

Abstract
Apical membrane antigen 1 (AMA1) is expressed in schizont-stage malaria parasites and sporozoites and is thought to be involved in the invasion of host red blood cells. AMA1 is an important vaccine candidate, as immunization with this antigen induces a protective immune response in rodent and monkey models of human malaria. Additionally, anti-AMA1 polyclonal and monoclonal antibodies inhibit parasite invasion in vitro. We have isolated a 20-residue peptide (R1) from a random peptide library that binds to native AMA1 as expressed by Plasmodium falciparum parasites. Binding of R1 peptide is dependent on AMA1 having the proper conformation, is strain specific, and results in the inhibition of merozoite invasion of host erythrocytes. The solution structure of R1, as determined by nuclear magnetic resonance spectroscopy, contains two structured regions, both involving turns, but the first region, encompassing residues 5 to 10, is hydrophobic and the second, at residues 13 to 17, is more polar. Several lines of evidence reveal that R1 targets a “hot spot” on the AMA1 surface that is also recognized by other peptides and monoclonal antibodies that have previously been shown to inhibit merozoite invasion. The functional consequence of binding to this region by a variety of molecules is the inhibition of merozoite invasion into host erythrocytes. The interaction between these peptides and AMA1 may further our understanding of the molecular mechanisms of invasion by identifying critical functional regions of AMA1 and aid in the development of novel antimalarial strategies.
Malaria is responsible for more than 1 million deaths annually. The clinical symptoms of this disease are caused by the erythrocytic stage of the parasite life cycle, and significant efforts have been devoted to arresting this stage of parasite growth. In particular, proteins involved in invasion of host red blood cells are attractive targets for vaccine and drug development, due to their accessibility to the host immune system. Although the molecular mechanisms of invasion are not well understood, ultrastructural studies have revealed a distinct set of steps involving parasite attachment, reorientation, and, ultimately, invasion of the host red blood cell (8). A number of proteins have been implicated in invasion, but in most cases their precise functions remain unknown.
Apical membrane antigen 1 (AMA1) is an important vaccine candidate that is expressed in mature stage parasites and is thought to be essential for invasion (37, 52). Plasmodium falciparum AMA1 (PfAMA1) is a type I integral membrane protein that is produced as an 83-kDa precursor and is localized initially to the micronemes, apical organelles of the parasite (3, 17). Eight conserved intramolecular disulfide bonds constrain this protein into three distinct domains (22). Shortly after synthesis, this precursor is cleaved to a 66-kDa product which is translocated onto the merozoite surface where much of the ectodomain is shed during invasion (23, 36).
A large body of evidence supports the role of AMA1 in invasion and its position as a leading vaccine candidate. The stage-specific expression and localization of AMA1 indicate a role in invasion, and AMA1 knockouts are not viable, implying that AMA1 is critical in the parasite life cycle (50). An AMA1 orthologue has been identified in other apicomplexan parasites, suggesting a conserved function across species (19). Immunological studies have shown that anti-AMA1 polyclonal and monoclonal antibodies block parasite invasion in vitro (27, 46, 48), as do Fab fragments and peptides that bind AMA1, ruling out inhibition due to steric hindrance or agglutination (27, 31, 47, 48). AMA1 is expressed in sporozoites, and anti-AMA1 antibodies block parasite invasion of liver cells, suggesting a role for AMA1 in both erythrocyte and hepatocyte invasion (14, 43). Immunization with AMA1 protected against parasite challenge in simian and rodent systems and passive transfer of anti-AMA1 antibodies also facilitated protection (2, 10, 11, 26, 35, 45). Interestingly, immunization with reduced and alkylated AMA1 was not protective, indicating that a protective immune response depended on correct disulfide bond formation (2). Inhibitory binders are therefore likely to target disulfide-dependent epitopes. AMA1 is also a target of the naturally acquired immune response, and anti-AMA1 antibodies isolated from individuals exposed to malaria block parasite invasion in vitro (21, 48). All of these data clearly indicate that AMA1 is a molecule essential to the parasite and a prime target for the development of strategies to combat malaria.
Although lacking the major structural polymorphisms of some malarial antigens, for example, MSP2 (44), AMA1 exhibits allelic variation that is indicative of immune selection (39, 40). This variation influences the effectiveness of the immune response, as inhibition and protection are strain specific (11, 18, 21, 39, 40). For development of an effective vaccine, immunization would need to be effective against multiple parasite strains. Simultaneous inoculation with two alleles has achieved some success in inducing dual strain protection (26). There is some evidence that AMA1 is involved in reorientation of the merozoite on the erythrocyte surface and that inhibitory antibodies may disrupt AMA1 processing, but the precise role of AMA1 in merozoite invasion, and hence the mechanism of inhibition by anti-AMA1 reagents, remains elusive (13, 34).
In this study we describe a novel 20-mer peptide, isolated from a phage-displayed random peptide library, that binds to AMA1 and is a much more potent inhibitor of merozoite invasion than the previously identified F1 peptide (31). This peptide binds in a strain-specific, reduction-sensitive manner and, together with other AMA1 binders, identifies a “hot spot” or unique region on AMA1 that is targeted by molecules that inhibit invasion of host red blood cells. Structural studies have identified the presence of a turn-like conformation in the R1 peptide which is also present in the F1 peptide and which may represent an important feature for AMA1 binding. An aim of this work was to use peptides and other inhibitory AMA1 binders as probes to identify functionally important regions of AMA1 and common features of inhibitory AMA1 binders. Ultimately, characterizing the interaction between AMA1 and inhibitory binders may improve our understanding of the role of AMA1 in invasion and aid in development of an effective vaccine or pharmaceuticals targeting AMA1.
MATERIALS AND METHODS
Materials.
Escherichia coli strains used were TG1, CJ236, and K91. The helper phage used was M13KO7 (Amersham Biosciences). Anti-AMA1 monoclonal antibodies (MAbs) 1F9 and 5G8, anti-AMA1 polyclonal rabbit antiserum and recombinant protein AMA1, merozoite surface protein 2 (MSP2) and MSP3, and ring-infected erythrocyte surface antigen (RESA) were produced as described previously (1, 9, 21). Anti-AMA1 MAb 4G2 was provided by Alan Thomas (Biomedical Primate Research Centre, Rijswijk, The Netherlands). Peptides were synthesized by AusPep Pty. Ltd. (Melbourne, Australia) to >70% purity. 1H nuclear magnetic resonance (NMR) spectra of the peptide (see below) showed a single set of resonances, indicating that any impurities were not peptidic.
Selection of AMA1-binding peptides.
A 20-mer phage-displayed random peptide library was constructed that contained >108 individual clones (7). Selection for phage displaying peptides with an affinity for AMA1 was carried out essentially as described elsewhere (1). Briefly, AMA1 (10 μg/ml) was immobilized on 96-well immunoplates (Nunc) and exposed to the naïve library. After unbound phage was washed away, bound phage were eluted using 0.1 M glycine-HCl (pH 2.2) and neutralized with 2 M Tris (pH 9). This panning procedure was repeated four times to enrich for phage displaying AMA1-specific peptides, with washing stringency increasing in each round.
PCR and DNA sequencing.
PCR was used to amplify the DNA insert coding for the 20-mer peptide, and DNA analysis was performed using automated dye terminator cycle sequencing (SUPAMAC; Centre for Proteome Research and Gene Product Mapping, Eveleigh, New South Wales, Australia), as described previously (31).
Determining phage titer.
Phages were serially diluted 10-fold in culture media, K91 E. coli cells were added and incubated for 30 min to allow phage infection, and 20 μl of 10−7 to 10−12 dilutions were plated out on selective media and incubated overnight at 37°C. Colonies produced were counted, and the phage concentration was subsequently determined in CFU per milliliter.
ELISA.
96-well microtiter plates (Maxisorb [Nunc] or preblocked Reacti-Bind neutravidin [high binding capacity; Pierce]) were coated with 2 to 5 μg/ml of target molecule diluted in coating buffer (15 mM Na2CO3, 34 mM NaHCO3, pH 9.6) or enzyme-linked immunosorbent assay (ELISA) wash buffer (phosphate-buffered saline [PBS], 0.05% Tween 20, 0.1% bovine serum albumin [BSA]) and incubated overnight at 4°C. Unbound protein or peptide was removed by washing with PBS, and any unbound surfaces were blocked using 10% milk powder in PBS. A total of 100 μl/well of the primary probe (phage, recombinant AMA1, MAb 4G2, or MAb 1F9) was then added and incubated at room temperature for 1 h with shaking. Bound phage was detected using anti-M13 antibodies conjugated to horseradish peroxidase (HRP) (Amersham Biosciences) (1:5,000). AMA1 was detected using polyclonal anti-AMA1 rabbit sera (1:2,000) followed by an anti-rabbit immunoglobulin G (IgG) HRP conjugate (1:1,000; Amersham Biosciences). MAbs 4G2 and 1F9 were detected using anti-rat (1:2,000; Jackson ImmunoResearch Laboratories) or anti-mouse (1:1,000; Amersham Biosciences) IgG HRP conjugate, respectively. Binding was visualized using either tetra-methylbenzidine (Sigma) or o-phenylenediamine (Sigma), and the absorbance was read at 450 or 490 nm, respectively. Inhibition ELISAs were carried out essentially as described above except that various concentrations of competing peptide were incubated with the primary probe for 15 min before being applied to the wells. All assays were performed in duplicate, and error bars indicate the standard deviation.
Western blotting.
Recombinant AMA1 and MSP3 (0.5 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 10% gels under nonreducing conditions and transferred to a polyvinylidene difluoride transfer membrane (PVDF-Plus; Millipore). Membranes were blocked overnight with 5% skim milk powder. After rinsing with PBS-0.05% Tween 20, membranes were probed with phage displaying the relevant peptide (1011 CFU/ml) and incubated at room temperature with gentle shaking for 1 h. Membranes were then rinsed with PBS-0.05% Tween 20. The secondary probe, anti-M13 monoclonal antibody conjugated to HRP (1:5,000; Amersham Biosciences) was then added and incubated for 1 h at room temperature. Following washes, detection was performed with enhanced chemiluminescent substrate (Pierce).
Immunofluorescence assay.
The 3D7 parasite line was cultured essentially as described previously (49). Parasites were synchronized by sorbitol lysis of all but ring stage parasites (29). Smears of synchronized schizont-stage 3D7 parasites were fixed in ice-cold 50% methanol-50% acetone and probed with 100 μl of R1 with a biotin moiety attached to the C terminus (10 μg/ml) and an anti-AMA1 monoclonal antibody, 1F9 (5 μg/ml). Slides were incubated for 45 min at room temperature and washed five times with 1.5 ml PBS-0.5% BSA (Sigma). Streptavidin-fluorescein conjugate (Molecular Probes) (5 μg/ml) was used to detect bound biotinylated R1 peptide. Bound MAb 1F9 was detected using 2 μg/ml anti-mouse IgG Alexa Fluor 568 (Molecular Probes). Following washes, fluorescence was visualized using fluorescence microscopy (Olympus BX50) with a digital camera.
Invasion inhibition assay.
The 3D7, D10, HB3, and W2mef parasite lines were cultured as described previously (49). Peptides were serially diluted in PBS, and 50 μl was added to sterile flat-bottomed microtiter wells (Nunc). To each well, 50 μl of complete culture media and 100 μl of synchronized schizont-stage parasites were added (4% hematocrit, 0.3% parasitemia) and plates were gassed, covered, and incubated for 40 to 42 h at 37°C. A 50-μl volume of parasites was then washed with 250 μl ice-cold PBS and incubated at −20°C for at least 2 h. Following thawing, relative parasitemia levels were determined by assaying for parasite lactate dehydrogenase activity (5, 26). Briefly, 100 μl of lactate dehydrogenase assay buffer (0.1 M Tris [pH 8.0] containing 50 mM sodium l-lactate, 0.25% Triton X-100, 5 μg of 3-acetylpyridine adenine dinucleotide, 0.1 units of diaphorase, and 20 μg of Nitro Blue Tetrazolium) (all reagents were from Sigma) was added to each well and incubated for 30 to 45 min in the dark at room temperature, with shaking. Absorbance was measured at 650 nm, and percent inhibition of invasion was calculated as follows:
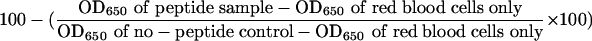 |
where OD650 is optical density at 650 nm. Assays were done in triplicate, and error bars represent the standard deviations.
Phage display of R1 fragments using the phagemid system.
Overlapping fragments of the R1 peptide were displayed on the surface of phage by use of a phagemid system (4). Mutagenesis was achieved essentially as previously described (42). Briefly, single-stranded uracilated pHENH6 vector was purified from CJ236 E. coli cells. An oligonucleotide (Genesearch) encoding the desired fragment was annealed to the DNA template and used to prime synthesis of double-stranded DNA. A 2-μl volume of double-stranded product was used to electroporate 38 μl of electrocompetent TG1 E. coli cells, and colonies harboring the desired mutant were identified by DNA sequencing. The addition of helper phage (M13K07; Amersham Biosciences) enabled production of phagemid displaying either full-length or fragmented R1 peptide.
NMR spectroscopy and structure determination.
The R1 peptide was dissolved to a final concentration of approximately 0.7 mM in 300 μl of 95% H2O-5% 2H2O containing 10 mM sodium acetate. The pH was adjusted to 4.5. Spectra were recorded at 5°C on Bruker AMX-500 and DRX-600 spectrometers with two-dimensional homonuclear total correlation spectroscopy (TOCSY) and double-quantum-filtered correlation spectroscopy (DQF-COSY) spectra at 500 MHz and a nuclear Overhauser effect (NOE) spectroscopy spectrum with a 250-ms mixing time at 600 MHz. A series of one-dimensional spectra over the temperature range 5 to 30°C, at 5°C intervals, was also collected to measure amide temperature coefficients. The water resonance was suppressed using the WATERGATE pulse sequence (38). Amide exchange rates were monitored by dissolving freeze-dried peptide in 2H2O at pH 4.9 and then recording a series of one-dimensional spectra at 5°C followed by 60-ms TOCSY and 250-ms NOE spectroscopy spectra at 600 MHz. Diffusion measurements were performed using a pulsed-field-gradient longitudinal eddy-current delay pulse sequence (12, 15), as implemented by Yao et al. (53). Spectra were processed using XWINNMR (version 3.5; Bruker Biospin) and analyzed using XEASY (version 1.3.13). All spectra were referenced to an impurity peak at 0.15 ppm.
The 3JHNHA coupling constants were measured from a DQF-COSY spectrum and converted to dihedral restraints as follows: for 3JHNHα > 8 Hz, φ = −120 ± 40°; for 3JHNHα < 6 Hz, φ = −60 ± 30°. If a positive φ angle could be excluded on the basis of NOE data (32), φ angles were restricted to the range −180 to 0°. No χ1 angle restraints and no hydrogen bonds were included as structural restraints. For structure calculations, intensities of NOE cross-peaks were measured in XEASY and calibrated using the CALIBA macro of the program CYANA (version 1.0.6) (20). Initial structures were calculated using torsion angle dynamics in CYANA (20). After the structures were optimized for a low-target function, the final constraint set was then used to calculate a new family of 100 structures by use of the standard simulated annealing script supplied with Xplor-NIH (41). A box of water was built around the peptide structure, which was then energy minimized. The 20 lowest-energy structures were selected for analysis in PROCHECK-NMR (30) and MOLMOL (28). Structural figures were prepared using MOLMOL and InsightII (Accelrys, San Diego, Calif.). The final structures had no experimental distance violations greater than 0.2 Å or dihedral angle violations greater than 5o.
RESULTS
When a library of phages displaying more than 5 × 108 different 20-mer peptides was panned on immobilized AMA1, four rounds of panning were sufficient to enrich a pool of phage with affinity for AMA1 (Fig. 1A). This phage pool showed little affinity for several unrelated proteins and was specific for PfAMA1, as it failed to bind to P. chabaudi adami AMA1 (PcAMA1), which shares 52% amino acid sequence identity with PfAMA1 (Fig. 1B). Sequence analysis of the DNA inserts from 20 individual clones from the final round of panning identified two unique peptide sequences (Fig. 2). In both phage ELISA and phage immunoblotting, R1 (VFAEFLPLFSKFGSRMHILK) and R3 (PVLRSGRCAELIQIGFRCRA) bound specifically to PfAMA1 and not to any of the other proteins tested (Fig. 2).
FIG. 1.

Selection of AMA1-binding peptides. (A) Equal amounts of phage (1 × 1011 CFU/ml) after each round of panning (round 0 [Rd 0] to Rd 4) binding to immobilized PfAMA1. Phage displaying peptides with an affinity for AMA1 are enriched after round 4. (B) Pooled phages, after the fourth round of panning, were incubated with wells coated with PfAMA1, PcAMA1, RESA, BSA, or MAb 1F9 or left uncoated. Round-4 phages are specific for PfAMA1.
FIG. 2.

R1 and R3 peptides are specific for AMA1. (A) Two clones, R1 and R3, were identified from the pooled round-4 phages. R1 and R3 phages were incubated with wells coated with AMA1, MSP3, MSP2, RESA, and BSA and were found to be specific for AMA1. (B) Volumes (0.5 μg each) of recombinant AMA1 and MSP3 were transferred to polyvinylidene difluoride membranes following sodium dodecyl sulfate-polyacrylamide gel electrophoresis and probed with R1 (left panel) and R3 (right panel) phage. Both R1 and R3 phages bind specifically to AMA1.
Since the R1 peptide exhibited slightly superior binding to AMA1 it was chosen for further analysis. R1 was synthesized and shown to retain its affinity for AMA1, as there was a dose-dependent reduction of binding of phage-displayed R1 to AMA1 when the peptide was included in the assay (Fig. 3A). R1 has a 50% inhibitory concentration of 200 nM in this assay format, and a peptide corresponding to a mutated form of R1 (R1[s]) showed no inhibition of the binding of phage-displayed R1 (Fig. 3A). R1 also inhibited the binding of phage displaying the 15-residue peptide F1 (GWRLLGFGPASSFSM), which has previously been shown to bind specifically to AMA1 (Fig. 3B) (31). Indeed, R1 was at least 10-fold more effective at inhibiting phage-displayed F1 peptide than synthetic F1 itself. Consistent with these data, R1 with a biotin moiety attached to the C or N terminus of the peptide was able to capture AMA1 in a dose-dependent manner when immobilized on neutravidin-coated plastic (Fig. 3C). Interestingly, R1 biotinylated at the N terminus was a weaker AMA1 binder, perhaps reflecting the fact that when peptides were isolated they were fused via their C terminus to the phage coat protein, and thus this region of the peptide is more amenable to modification than the N terminus. R1 was also a more potent AMA1 capture agent than C-terminally biotinylated F1 peptide. Taken together, these data suggest that R1 binds to AMA1 with higher affinity than F1 and that both peptides recognize similar regions on AMA1.
FIG. 3.

Synthetic R1 peptide binds AMA1. (A) Binding of R1 phage to wells coated with AMA1 in the presence of increasing concentrations of synthetic R1 peptide and a synthetic mutated version of the R1 peptide (R1[s]). R1 peptide binds AMA1, but the mutated version does not. (B) The synthetic peptides R1, F1, and F1(s), a scrambled version of the F1 peptide, were tested for their ability to block binding of F1 phage to AMA1. R1 peptide inhibits the interaction between AMA1 and F1 phage more effectively than F1 peptide itself, indicating that R1 and F1 peptides bind similar sites on AMA1. (C) Recombinant AMA1 was incubated with wells coated with R1 and F1 peptides biotinylated at the C or N terminus. AMA1 binds more strongly to C-terminally immobilized R1 peptide.
A comparison of R1 binding to recombinant AMA1 from four different lines of P. falciparum demonstrated that R1 recognizes AMA1 from the D10 line of P. falciparum, as well as 3D7 PfAMA1, the form of the antigen used for panning. R1 had little affinity for AMA1 from HB3 or W2mef lines of parasites (Fig. 4A). AMA1 from 3D7 and D10 share >98% identity but are more distantly related to either HB3- or W2mef-derived AMA1. This suggests that the polymorphisms previously documented in AMA1 studies (33) play a role in binding not only antibodies but also phage-derived peptides. Importantly, the lack of binding to reduced and alkylated 3D7 PfAMA1 suggested that R1 recognizes a conformational epitope on the AMA1 surface (Fig. 4A). R1, which was selected on recombinant AMA1, also bound to the parasite antigen. When R1 binding was analyzed by immunofluorescence microscopy a punctate fluorescence pattern typical for AMA1 in the micronemes was observed. The staining with R1 partially colocalized with 1F9, a previously described MAb to AMA1 (Fig. 4B).
FIG. 4.

R1 binds strain-specifically and recognizes the parasite antigen. (A) Phage displaying the R1 peptide binding to immobilized AMA1 derived from a number of parasite lines. Clearly, AMA1 recognition is highly specific, as R1 phage can only interact with the 3D7 and D10 PfAMA1 lines. R&A AMA1, reduced and alkylated AMA1. (B) Immunofluorescence assays showing biotinylated R1 peptide binding to parasite-infected red blood cells. The punctate fluorescence pattern characteristic of AMA1 binding shows partial colocalization with anti-AMA1 MAb 1F9.
Evidence that these molecules bind to an overlapping site comes from the observation that R1 peptide was able to partially block the binding of MAbs 4G2 and 1F9 to AMA1 (Fig. 5A and B). Moreover, the almost complete inhibition of phage displaying R1 binding to AMA1 by MAbs 4G2 and 1F9 also argues for a common binding footprint for these molecules. Little effect was observed following the addition of anti-AMA1 MAb 5G8, an antibody that binds to a linear epitope in the prodomain of AMA1 (data not shown) (9). Since MAb 4G2 and F1 peptide have been reported to block merozoite invasion (27, 31) and recent data indicate that MAb 1F9 also inhibits this process (A. M. Coley, K. Parisi, R. Masciantonio, J. Hoeck, J. L. Cesey, V. Murphy, K. S. Harris, R. F. Anders, and M. Foley, manuscript in preparation), R1 should also prevent merozoite invasion of erythrocytes. Incubation of soluble R1 with P. falciparum parasites in vitro resulted in a dramatic decrease in the invasion of host erythrocytes by 3D7 and D10 merozoites (Fig. 5C). Consistent with the binding data, R1 had little impact on invasion of the HB3 and W2mef parasite lines. Inhibition of invasion by R1 was dose dependent, the concentration of peptide that inhibited 50% of merozoites being 4 μM. Thus, R1 is at least fivefold more potent than the F1 peptide (31) at inhibiting merozoite invasion.
FIG. 5.

R1 peptide binds a site similar to that bound by inhibitory MAbs 4G2 and 1F9 and blocks parasite invasion of red blood cells. Anti-AMA1 MAbs 4G2 (0.01 μg/ml) (A) and 1F9 (0.05 μg/ml) (B) were allowed to bind immobilized AMA1 in the presence of increasing concentrations of synthetic peptide. The interaction of each of these MAbs with AMA1 is inhibited by the addition of R1 peptide. (C) Percent inhibition of invasion of the 3D7, D10, HB3, and W2mef parasite lines following addition of increasing concentrations of R1 peptide.
Since the structure of the F1 peptide has been described previously (25) it was of interest to determine the structure of R1 to identify molecular features that might correlate with inhibition of merozoite invasion. Limited chemical shift dispersion in 1H NMR spectra of R1 in aqueous solution implied that it samples a range of conformations, as might be expected for a peptide of this size that lacks disulfide or other cross-links. On the other hand, the only Pro in R1, at position 7, showed no cis-trans isomerization, implying that the conformation was constrained in at least this region of the peptide. 1H chemical shifts were assigned for all backbone and most side chain protons (see Table S1 in the supplementary material). Translational diffusion coefficients of 9.10 × 10−11 m2 s−1 at 5°C and 1.76 × 10−10 m2 s−1 at 25°C were similar, allowing for viscosity and temperature effects, to those for peptides studied by Keizer et al. (25) and Yao et al. (53), indicating that R1 was monomeric under the solution conditions used for this study. 3JHNHα coupling constants extracted from a DQF-COSY spectrum showed that only Arg15 and Ile18 had 3JHNHα > 8 Hz, while Ser14 was the only residue with 3JHNHα < 6 Hz. The NOEs were predominantly intraresidue or sequential, with very few medium-range and no long-range ( i- j > 4) NOEs. Temperature coefficients for the backbone amide protons of R1 were all >4 ppb/K (see Table S1 in the supplementary material), indicating that the amides were largely solvent exposed (6). However, amide exchange experiments implied that most amides were partially protected from solvent. All of the backbone amides were still visible after 15.5 h in a TOCSY; this was somewhat surprising, as this peptide did not adopt a well-defined structure over its entire length.
Initial structures were calculated using distance and dihedral angle restraints in CYANA (20) before optimization in Xplor-NIH (41). All structures were further refined and energy minimized in a box of water. The angular order parameter (S) measures the precision of torsion angles in a family of structures, with values of S > 0.8 generally indicating that an angle is well defined. For R1, φ and ψ angles were generally not well defined according to this parameter (see Fig. S1 in the supplementary material) but the side chain c1 angles for Leu6, Phe9, and Ile18 were exceptionally well ordered. It appears that R1 does not adopt a stable conformation over the majority of residues, as reflected in a global backbone root mean square deviation value > 2 Å and a lack of medium-range NOEs, but has regions of local structure over residues 5 to 10 and 13 to 17 (Fig. 6). When the family of structures was superimposed over residues 5 to 10 and 13 to 17, pairwise backbone root mean square deviations of 1.43 Å and 1.16 Å, respectively, were obtained (see Table S2 in the supplementary material). In the first of these structured regions, Pro7-Ser10 adopted a turn-like structure, with Leu8 and Phe9 occupying the i+1 and i+2 positions, while a slightly distorted type I β-turn encompassing Gly13-Met16 was evident in the second structured region (Fig. 6). The backbone φ and ψ angles and coupling constants for Ser14 and Arg15 and the intense amide-amide NOE between Arg15 and Met 16 supported the presence of such a turn. Pro and Gly, which occur in both structured regions of R1, are highly favored in turns (24).
FIG. 6.

Structure of R1. Stereo views of the family of the 20 lowest-energy structures, showing regions covering residues Glu4 to Lys11 (A) and Gly13 to Ile18 (B). Structures were superimposed over the backbone heavy atoms (N, Cα, C′) of residues 5 to 10 (A) and 13 to 17 (B), respectively. In both structured regions, residues in the second and third positions of the turn-like conformations are labeled (Ser14-Arg15 and Leu8-Phe9). (C) Connolly surface representation of R1. The closest-to-average structure from the family of 20 structures is shown. Hydrophobic residues Phe5, Leu6, Leu8, and Phe9 all interact with Pro7.
It is interesting that residues 1 to 5 contain an FXP motif that was also present in the type I β-turn of F1 (25). Furthermore, alanine-scanning mutagenesis of F1 peptide has identified these residues as critical for binding to AMA1 and inhibition of merozoite invasion of erythrocytes (31). The presence of a FXP motif in the turn-like conformation in R1 raised the possibility that the information for AMA1 binding in R1 may reside in the well-structured region (residues 5 to 10) of R1. To examine this possibility, a phage displaying a shorter R1 fragment containing this FXP motif (residues 5 to 14) was constructed and evaluated for binding to AMA1. In addition, two other fragments corresponding to residues 1 to 7 and 10 to 20 were also displayed on phage to achieve complete overlap of the R1 peptide. None of these three phage-displayed regions by itself was able to bind to AMA1 (Fig. 7), whereas phage displaying the intact R1 peptide demonstrated the expected high-level binding to AMA1. These data suggest that residues important for AMA1 binding are spread throughout the entire R1 peptide and that high-affinity binding cannot be achieved by a short fragment of R1.
FIG. 7.

Overlapping fragments of R1 are unable to bind AMA1. Phagemids displaying overlapping fragments of R1 were incubated with immobilized AMA1. Phagemids are standardized for peptide expression.
DISCUSSION
In this study we describe a novel peptide (R1), derived from a random 20-mer sequence library, with high affinity for the malarial antigen AMA1. This peptide was also found to inhibit merozoite invasion of malaria parasites cultured in vitro. R1 was found to have a less-well-defined global structure in solution compared with previously described AMA1-binding 15-mer peptides that also inhibited merozoite invasion (25). Interestingly, however, both R1 and F1 peptides appear to bind to the same region of AMA1 and this region overlaps with the binding site for two monoclonal antibodies that also inhibit merozoite invasion.
R1 was one of two peptides isolated after four rounds of panning of a 20-mer random peptide library on recombinant AMA1. It also bound to native AMA1 as expressed on parasites, and this interaction was highly specific in that it bound to AMA1 from the D10 and 3D7 lines of P. falciparum but not the W2mef or HB3 lines. In competition experiments the R1 peptide blocked the binding of F1 phage to AMA1 and partially blocked the binding of MAbs 4G2 and 1F9. Interestingly, both F1 and MAb 4G2 have previously been shown to have different strain specificities with respect to R1 (27, 31), indicating that, although similar, the site recognized by these AMA1 binders is not identical. Consistent with previous reports that F1 peptide and MAb 4G2 inhibit parasite invasion (27, 31) and recent evidence that MAb 1F9 is also inhibitory (Coley et al., manuscript in preparation), R1 was found to block merozoite invasion of erythrocytes. Indeed, in this study R1 was at least fivefold more potent as an inhibitor than F1. Taken together, these results argue for the presence of a region or hot spot on the surface of AMA1 that is targeted by molecules of different classes. Given that the binding of these molecules results in a decrease in merozoite invasion it can be inferred that this hot spot is critical for the function of AMA1, perhaps representing a cleft that is involved in interactions with other proteins during invasion.
As yet, no three-dimensional structure of PfAMA1 is available, and therefore it is not possible to map the binding site on the surface of the protein, although previous work has identified domain I of AMA1 as critical for the 1F9-AMA1 interaction (9). Epidemiological studies showing that certain residues in domain I are polymorphic and that those polymorphisms appear to have been selected and maintained by an inhibitory immune response suggest that these residues are at least located close to the region bound by inhibitory molecules. Thus, we suggest that the hot spot probably comprises a hydrophobic region that is necessary for some function and therefore that the residues making up this region cannot easily be changed without affecting parasite fitness, as AMA1 appears to be essential. The polymorphic residues may surround this site, and alterations of these amino acids may prevent inhibitory antibodies from binding. This would be consistent with the strain-specific nature of the inhibitory immune response against AMA1 in humans and in primates (11, 18, 21, 39, 40). Given that the polymorphic residues in AMA1 tend to cluster in domain I and to a lesser extent in domain 3 it is possible that the residues that make up the hot spot are located primarily in domain I, but residues in domain 3 may also play a role in binding inhibitory antibodies.
Thus, the region on AMA1 appears to be larger than a single epitope and may be large enough to accommodate several nonidentical but overlapping epitopes. This raises the intriguing issue of whether antibodies exist in malaria-infected individuals that bind close to this hot spot and block the binding of inhibitory antibodies. Such a situation exists for MSP1; antibodies have been described that block the binding of inhibitory antibodies to MSP1 (16, 44, 51). It has been argued that these blocking antibodies will limit the efficacy of an MSP1-based vaccine, and efforts are focused on constructing MSP1 variants lacking these epitopes (51).
R1 and F1 peptides are able to both bind to AMA1 and inhibit merozoite invasion despite the differences in sequence and overall structures. A bulky hydrophobic residue at the apex of the turn structure may also be important for the binding of both peptides. R1 contains two regions of ordered structure, Phe5-Ser10 and Gly13-His17. Pro7 appears to be the nucleus of the first region, as it makes contact with Phe5, Leu6, Leu8, and Phe9. This highly hydrophobic cluster, FLPLF, has similarities to the L(I)GFGP region of F1, which formed a well-defined β-turn (25), and indeed a turn-like conformation is observed for Pro7-Ser10 in R1. The absence of Gly in this region of R1 accounts for the fact that it does not contain the well-defined type I β-turn found in F1. Also in contrast to F1, this region of R1 does not contain all the residues essential for AMA1 binding activity, as none of the three fragments that span the R1 sequence binds to AMA1. The other structured region of R1, encompassing residues 13 to 17, is much more polar (GSRMH) compared to residues 5 to 10 and was found to contain a slightly distorted type I β-turn from Gly13-Met16. The lack of activity of truncated analogues of R1 containing either structured region alone suggests that there may be some interaction between these two structured regions. Although this interaction is not evident in solution, it may be stabilized when R1 binds to AMA1. Evidence that there may be some interaction even when free in solution comes from the amide exchange data, which indicated that, unexpectedly, most backbone amides of R1 were protected from rapid exchange with solvent. Generally speaking, small linear peptides such as R1 would be expected to have no protected backbone amides. Further studies will be directed at probing the roles of individual residues in these structured regions of R1 on its structure and affinity for AMA1 as well as at defining the structure of R1 bound to AMA1 or fragments thereof.
Using the information obtained in this study it may be possible to develop novel therapeutic molecules that would inhibit invasion of merozoites by targeting AMA1. Such molecules could be protein based, consisting of an inhibitory core and a larger scaffold, such as the Fc portion of an immunoglobulin molecule, to manipulate properties such as bioavailability and residence time in the bloodstream. Alternatively, our observations could help in the rational design of peptidomimetics based on the turn-like structures that may result in a relatively inexpensive antimalarial therapy. The peptides and MAbs that bind to the AMA1 hot spot could be used to analyze the fine specificity of the antibody response following malaria infection or immunization with AMA1-containing vaccines. Furthermore, characterization of this hot spot may enable development of a vaccine incorporating this region of AMA1, skewing the immune system towards the production of inhibitory antibodies.
Supplementary Material
Acknowledgments
This work was supported in part by the National Health and Medical Research Council of Australia and the World Health Organization and a grant from the National Institutes of Health (NIH R01AI59229).
We thank Alan Thomas for kindly providing MAb 4G2. We thank Ronelle Welton and Vince Murphy for providing various strains of recombinant AMA1.
Editor: W. A. Petri, Jr.
Footnotes
Supplementary material for this article may be found at http://iai.asm.org.
REFERENCES
- 1.Adda, C. G., L. Tilley, R. F. Anders, and M. Foley. 1999. Isolation of peptides that mimic epitopes on a malarial antigen from random peptide libraries displayed on phage. Infect. Immun. 67:4679-4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anders, R. F., P. E. Crewther, S. Edwards, M. Margetts, M. Matthew, B. Pollock, and D. Pye. 1998. Immunization with recombinant AMA1 protects mice against infection with Plasmodium chabaudi. Vaccine 16:240-247. [DOI] [PubMed] [Google Scholar]
- 3.Bannister, L. H., J. M. Hopkins, A. R. Dluzewski, G. Margos, I. T. Williams, M. J. Blackman, C. H. Kocken, A. W. Thomas, and G. H. Mitchell. 2003. Plasmodium falciparum apical membrane antigen 1 (PfAMA1) is translocated within micronemes along subpellicular microtubules during merozoite development. J. Cell Sci. 116:3825-3834. [DOI] [PubMed] [Google Scholar]
- 4.Barbas, C. F., III, D. R. Burton, J. K. Scott, and G. J. Silverman. 2001. Phage display: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 5.Basco, L. K., F. Marquet, M. M. Makler, and J. Lebras. 1995. Plasmodium falciparum and Plasmodium vivax—lactate dehydrogenase activity and its application for in vitro drug susceptibility assay. Exp. Parasitol. 80:260-271. [DOI] [PubMed] [Google Scholar]
- 6.Baxter, N. J., and M. P. Williamson. 1997. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9:359-369. [DOI] [PubMed] [Google Scholar]
- 7.Casey, J. L., A. M. Coley, R. F. Anders, V. J. Murphy, K. S. Humberstone, A. W. Thomas, and M. Foley. 2004. Antibodies to malaria peptide mimics inhibit Plasmodium falciparum invasion of erythrocytes. Infect. Immun. 72:1126-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chitnis, C. E., and M. J. Blackman. 2000. Host cell invasion by malaria parasites. Parasitol. Today 16:411-415. [DOI] [PubMed] [Google Scholar]
- 9.Coley, A. M., N. V. Campanale, J. L. Casey, A. N. Hodder, P. E. Crewther, R. F. Anders, L. M. Tilley, and M. Foley. 2001. Rapid and precise epitope mapping of monoclonal antibodies against Plasmodium falciparum AMA1 by combined phage display of fragments and random peptides. Protein Eng. 14:691-698. [DOI] [PubMed] [Google Scholar]
- 10.Collins, W. E., R. F. Anders, M. Pappaioanou, G. H. Campbell, G. V. Brown, D. J. Kemp, R. L. Coppel, J. C. Skinner, P. M. Andrysiak, and J. M. Favaloro. 1986. Immunization of Aotus monkeys with recombinant proteins of an erythrocyte surface antigen of Plasmodium falciparum. Nature 323:259-262. [DOI] [PubMed] [Google Scholar]
- 11.Crewther, P. E., M. L. Matthew, R. H. Flegg, and R. F. Anders. 1996. Protective immune responses to apical membrane antigen 1 of Plasmodium chabaudi involve recognition of strain-specific epitopes. Infect. Immun. 64:3310-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dingley, A. J., J. P. Mackay, B. E. Chapman, M. B. Morris, P. W. Kuchel, B. D. Hambly, and G. F. King. 1995. Measuring protein self-association using pulsed-field-gradient NMR spectroscopy: application to myosin light chain 2. J. Biomol. NMR 6:321-328. [DOI] [PubMed] [Google Scholar]
- 13.Dutta, S., J. D. Haynes, J. K. Moch, A. Barbosa, and D. E. Lanar. 2003. Invasion-inhibitory antibodies inhibit proteolytic processing of apical membrane antigen 1 of Plasmodium falciparum merozoites. Proc. Natl. Acad. Sci. USA 100:12295-12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Florens, L., M. P. Washburn, J. D. Raine, R. M. Anthony, M. Grainger, J. D. Haynes, J. K. Moch, N. Muster, J. B. Sacci, D. L. Tabb, A. A. Witney, D. Wolters, Y. Wu, M. J. Gardner, A. A. Holder, R. E. Sinden, J. R. Yates, and D. J. Carucci. 2002. A proteomic view of the Plasmodium falciparum life cycle. Nature 419:520-526. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs, S. J., and C. S. Johnson. 1991. A PFG NMR experiment for accurate diffusion and flow studies in the presence of eddy currents. J. Magn. Reson. 93:395-402. [Google Scholar]
- 16.Guevara Patino, J. A., A. A. Holder, J. S. McBride, and M. J. Blackman. 1997. Antibodies that inhibit malaria merozoite surface protein 1 processing and erythrocyte invasion are blocked by naturally acquired human antibodies. J. Exp. Med. 186:1689-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Healer, J., S. Crawford, S. Ralph, G. McFadden, and A. F. Cowman. 2002. Independent translocation of two micronemal proteins in developing Plasmodium falciparum merozoites. Infect. Immun. 70:5751-5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Healer, J., V. Murphy, A. N. Hodder, R. Masciantonio, A. W. Gemmill, R. F. Anders, A. F. Cowman, and A. Batchelor. 2004. Allelic polymorphisms in apical membrane antigen 1 are responsible for evasion of antibody-mediated inhibition in Plasmodium falciparum. Mol. Microbiol. 52:159-168. [DOI] [PubMed] [Google Scholar]
- 19.Hehl, A. B., C. Lekutis, M. E. Grigg, P. J. Bradley, J. F. Dubremetz, E. Ortega-Barria, and J. C. Boothroyd. 2000. Toxoplasma gondii homologue of Plasmodium apical membrane antigen 1 is involved in invasion of host cells. Infect. Immun. 68:7078-7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrmann, T., P. Güntert, and K. Wüthrich. 2002. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319:209-227. [DOI] [PubMed] [Google Scholar]
- 21.Hodder, A. N., P. E. Crewther, and R. F. Anders. 2001. Specificity of the protective antibody response to apical membrane antigen 1. Infect. Immun. 69:3286-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodder, A. N., P. E. Crewther, M. L. Matthew, G. E. Reid, R. L. Moritz, R. J. Simpson, and R. F. Anders. 1996. The disulfide bond structure of Plasmodium apical membrane antigen 1. J. Biol. Chem. 271:29446-29452. [DOI] [PubMed] [Google Scholar]
- 23.Howell, S. A., I. Wells, S. L. Fleck, C. Kettleborough, C. R. Collins, and M. J. Blackman. 2003. A single malaria merozoite serine protease mediates shedding of multiple surface proteins by juxtamembrane cleavage. J. Biol. Chem. 278:23890-23898. [DOI] [PubMed] [Google Scholar]
- 24.Hutchinson, E. G., and J. M. Thornton. 1994. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 3:2207-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keizer, D. W., L. A. Miles, F. Li, M. Nair, R. F. Anders, A. M. Coley, M. Foley, and R. S. Norton. 2003. Structures of phage-display peptides that bind to the malarial surface protein, apical membrane antigen 1, and block erythrocyte invasion. Biochemistry 42:9915-9923. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy, M. C., J. Wang, Y. L. Zhang, A. P. Miles, F. Chitsaz, A. Saul, C. A. Long, L. H. Miller, and A. W. Stowers. 2002. In vitro studies with recombinant Plasmodium falciparum apical membrane antigen 1 (AMA1): production and activity of an AMA1 vaccine and generation of a multiallelic response. Infect. Immun. 70:6948-6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kocken, C. H., A. M. van der Wel, M. A. Dubbeld, D. L. Narum, F. M. van de Rijke, G. J. van Gemert, X. van der Linde, L. H. Bannister, C. Janse, A. P. Waters, and A. W. Thomas. 1998. Precise timing of expression of a Plasmodium falciparum-derived transgene in Plasmodium berghei is a critical determinant of subsequent subcellular localization. J. Biol. Chem. 273:15119-15124. [DOI] [PubMed] [Google Scholar]
- 28.Koradi, R., M. Billeter, and K. Wüthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51-55, 29-32. [DOI] [PubMed] [Google Scholar]
- 29.Lambros, C., and J. P. Vanderberg. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418-420. [PubMed] [Google Scholar]
- 30.Laskowski, R. A., J. A. Rullmannn, M. W. MacArthur, R. Kaptein, and J. M. Thornton. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8:477-486. [DOI] [PubMed] [Google Scholar]
- 31.Li, F., A. Dluzewski, A. M. Coley, A. Thomas, L. Tilley, R. F. Anders, and M. Foley. 2002. Phage-displayed peptides bind to the malarial protein apical membrane antigen 1 and inhibit the merozoite invasion of host erythrocytes. J. Biol. Chem. 277:50303-50310. [DOI] [PubMed] [Google Scholar]
- 32.Ludvigsen, S., and F. M. Poulsen. 1992. Positive theta-angles in proteins by nuclear magnetic resonance spectroscopy. J. Biomol. NMR 2:227-233. [DOI] [PubMed] [Google Scholar]
- 33.Marshall, V. M., L. Zhang, R. F. Anders, and R. L. Coppel. 1996. Diversity of the vaccine candidate AMA1 of Plasmodium falciparum. Mol. Biochem. Parasitol. 77:109-113. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell, G. H., A. W. Thomas, G. Margos, A. R. Dluzewski, and L. H. Bannister. 2004. Apical membrane antigen 1, a major malaria vaccine candidate, mediates the close attachment of invasive merozoites to host red blood cells. Infect. Immun. 72:154-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narum, D. L., S. A. Ogun, A. W. Thomas, and A. A. Holder. 2000. Immunization with parasite-derived apical membrane antigen 1 or passive immunization with a specific monoclonal antibody protects BALB/c mice against lethal Plasmodium yoelii yoelii YM blood-stage infection. Infect. Immun. 68:2899-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narum, D. L., and A. W. Thomas. 1994. Differential localization of full-length and processed forms of Pf83/AMA1 an apical membrane antigen of Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 67:59-68. [DOI] [PubMed] [Google Scholar]
- 37.Peterson, M. G., V. M. Marshall, J. A. Smythe, P. E. Crewther, A. Lew, A. Silva, R. F. Anders, and D. J. Kemp. 1989. Integral membrane protein located in the apical complex of Plasmodium falciparum. Mol. Cell. Biol. 9:3151-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piotto, M., V. Saudek, and V. Sklenar. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2:661-665. [DOI] [PubMed] [Google Scholar]
- 39.Polley, S. D., and D. J. Conway. 2001. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics 158:1505-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polley, S. D., W. Chokejindachai, and D. J. Conway. 2003. Allele frequency-based analyses robustly map sequence sites under balancing selection in a malaria vaccine candidate antigen. Genetics 165:555-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwieters, C. D., J. J. Kuszewski, N. Tjandra, and G. M. Clore. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160:65-73. [DOI] [PubMed] [Google Scholar]
- 42.Sidhu, S. S., H. B. Lowman, B. C. Cunningham, and J. A. Wells. 2000. Phage display for selection of novel binding peptides. Methods Enzymol. 328:333-363. [DOI] [PubMed] [Google Scholar]
- 43.Silvie, O., J. F. Franetich, S. Charrin, M. S. Mueller, A. Siau, M. Bodescot, E. Rubinstein, L. Hannoun, Y. Charoenvit, C. H. Kocken, A. W. Thomas, G. J. van Gemert, R. W. Sauerwein, M. J. Blackman, R. F. Anders, G. Pluschke, and D. Mazier. 2004. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J. Biol. Chem. 279:9490-9496. [DOI] [PubMed] [Google Scholar]
- 44.Smythe, J. A., R. L. Coppel, K. P. Day, R. K. Martin, A. M. Oduola, D. J. Kemp, and R. F. Anders. 1991. Structural diversity in the Plasmodium falciparum merozoite surface antigen 2. Proc. Natl. Acad. Sci. USA 88:1751-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stowers, A. W., M. C. Kennedy, B. P. Keegan, A. Saul, C. A. Long, and L. H. Miller. 2002. Vaccination of monkeys with recombinant Plasmodium falciparum apical membrane antigen 1 confers protection against blood-stage malaria. Infect. Immun. 70:6961-6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor, H. M., M. Grainger, and A. A. Holder. 2002. Variation in the expression of a Plasmodium falciparum protein family implicated in erythrocyte invasion. Infect. Immun. 70:5779-5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas, A. W., J. A. Deans, G. H. Mitchell, T. Alderson, and S. Cohen. 1984. The Fab fragments of monoclonal IgG to a merozoite surface antigen inhibit Plasmodium knowlesi invasion of erythrocytes. Mol. Biochem. Parasitol. 13:187-199. [DOI] [PubMed] [Google Scholar]
- 48.Thomas, A. W., J. F. Trape, C. Rogier, A. Goncalves, V. E. Rosario, and D. L. Narum. 1994. High prevalence of natural antibodies against Plasmodium falciparum 83-kilodalton apical membrane antigen (Pf83/AMA1) as detected by capture-enzyme-linked immunosorbent assay using full-length baculovirus recombinant Pf83/AMA1. Am. J. Trop. Med. Hyg. 51:730-740. [DOI] [PubMed] [Google Scholar]
- 49.Trager, W., and J. B. Jensen. 1976. Human malaria parasites in continuous culture. Science 193:673-675. [DOI] [PubMed] [Google Scholar]
- 50.Triglia, T., J. Healer, S. R. Caruana, A. N. Hodder, R. F. Anders, B. S. Crabb, and A. F. Cowman. 2000. Apical membrane antigen 1 plays a central role in erythrocyte invasion by Plasmodium species. Mol. Microbiol. 38:706-718. [DOI] [PubMed] [Google Scholar]
- 51.Uthaipibull, C., B. Aufiero, S. E. Syed, B. Hansen, J. A. Guevara Patino, E. Angov, I. T. Ling, K. Fegeding, W. D. Morgan, C. Ockenhouse, B. Birdsall, J. Feeney, J. A. Lyon, and A. A. Holder. 2001. Inhibitory and blocking monoclonal antibody epitopes on merozoite surface protein 1 of the malaria parasite Plasmodium falciparum. J. Mol. Biol. 307:1381-1394. [DOI] [PubMed] [Google Scholar]
- 52.Waters, A. P., A. W. Thomas, J. A. Deans, G. H. Mitchell, D. E. Hudson, L. H. Miller, T. F. McCutchan, and S. Cohen. 1990. A merozoite receptor protein from Plasmodium knowlesi is highly conserved and distributed throughout Plasmodium. J. Biol. Chem. 265:17974-17979. [PubMed] [Google Scholar]
- 53.Yao, S., G. J. Howlett, and R. S. Norton. 2000. Peptide self-association in aqueous trifluoroethanol monitored by pulsed field gradient NMR diffusion measurements. J. Biomol. NMR 16:109-119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.