 days after infection, one experimental and one control group of mice were sacrificed, and the cecal tissue was taken for histopathology and T-RFLP analysis. As in the 30-day infection experiment, wild-type C57BL/6 animals did not develop significant inflammation or hyperplasia in the setting of H. hepaticus colonization (data not shown).
Figure 5 shows representative MspI T-RFLP traces from cecal samples of mice at 2, 8, 14, and 42 days after infection with H. hepaticus. At all time points, a TRF corresponding to the H. hepaticus-specific TRF was visible in each tracing. This TRF was not seen in any of the uninfected control animals (data not shown). To provide an estimate of the level of colonization by H. hepaticus at each time point, the fraction of the total represented by the H. hepaticus-specific TRF was calculated. At the earliest time point, the H. hepaticus TRF represented only a small fraction (<1%) of the total TRF signal (calculated by peak height) in the tracing. Over time, however, the H. hepaticus-specific TRF became the dominant TRF seen, eventually representing ∼50% of the total signal in the trace. This result was consistent over all of the animals examined. Figure 6A plots the relative fraction represented by the H. hepaticus-specific TRF in each animal at each time point. The H. hepaticus-specific TRF is initially a minor component of the total community at 2 days after infection but becomes a major component at 8 days after infection and the dominant component by 14 days. This predominance of the H. hepaticus-specific TRF remained at 42 days after infection. In contrast, there was no change in the appearance of the T-RFLP profiles for the uninfected animals over the entire 42-day time course (data not shown).
FIG. 5.

Representative T-RFLP traces temporally monitoring the establishment of colonization in the murine cecum by H. hepaticus. Two days after infection, H. hepaticus is only a minor fraction (pi) of the total cecal microbiota but by 8 days after infection becomes a major component (pi = 0.30) of the total. By 14 days after infection, H. hepaticus is the dominant member of the mucosa-associated microbiota, and this dominance persists through 42 days after infection. For each trace, the number of peaks (S), Shannon-Weiner diversity index (H), and Shannon evenness (H/Hmax) are shown.
FIG. 6.

Summary of the temporal monitoring of the colonization of the cecae of mice by H. hepaticus. A. The fraction of the total community represented by H. hepaticus (pi, as calculated by T-RFLP analysis) is plotted for the three mice in each experimental group at 2, 8, 14, and 42 days after infection. H. hepaticus is initially a minor component of the mucosa-associated microbiota 2 days after infection but becomes the predominant member of the community by 14 days after infection. B. The Shannon diversity index (H) plotted for the three mice in each experimental group at 2, 8, 14, and 42 days after infection. As the pi of H. hepaticus increases, this is accompanied by a corresponding decrease in the diversity of the mucosa-associated microbiota. Comparisons for all pairs of time points were performed by analysis of variance using Tukey-Kramer HSD. Time points not connected by the same letter are significantly different with an alpha level set to 0.05.
A plot of the Shannon-Weiner diversity index during the time course reveals that as H. hepaticus becomes increasingly dominant as a component of the mucosa-associated microbiota, the overall diversity of the community decreases (Fig. 6B).
DISCUSSION
Colonization is the initial step in the pathogenesis of many enteric bacterial pathogens. A great deal of insight has been gained on the genetic adaptations that enteric pathogens have evolved to permit successful colonization and the eventual development of disease (12, 13). A key emphasis has been placed on examining the interaction between pathogenic bacteria and host cells. An aspect of bacterial pathogenesis that has been less studied is the interaction between pathogenic bacteria and the preexisting microbiota that inhabits a particular ecologic niche within the host. The indigenous microbiota of the host have been postulated to interfere with the invasion by pathogenic organisms, so-called “colonization resistance” (6, 17). It has been observed that certain bacteria residing in the GI tract, whether naturally occurring or experimentally administered, can protect the host from pathogenic bacteria (1, 24). It is likely that a number of mechanisms, including nutrient depletion, competition for binding sites on the mucosal epithelium, and the production of inhibitory substances contribute to colonization resistance (7, 18, 19).
One of the reasons that the interaction between pathogenic bacteria and the indigenous host microbiota has not been studied in detail is that the study of complex microbial communities has been difficult. The intestinal tract of mammals is inhabited by a large and phylogenetically diverse community of microorganisms, many of them obligate anaerobes. Over the past decade, molecular-based approaches have revealed enormous phylogenetic diversity in the microbial world that is not yet represented in culture (34). These non-culture-based techniques, which generally involve the retrieval of the DNA sequence of the small subunit rRNA gene (16S rRNA-encoding gene in the case of bacteria), were initially developed to examine microbial diversity in soil and aquatic environments. More recently, these techniques have been used to examine the indigenous microbiota of mammals. Earlier culture-based examinations of the biota of the mammalian gastrointestinal tract suggested that the majority of morphotypes seen by microscopic examination could be cultivated in the laboratory (33). More recent culture-independent analysis of the microbial ecology of the gastrointestinal tract has suggests that the overall diversity is greater than previously estimated (4, 11, 22, 25, 42, 48). This is due in large part to the fact that sequence-based methodologies can discriminate between bacterial isolates that may have identical morphologies and similar in vitro characteristics.
Initially, culture-independent studies on the mammalian gastrointestinal tract cataloged the species richness that is encountered in that environment (29, 37, 42, 48). More recently, these techniques have also been used to follow changes in the intestinal microbiota over time and to compare the resident microbiota between individuals. These studies have revealed that individuals posses a community of microbes that can vary extensively from individual to individual, and within an individual, it can vary with anatomic location (11, 51). We have used these techniques to monitor the changes in the fecal microbiota that can occur in the setting of antibiotic-associated diarrhea (50).
H. hepaticus is a murine pathogen that has been found to be widespread in research mouse colonies. Depending on the strain of mouse, H. hepaticus infection is associated with biliary tract or lower gastrointestinal tract disease. In many strains of mice colonized with H. hepaticus, hepatic disease is subclinical and may be accompanied by subclinical enteritis (generally typhlitis or colitis) (41). However, in mice with altered immune function, the typhlitis/colitis can be severe, leading to rectal prolapse, weight loss, and death. This murine typhlitis/colitis in the setting of altered immune function has been employed as a model for inflammatory bowel disease (14).
In the experiments presented here, we used culture-independent community analysis to monitor changes in the mucosa-associated microbiota of the cecum during murine infection with H. hepaticus. Wild-type animals were chosen for this infection study to avoid any changes in the microbial community structure secondary to the development of typhlocolitis. In this way, any alterations in the mucosa-associated microbiota could be attributed to colonization by H. hepaticus alone and not by changes in the mucosal environment due to the development of an active inflammatory response.
T-RFLP analysis proved to be a useful method for estimating the abundance of H. hepaticus in the mucosa-associated community. Previous studies using culture (16) and a quantitative PCR assay targeting the cytolethal distending toxin of H. hepaticus (20) suggest that the organism is encountered in high numbers in the cecum of colonized mice. Although there are potential biases that result from using PCR to interrogate an entire community (46), the T-RFLP data presented here suggest that H. hepaticus becomes the dominant member of the mucosa-associated microbiota of the cecum in infected animals. This conclusion is based on the assumption that peak height by TRF is proportional to the abundance of a particular 16S rRNA-encoding gene species in the community and thus the relative abundance of that particular bacterial species. Clone library analysis also confirmed that H. hepaticus readily colonizes the cecal mucosa and becomes the predominant bacterial species present.
It has been previously suggested that T-RFLP analysis is appropriate for monitoring changes in a given community (28). The use of T-RFLP also revealed that the process of colonization of the cecal mucosa by H. hepaticus was associated with reproducible shifts in the overall structure of the microbial community. In the time course experiment, the kinetics by which H. hepaticus increased as a component of the community were similar from animal to animal, as were the changes in the non-H. hepaticus TRFs. Additionally, the C57BL/6 mice in the first experiment came from a different colony than the C57BL/6 mice used in the time course experiment. There were detectable differences in the TRF profiles between uninfected animals from each of these colonies (data not shown). However, the final shifts in the community resulting from H. hepaticus colonization were very similar, suggesting that H. hepaticus challenge applies a reproducible ecologic stress on the existing microbiota.
We used indices of community diversity, traditionally applied to macroecologic communities such as wetlands and forests, to quantify the changes observed using T-RLFP analysis in the community structure of the mucosa-associated microbiota following colonization with H. hepaticus. H. hepaticus colonization resulted in a significant decrease in the diversity of this community. This effect appears to be primarily due to the relative dominance that H. hepaticus assumes within the community. Reanalysis of the T-RFLP profiles following removal of H. hepaticus from the mucosal communities of infected animals revealed that the diversity of the remainder of the community was not significantly different from that seen in uninfected animals. This suggests that H. hepaticus has minimal interactions with other members of the indigenous microbiota, at least not interactions detectable by analysis of the entire community by T-RFLP profiling.
Reanalysis of the clone libraries with suppression of H. hepaticus-specific clones in general supports the findings of the repeat T-RFLP analysis. However, the finer-scale resolution of taxonomic information provided by 16S rRNA-encoding gene sequence analysis supplies some additional insights into the community dynamics encountered in animals infected with H. hepaticus. LIBSHUFF analysis provides a measure of the difference between two 16S rRNA-encoding gene clone libraries based on the ability of one library to “cover” the diversity seen in a second library (40). LIBSHUFF analysis of the libraries when H. hepaticus 16S rRNA-encoding gene clones are included indicates that the library from the uninfected animal is unable to provide coverage of the library from the H. hepaticus-infected animal (P = 0.002), but in the reverse case, the library from the infected animal provides at least partial coverage of the library from the uninfected animal (P = 0.076). These results are consistent with the idea that the library from the uninfected animal is a subset of the library from the infected animal (40), and this is supported by visual inspection of the phylogenetic tree. However, LIBSHUFF analysis following the removal of 16S rRNA-encoding gene clones with homology to H. hepaticus provides additional information regarding the diversity of the indigenous microbiota in H. hepaticus-infected animals. This reanalysis shows that the library from the uninfected animal easily provides coverage of the library from the infected animal (P = 0.31) when H. hepaticus is removed. However, there is a trend (P = 0.051) towards inadequate coverage of the library from the uninfected animal by the library from the infected animal. Therefore, while the most obvious changes in diversity, as measured by T-RFLP and clone library analysis, are due to the dominance that H. hepaticus assumes within the community, clone library analysis suggests that more subtle perturbations of the indigenous microbiota may also be occurring.
At first glance, it is somewhat surprising that H. hepaticus was able to “invade” an established, diverse ecosystem with such ease. However, H. hepaticus, a bona fide murine pathogen, is excluded from most specific-pathogen-free mouse colonies (47). Thus, experimentally introduced H. hepaticus may be filling an underutilized or possibly “empty” ecologic niche in the gastrointestinal tract. In part, this can explain the apparent lack of interaction (e.g., direct competition) between H. hepaticus and other members of the mucosal microbiota. It has been proposed that successful introduction of an “invasive species” to an established ecosystem is a function of how different an invader is from established species (44).
To our knowledge, this is the first time that culture-independent techniques have been used to monitor the invasion of a complex mammal-associated microbial community by a pathogen. Our results demonstrate the power of this type of analysis on revealing details of the relationship between a pathogen and the existing microbiota of the gut. Given the importance of the indigenous GI microbiota in both health and disease, the development and use of methods by which this complex population can be monitored will aid in the study of a wide variety of gastrointestinal conditions including gastroenteritis, antibiotic-associated diarrhea, and inflammatory bowel disease.
Acknowledgments
This work was supported by a Michigan State University Intramural Research Grant Program New Investigator Award to V.B.Y., a Crohn's and Colitis Foundation of America (CCFA) Senior Investigator Award to V.B.Y., summer undergraduate research fellowships from the CCFA and the American Society for Microbiology to C.J.K., and funding from the Michigan State University Center for Microbial Pathogenesis.
We thank Jennifer Ellis-Cortez for expert technical assistance.
Editor: V. J. DiRita
REFERENCES
- 1.Asahara, T., K. Shimizu, K. Nomoto, T. Hamabata, A. Ozawa, and Y. Takeda. 2004. Probiotic bifidobacteria protect mice from lethal infection with Shiga toxin-producing Escherichia coli O157:H7. Infect. Immun. 72:2240-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avaniss-Aghajani, E., K. Jones, D. Chapman, and C. Brunk. 1994. A molecular technique for identification of bacteria using small subunit ribosomal RNA sequences. BioTechniques 17:144-146, 148-149. [PubMed] [Google Scholar]
- 3.Avaniss-Aghajani, E., K. Jones, A. Holtzman, T. Aronson, N. Glover, M. Boian, S. Froman, and C. F. Brunk. 1996. Molecular technique for rapid identification of mycobacteria. J. Clin. Microbiol. 34:98-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backhed, F., R. E. Ley, J. L. Sonnenburg, D. A. Peterson, and J. I. Gordon. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915-1920. [DOI] [PubMed] [Google Scholar]
- 5.Blackwood, C. B., T. Marsh, S. H. Kim, and E. A. Paul. 2003. Terminal restriction fragment length polymorphism data analysis for quantitative comparison of microbial communities. Appl. Environ. Microbiol. 69:926-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohnhoff, M., B. L. Drake, and C. P. Miller. 1954. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc. Soc. Exp. Biol. Med. 86:132-137. [DOI] [PubMed] [Google Scholar]
- 7.Bourlioux, P., B. Koletzko, F. Guarner, and V. Braesco. 2003. The intestine and its microflora are partners for the protection of the host: report on the Danone Symposium “The Intelligent Intestine,” held in Paris, June 14, 2002. Am. J. Clin. Nutr. 78:675-683. [DOI] [PubMed] [Google Scholar]
- 8.Bruce, K. D., and M. R. Hughes. 2000. Terminal restriction fragment length polymorphism monitoring of genes amplified directly from bacterial communities in soils and sediments. Mol. Biotechnol. 16:261-269. [DOI] [PubMed] [Google Scholar]
- 9.Clement, B. G., L. E. Kehl, K. L. DeBord, and C. L. Kitts. 1998. Terminal restriction fragment patterns (TRFPs), a rapid, PCR-based method for the comparison of complex bacterial communities. J. Microbiol. Methods 31:135-142. [Google Scholar]
- 10.Cole, J. R., B. Chai, T. L. Marsh, R. J. Farris, Q. Wang, S. A. Kulam, S. Chandra, D. M. McGarrell, T. M. Schmidt, G. M. Garrity, and J. M. Tiedje. 2003. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckburg, P. B., E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman. 2005. Diversity of the human intestinal microbial flora. Science 308:1635-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falkow, S. 2004. Molecular Koch's postulates applied to bacterial pathogenicity—a personal recollection 15 years later. Nat. Rev. Microbiol. 2:67-72. [DOI] [PubMed] [Google Scholar]
- 13.Finlay, B. B., and S. Falkow. 1997. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 61:136-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foltz, C. J., J. G. Fox, R. Cahill, J. C. Murphy, L. Yan, B. Shames, and D. B. Schauer. 1998. Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter 3:69-78. [DOI] [PubMed] [Google Scholar]
- 15.Forney, L. J., X. Zhou, and C. J. Brown. 2004. Molecular microbial ecology: land of the one-eyed king. Curr. Opin. Microbiol. 7:210-220. [DOI] [PubMed] [Google Scholar]
- 16.Fox, J. G., F. E. Dewhirst, J. G. Tully, B. J. Paster, L. Yan, N. S. Taylor, M. J. Collins, Jr., P. L. Gorelick, and J. M. Ward. 1994. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 32:1238-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freter, R. 1955. The fatal enteric cholera infection in the guinea pig, achieved by inhibition of normal enteric flora. J. Infect. Dis. 97:57-65. [DOI] [PubMed] [Google Scholar]
- 18.Freter, R. 1962. In vivo and in vitro antagonism of intestinal bacteria against Shigella flexneri. II. The inhibitory mechanism. J. Infect. Dis. 110:38-46. [DOI] [PubMed] [Google Scholar]
- 19.Freter, R., and A. Ozawa. 1963. Explanation for limitation of populations of Escherichia coli in broth cultures. J. Bacteriol. 86:904-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge, Z., D. A. White, M. T. Whary, and J. G. Fox. 2001. Fluorogenic PCR-based quantitative detection of a murine pathogen, Helicobacter hepaticus. J. Clin. Microbiol. 39:2598-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Good, I. J. 1953. The population frequencies of species and the estimation of population parameters. Biometrica 40:237-264. [Google Scholar]
- 22.Hayashi, H., M. Sakamoto, and Y. Benno. 2002. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol. Immunol. 46:535-548. [DOI] [PubMed] [Google Scholar]
- 23.Heck, K. J. J., and G. V. Belle. 1975. Explicit calculation of the rarefaction diversity measurement and the determination of sufficient sample size. Ecology 56:1459-1461. [Google Scholar]
- 24.Heczko, U., A. Abe, and B. B. Finlay. 2000. Segmented filamentous bacteria prevent colonization of enteropathogenic Escherichia coli O103 in rabbits. J. Infect. Dis. 181:1027-1033. [DOI] [PubMed] [Google Scholar]
- 25.Hold, G. L., S. E. Pryde, V. J. Russell, E. Furrie, and H. J. Flint. 2002. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol. Ecol. 39:33-39. [DOI] [PubMed] [Google Scholar]
- 26.Holdeman, L. V., I. J. Good, and W. E. Moore. 1976. Human fecal flora: variation in bacterial composition within individuals and a possible effect of emotional stress. Appl. Environ. Microbiol. 31:359-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hooper, L. V. 2004. Bacterial contributions to mammalian gut development. Trends Microbiol. 12:129-134. [DOI] [PubMed] [Google Scholar]
- 28.Kibe, R., M. Sakamoto, H. Hayashi, H. Yokota, and Y. Benno. 2004. Maturation of the murine cecal microbiota as revealed by terminal restriction fragment length polymorphism and 16S rRNA gene clone libraries. FEMS Microbiol. Lett. 235:139-146. [DOI] [PubMed] [Google Scholar]
- 29.Leser, T. D., J. Z. Amenuvor, T. K. Jensen, R. H. Lindecrona, M. Boye, and K. Moller. 2002. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 68:673-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, W. T., T. L. Marsh, H. Cheng, and L. J. Forney. 1997. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 63:4516-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marsh, T. L. 1999. Terminal restriction fragment length polymorphism (T-RFLP): an emerging method for characterizing diversity among homologous populations of amplification products. Curr. Opin. Microbiol. 2:323-327. [DOI] [PubMed] [Google Scholar]
- 32.McCracken, V. J., and R. G. Lorenz. 2001. The gastrointestinal ecosystem: a precarious alliance among epithelium, immunity and microbiota. Cell. Microbiol. 3:1-11. [DOI] [PubMed] [Google Scholar]
- 33.Moore, W. E., and L. V. Holdeman. 1974. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl. Microbiol. 27:961-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-740. [DOI] [PubMed] [Google Scholar]
- 35.Pryde, S. E., S. H. Duncan, G. L. Hold, C. S. Stewart, and H. J. Flint. 2002. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217:133-139. [DOI] [PubMed] [Google Scholar]
- 36.Ricke, P., S. Kolb, and G. Braker. 2005. Application of a newly developed ARB software-integrated tool for in silico terminal restriction fragment length polymorphism analysis reveals the dominance of a novel pmoA cluster in a forest soil. Appl. Environ. Microbiol. 71:1671-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salzman, N. H., H. De Jong, Y. Paterson, H. J. Harmsen, G. W. Welling, and N. A. Bos. 2002. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology 148:3651-3660. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt, T. M., and D. A. Relman. 1994. Phylogenetic identification of uncultured pathogens using ribosomal RNA sequences. Methods Enzymol. 235:205-222. [DOI] [PubMed] [Google Scholar]
- 39.Seguritan, V., and F. Rohwer. 2001. FastGroup: a program to dereplicate libraries of 16S rDNA sequences. BMC Bioinformatics 2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singleton, D. R., M. A. Furlong, S. L. Rathbun, and W. B. Whitman. 2001. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl. Environ. Microbiol. 67:4374-4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solnick, J. V., and D. B. Schauer. 2001. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin. Microbiol. Rev. 14:59-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suau, A., R. Bonnet, M. Sutren, J. J. Godon, G. R. Gibson, M. D. Collins, and J. Dore. 1999. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 65:4799-4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suerbaum, S., C. Josenhans, T. Sterzenbach, B. Drescher, P. Brandt, M. Bell, M. Droge, B. Fartmann, H. P. Fischer, Z. Ge, A. Horster, R. Holland, K. Klein, J. Konig, L. Macko, G. L. Mendz, G. Nyakatura, D. B. Schauer, Z. Shen, J. Weber, M. Frosch, and J. G. Fox. 2003. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. USA 100:7901-7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tilman, D. 2004. Niche tradeoffs, neutrality, and community structure: a stochastic theory of resource competition, invasion, and community assembly. Proc. Natl. Acad. Sci. USA 101:10854-10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vollaard, E. J., and H. A. Clasener. 1994. Colonization resistance. Antimicrob. Agents Chemother. 38:409-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Wintzingerode, F., U. B. Gobel, and E. Stackebrandt. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21:213-229. [DOI] [PubMed] [Google Scholar]
- 47.Whary, M. T., J. H. Cline, A. E. King, C. A. Corcoran, S. Xu, and J. G. Fox. 2000. Containment of Helicobacter hepaticus by use of husbandry practices. Comp. Med. 50:78-81. [PubMed] [Google Scholar]
- 48.Wilson, K. H., and R. B. Blitchington. 1996. Human colonic biota studied by ribosomal DNA sequence analysis. Appl. Environ. Microbiol. 62:2273-2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young, V. B., K. A. Knox, J. S. Pratt, J. S. Cortez, L. S. Mansfield, A. B. Rogers, J. G. Fox, and D. B. Schauer. 2004. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 72:2521-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young, V. B., and T. M. Schmidt. 2004. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin. Microbiol. 42:1203-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zoetendal, E. G., A. D. Akkermans, and W. M. De Vos. 1998. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl. Environ. Microbiol. 64:3854-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]