

Abstract
Transcription by RNA polymerase can stimulate localized DNA supercoiling in Escherichia coli. In vivo, there is extensive experimental support for a “twin-domain” model in which positive DNA supercoils are generated ahead of a translocating RNA polymerase complex and negative supercoils are formed behind it. Negative supercoils accumulate in the template DNA because the positive supercoils are preferentially removed by cellular topoisomerase action. Yet, in vitro, clear and convincing support for the twin-domain mechanism has been lacking. In this article, we reconcile this inconsistency by showing that, in a defined in vitro system with plasmid DNA templates, a variety of sequence-specific DNA-binding proteins, such as the bacteriophage λ O replication initiator or the E. coli lactose or galactose repressors, strikingly stimulate transcription-coupled DNA supercoiling. We demonstrate further that this stimulation requires the presence in the DNA template of a recognition sequence for the relevant DNA-binding protein and depends on the production of long RNA chains by an RNA polymerase. Our data are most consistent with a model in which specific DNA-binding proteins facilitate a twin-domain mechanism to enhance DNA supercoiling during transcription. More precisely, we suggest that some nucleoprotein complexes, perhaps those that contain sharply bent DNA, can form barriers that impede the diffusion and merger of independent chromosomal supercoil domains. Localization of DNA supercoils by nucleoprotein complexes may serve as a general mechanism for modulating DNA transactions that are sensitive to DNA superhelicity.
A number of DNA transactions in Escherichia coli are greatly facilitated by negative supercoiling of the bacterial genome (1). For example, opening of the DNA duplex during initiation of transcription at certain promoters and during initiation of DNA replication at the chromosomal origin is energetically favored when the DNA template is negatively supercoiled (see refs. 1 and 2 for reviews). The global DNA supercoiling level in E. coli is principally set through the opposing actions of DNA gyrase and DNA topoisomerase I (Topo I) (3, 4), although recent findings indicate that DNA Topo IV also participates in the relaxation of negatively supercoiled DNA (5).
Several studies have demonstrated that the process of transcription of cellular DNA can enhance negative DNA supercoiling locally (3, 6–8). Liu and Wang formulated the “twin-supercoiled-domain” model (9) to explain this effect of transcription on DNA supercoiling. Extensive experimental support has been obtained in vivo (reviewed in refs. 1, 10, and 11) for the idea that positive DNA supercoils are generated in front of an advancing transcription complex while an equivalent number of negative DNA supercoils are formed behind it. Formation of these twin supercoiled domains occurs only when the translocating RNA polymerase and associated macromolecules are unable to rotate freely about the DNA helical axis. Under this condition, the template DNA rotates instead. The available evidence indicates that DNA gyrase rapidly converts positive DNA supercoils generated by transcription into negative supercoils (1, 6, 12).
More recent studies of the mechanism of transcription-coupled DNA supercoiling in vitro, however, have yielded results that seemingly are inconsistent with the twin-domain model. Hypernegatively supercoiled [(− −) SC] plasmid DNA isolated from topA mutants was found to contain R loops and, moreover, formation of (− −) SC DNA during transcription in vitro in the presence of DNA gyrase was strongly linked to the production of R loops (13).
In this article, we show that the interaction of site-specific DNA-binding proteins with a DNA template can evoke a remarkable stimulation of negative DNA supercoiling during transcription. Our data suggests that such DNA-binding proteins enhance DNA supercoiling by facilitating the twin-domain mechanism rather than operating through a mechanism that involves R loops. We propose that a variety of nucleoprotein complexes acts as barriers that hinder diffusion of DNA supercoils, thereby slowing the merger and annihilation of the twin-supercoiled domains generated during transcription. By this means, DNA-binding proteins may localize, in the vicinity of their recognition sequences, DNA supercoils that arise from transcription. We discuss the possibility that localized supercoiling, induced by transcription or other DNA translocation events, may enable certain DNA transactions within the supercoiled domain.
Materials and Methods
Purified Proteins.
E. coli RNA polymerase was purified by the method of Hager et al. (14). T7 RNA polymerase was purified as described (15) from an E. coli strain, BL21/pAR1219 (generously provided by F. W. Studier, Brookhaven National Laboratory, Upton, NY), which overexpresses this enzyme. Bacteriophage λ O protein and E. coli DNA gyrase and HU protein were purified as described (16, 17). Preparations of purified E. coli DNA Topo I and E. coli RNase H were gifts of R. DiGate (Univ. of Maryland, Baltimore) and R. Crouch (National Institutes of Health), respectively. λ cI repressor was a gift of C. Pabo (Massachusetts Institute of Technology, Cambridge). E. coli gal repressor (GalR) and lac repressor (LacI) were kindly provided by S. Adhya (National Institutes of Health). EcoRI-Gln-111 restriction endonuclease was a generous gift of P. Modrich (Duke University, Durham, NC). All restriction enzymes, T4 DNA ligase, T4 DNA polymerase, and calf intestinal alkaline phosphatase were purchased from New England Biolabs.
Plasmid DNA Templates.
All plasmids were derived from plasmid pUC18 (18), except for plasmid pRLM409, which was derived from plasmid pUC8 (19) by means of an intermediate, plasmid pRLM156 (20). The construction details for all plasmids, as well as the maps of plasmids pRLM389, pRLM411, and pRLM419, are published as supporting information on the PNAS web site, www.pnas.org.
In Vitro Transcription/Supercoiling (T-S) Assay.
The standard in vitro T-S reaction mixture (300 μl) contained 40 mM Hepes/KOH (pH 7.6), 11 mM magnesium acetate, 100 mM potassium glutamate, 1 mM DTT, 4 mM ATP, 0.5 mM each of GTP, CTP, and UTP, 2.5 μg of supercoiled plasmid DNA, E. coli DNA gyrase (80 nM), and either T7 RNA polymerase (20 nM) or E. coli RNA polymerase (33 nM). Where specified, additional proteins were added to the standard T-S reaction mixture. All components were assembled on ice and incubated, unless specified otherwise, for 10 min at 30°C. T-S reactions were terminated by extraction with an equal volume of phenol. The DNA samples were precipitated with ethanol and dissolved in 60 μl of 10 mM Tris⋅HCl buffer (pH 8.0). Each sample was supplemented with 5 μg of RNase A and 1 unit of RNase H and incubated for 30 min at 37°C. Each DNA sample was extracted once with phenol and a one-quarter portion was analyzed by electrophoresis at 1 V/cm for 16 h in a 1% agarose gel in TAE buffer, pH 7.8, (21) containing 5 μg/ml chloroquine. Some DNA samples were also analyzed by two-dimensional agarose gel electrophoresis under the conditions described previously, except that the TAE running buffer contained 10 μg/ml of chloroquine in the first dimension and 50 μg/ml of chloroquine in the second dimension. Agarose gels were stained with ethidium bromide and photographed.
Results
The Bacteriophage λ O Replication Initiator Protein Stimulates Supercoiling of DNA Templates During Transcription.
It seemed possible that the failure to date to detect transcription-coupled DNA supercoiling in vitro, occurring by way of the twin-domain model (9), may stem in part from the general use of small, circular plasmid DNA templates. On such templates, rotational diffusion of the DNA segment that connects the negatively and positively supercoiled domains should lead to merger of the two domains and to rapid annihilation of any supercoils generated by transcription. We therefore considered the possibility that nucleoprotein complexes could potentially function as barriers that impede supercoil diffusion and slow merger of the two domains. To examine this idea more rigorously, it was necessary to construct a series of plasmid DNA templates (Fig. 1) that permits strict control over the initiation and termination of transcription. We first inserted into each DNA template one or more promoters (Fig. 1, filled arrowheads) that allow specific initiation of transcription by phage T7 RNA polymerase or by E. coli RNA polymerase. T7 promoters proved to be particularly useful for these studies, because the T7 RNA polymerase provides significantly higher site-specificity for initiation of transcription than does the bacterial enzyme. We also added one or multiple Rho-independent transcription terminator sequences (Fig. 1, winged triangles) to each plasmid. The presence of multiple terminators enabled us to restrict transcription to selected regions of the circular DNA template and to modulate the length of RNA transcripts produced. Finally, we also inserted into most plasmids one or more recognition sites for a sequence-specific DNA-binding protein.
Figure 1.

Plasmids used in assays of transcription-coupled DNA supercoiling. A set of plasmids derived from plasmid pUC18 or pUC8 (pRLM409) was constructed for use in analysis of the impact of transcription and site-specific DNA-binding proteins on DNA supercoiling in vitro. Large filled arrowhead, promoter for T7 RNA polymerase (T7P); winged triangles, Rho-independent E. coli rrnB T1 or plasmid pBR322 P4 transcription terminators; rectangle, bacteriophage λ replication origin (oriλ) with four iterons (O protein-recognition sites, cross-hatched area) and associated A + T-rich region (open area). For plasmid pRLM409, the locations of the λ pR and pL promoters are indicated. The directions of transcription on these plasmid templates from T7 and λ promoters are indicated by arrowheads or arrows, respectively. Only transcription in the clockwise direction is terminated by the rrnB and pBR322 transcription-termination elements present on these plasmids.
The T7 promoter present on plasmid pRLM375 is oriented such that transcription proceeds in a counterclockwise direction. Transcription of this template by T7 RNA polymerase in vitro for 10 min produces very long RNA transcripts that average greater than 4 kb in length (data not shown). This is the expected result, because each of the transcription terminators on pRLM375 is oriented such that only clockwise transcription is terminated. The input template DNA had an initial superhelical density, σ, of approximately −0.06. In the presence of the DNA intercalator chloroquine (5 μg/ml), this DNA was largely relaxed and migrated during agarose gel electrophoresis as if it contained a few (−) supercoils (Fig. 2A, lane 8). Incubation of pRLM375 DNA with DNA gyrase alone for 10 min introduced about 5 or 6 additional (−) supercoils (Fig. 2A, lane 7). As anticipated from earlier studies (9, 13), if the same DNA template in addition was transcribed with T7 RNA polymerase (i.e., incubated in the standard T-S reaction), a limited amount of (−−) SC DNA (estimated σ < − 0.11) was formed (Fig. 2A, lane 1).
Figure 2.

λ O protein stimulates transcription-coupled DNA supercoiling by E. coli DNA gyrase. T-S assays were performed as described under Materials and Methods, except that the reaction mixtures were supplemented with λ O protein as specified. The final topological states of the plasmid DNA templates were assessed by electrophoresis of the deproteinized DNA samples in 1% agarose gels containing 5 μg/ml of chloroquine. (A–C) Plasmid DNA templates pRLM375, pRLM384, and pRLM352, respectively, were transcribed by T7 RNA polymerase. (D) plasmid pRLM409 DNA was transcribed by E. coli RNA polymerase. For all images, the DNA samples applied to lanes 1–6 were from T-S reactions that contained 0, 25, 50, 100, 200, or 400 nM λ O protein (as dimer), respectively. The untreated input DNA templates were applied to lane 7 (B–D) or to lane 8 (A). The DNA sample applied to lane 7 of A is from a standard assay mixture that included DNA gyrase but no T7 RNA polymerase.
At the start, we selected the λ O replication initiator as a test protein for probing the effect of specific DNA-binding proteins on transcription-coupled DNA supercoiling. O, which binds as a dimer to each of 4 iterons present in oriλ, was chosen because it may play a role in the process of transcriptional activation of λ DNA replication (17). Strikingly, supplementation of the pRLM375 T-S reaction mixture with rising quantities of λ O protein produced increasingly large amounts of (−−) SC DNA (Fig. 2A, lanes 2–6). This massive stimulation by O required the presence of O-binding sites in the DNA template. When plasmid pRLM384 (Fig. 1C), which is identical to pRLM375 except that it does not contain an oriλ sequence, was used as the template, supplementation of the T-S reaction mixture with λ O was without noticeable effect on the generation of (−−) SC DNA (Fig. 2B, lanes 2–6). Moreover, O protein had no effect on DNA supercoiling when the transcripts produced by T7 RNA polymerase were short (≈250 bases), as is the case during clockwise transcription of pRLM352 DNA (Fig. 2C, lanes 2–6), regardless of the amount of RNA produced (data not shown). The impact of O on transcription-induced DNA supercoiling was not restricted to T7 RNA polymerase. O was even more potent when E. coli RNA polymerase was the transcribing enzyme (Fig. 2D, lanes 2–6).
With a related approach, we found that O protein is also capable of enhancing production of positively supercoiled DNA when a Topo that relaxes negative supercoils, E. coli Topo I, is substituted for DNA gyrase in the transcription reaction mixture (Fig. 3 A and B, lanes 2 and 3). O failed to stimulate positive DNA supercoiling, however, when T7 RNA polymerase was omitted from the reaction mixture (Fig. 3 A and B, lanes 4 and 5). Thus, stimulation of negative as well as positive DNA supercoiling by O strictly depends on transcription of the template DNA by RNA polymerase. The stimulation of positive DNA supercoiling by Topo I, however, is not as striking as the stimulation of negative DNA supercoiling by DNA gyrase.
Figure 3.
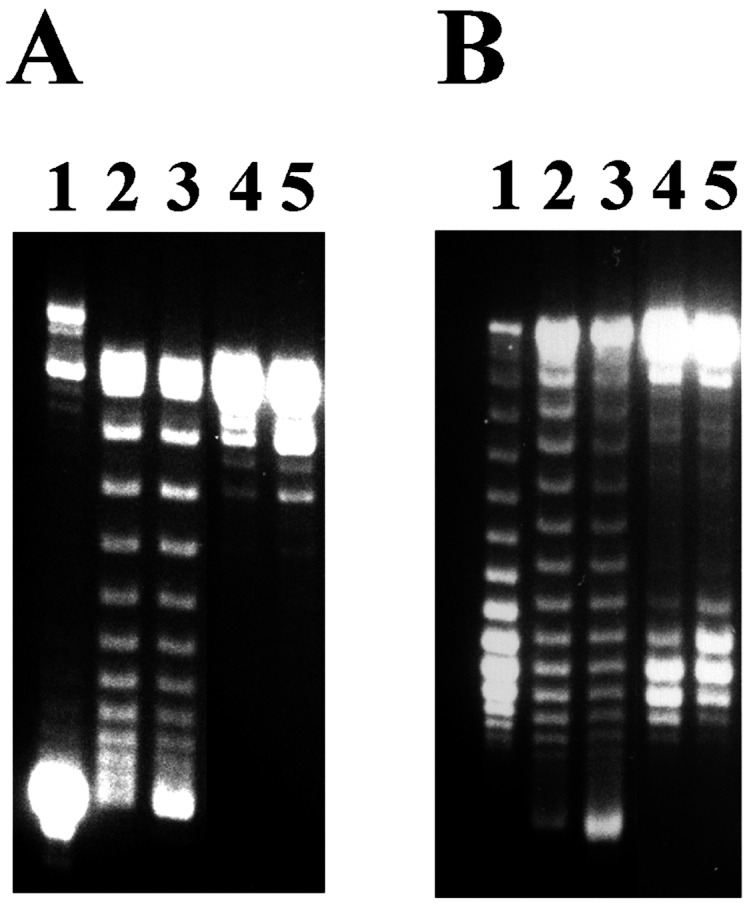
λ O stimulates positive supercoiling of DNA during transcription in the presence of E. coli DNA Topo I. T-S assays with plasmid pRLM375 DNA were performed as described under Materials and Methods, except that DNA gyrase was replaced with 67 nM E. coli DNA Topo I. The topological state of each deproteinized DNA sample was assessed by agarose gel electrophoresis both in the absence (A) or presence (B) of 2 μg/ml of chloroquine. Untreated plasmid DNA template was applied to lane 1. The products of T-S reactions that contained both T7 RNA polymerase and Topo I are depicted in lanes 2 (no O protein) and 3 (100 nM λ O protein was present in the T-S reaction mixture). The products of T-S reactions that contained Topo I but no T7 RNA polymerase were applied to lanes 4 (no O protein) and 5 (100 nM λ O protein was present in the T-S reaction mixture).
λ O-Protein-Mediated Stimulation of Transcription-Coupled DNA Supercoiling Does Not Depend on the Formation of R Loops.
It has been shown that DNA supercoiling obtained during transcription with purified RNA polymerase and DNA gyrase, in the absence of Topo I, largely depends on the formation of R loops (13, 22). Consistent with this latter finding, we found that inclusion of E. coli RNase H, which specifically degrades the RNA chain of an R loop, in the T-S reaction mixture appreciably reduced the formation of (− −) SC DNA (unpublished data; also compare lane 5 of Fig. 4A with lane 1 of Fig. 2A). We therefore tested the effect of E. coli HU protein on the T-S reaction, because HU is known to bind DNA and constrain (−) supercoils (23), an activity that would be anticipated to reduce the stability of R loops. HU protein interfered significantly with the generation of (− −) SC DNA in vitro (Fig. 4A, lane 1; also compare lanes 1 and 5 of Fig. 5C), without reducing the level of transcription (data not shown). It is likely, therefore, that, in the absence of O protein, R loops play a central role in the transcription-coupled DNA supercoiling process in the standard T-S reaction.
Figure 4.

λ O protein overrides the inhibitory effects of RNase H and HU protein on transcription-coupled DNA supercoiling. (A) T-S assays with pRLM375 template DNA were performed and analyzed as described under Materials and Methods and in the legend to Fig. 2. The DNA samples applied to lanes 1–4 were from T-S reaction mixtures that contained 720 nM E. coli HU protein, whereas the DNA samples applied to lanes 5–8 were from reaction mixtures that contained 137 nM E. coli RNase H. Additionally, λ O protein was present at 50 (lanes 2 and 6), 100 (lanes 3 and 7), or 200 nM (lanes 4 and 8) in the T-S reaction mixtures. (B) Analysis of template DNA supercoiling by two-dimensional chloroquine-agarose gel electrophoresis. Gels I and II: The plasmid DNA samples were obtained from T-S reaction mixtures that contained λ O protein, as described above in detail for lanes 3 and 7 (A), respectively. The spots marked a–c on these gels denote, respectively, the migration positions of nicked circular DNA, input negatively supercoiled template DNA, and (− −) SC DNA.
Figure 5.

Stimulation of transcription-coupled DNA supercoiling by other sequence-specific DNA-binding proteins. Plasmid DNA samples were analyzed by agarose gel electrophoresis in chloroquine after serving as templates in T-S reactions performed as described under Materials and Methods. (A) Plasmid pRLM419 (Fig. 7) was the DNA template. As indicated at the top of the image, the T-S reaction mixtures also contained Gal repressor (120 nM GalR), HU protein (720 nM), and/or d-galactose (30 mM gal). Untreated pRLM419 DNA was applied to lane 9. (B) Plasmid pRLM420 (see Supporting Methods, which is published on the PNAS web site) was the DNA template. As specified, the T-S reaction mixtures also contained Lac repressor (lanes 2 and 4, 20 nM LacI; lanes 3 and 5, 40 nM LacI) and/or isopropyl β-d-thiogalactoside (IPTG) (3 mM). Additionally, all reaction mixtures contained 720 nM HU protein. Untreated pRLM420 DNA was applied to lane 6. (C) Plasmid pRLM375 was the DNA template. As specified at the top of the image, the T-S reaction mixtures also contained the N-terminal domain of λ O (lanes 2 and 6, 75 nM λ ON; lanes 3 and 7, 150 nM λ ON; lanes 4 and 8, 300 nM λ ON) and/or HU protein (720 nM). Untreated pRLM375 DNA was applied to lane 9. (D) Plasmid pRLM411 DNA (Fig. 7), which contains a single recognition site for each EcoRI and λ O protein, was the DNA template. The T-S reaction mixtures, as indicated at the top of the image, also contained EcoRI Gln-111 protein (50 nM), λ O protein (100 nM), and/or 720 nM HU protein. Untreated pRLM411 DNA was applied to lane 9.
If the capacity of O protein to stimulate transcription-coupled DNA supercoiling is independent of R-loop formation, then O should stimulate DNA supercoiling even when protein inhibitors of R-loop formation, such as RNase H and HU protein, are present. Indeed, addition of O protein to T-S reaction mixtures containing significant levels of HU protein or RNase H (Fig. 4A, lanes 2–4 and 6–8, respectively) strongly stimulated formation of (− −) SC DNA. This same result was obtained when O protein was present in T-S reaction mixtures that contained both RNase H and HU protein (data not shown). We conclude that stimulation by O protein of DNA supercoiling is apparently not coupled to the formation of R loops during transcription.
Magnitude of DNA Supercoiling in O-Enhanced Transcription-Supercoiling Reactions.
We used two-dimensional electrophoresis in agarose gels in the presence of chloroquine to estimate the superhelical density of the product DNA molecules that accumulate during T-S reactions. Under our electrophoretic conditions, the input (−) SC plasmid DNA, which contained an average of 23 negative supercoils, migrated as a series of spots that was located below and to the right of position b on the DNA topoisomer curve (Fig. 4B, data not shown). Surprisingly, the principal DNA product of T-S reactions carried out in the presence of O protein failed to resolve into individual DNA topoisomers, even if R-loop formation was inhibited by the inclusion of HU protein or RNase H in the standard T-S reaction mixture (Fig. 4 B I and II, spot c, respectively). This situation remained the case even when the second-dimension agarose gel contained chloroquine at 250 μg/ml (data not shown). Our measurements indicate that the DNA molecules that migrate in spot c of Fig. 4B have a highly nonphysiological superhelical density of at least −0.15 (i.e., contain more than 56 negative supercoils) and probably significantly greater. Thus, in a short 10-min T-S reaction, the presence of O has yielded DNA molecules that have negative superhelical densities about three-fold higher than the normal physiological level.
Many Sequence-Specific DNA-Binding Proteins Can Stimulate Transcription-Coupled DNA Supercoiling.
We next investigated whether the potent capacity of O to enhance transcription-coupled DNA supercoiling is a property that is unique to the λ replication initiator. We first tested the well characterized E. coli GalR and LacI repressors. Plasmids that contain two gal-operon or two lac-operon operators, needed for effective DNA-looping and repression in vivo, were constructed and used as templates in T-S reactions. When the DNA template contained gal operators, addition of GalR to the standard T-S reaction mixture brought about a large increase in the amount of (− −) SC DNA produced (Fig. 5A, lane 2). This result was obtained regardless whether HU protein, which is required for effective repression by GalR in vivo (24), was included in the T-S reaction mixture (Fig. 5A, lane 4). Control experiments indicated that the enhancement of DNA supercoiling was specific for the GalR-gal operator protein–DNA interaction. GalR failed to stimulate DNA supercoiling when its inducer, galactose, was present (Fig. 5A, lanes 5–8) and when lac operators were substituted for the gal operators in the DNA template (data not shown). Likewise, Lac repressor stimulated supercoiling of the lac operator-containing DNA template during transcription (Fig. 5B, lanes 1–3) and this effect was sensitive to the presence of a specific inducer, isopropyl β-d-thiogalactoside, which lowers the affinity of LacI for its specific recognition sites (Fig. 5B, lanes 4 and 5).
We sought to understand how binding of a protein tightly to its recognition site on a circular DNA molecule facilitates supercoiling of that molecule when it is transcribed in the presence of DNA gyrase. It seemed possible that the mass of the DNA-bound protein(s) may influence the efficiency of DNA supercoiling. We tested this idea by using a smaller N-terminal fragment of λ O (λ ON) that retains the capacity of O to dimerize and bind specifically to DNA. This O fragment, containing amino acid residues 19–139, was found to be as potent as full-length O (299 residues) at stimulating transcription-coupled DNA supercoiling (Fig. 5C) after binding to the four O recognition sites in oriλ. Additionally, assembly of a large oriλ:O⋅P⋅DnaB preinitiation complex (25) on plasmid pRLM375 provided no further enhancement of DNA supercoiling in the standard T-S reaction (data not shown). Thus, nucleoprotein assemblies that range in mass from 110 kDa to as much as 1200 kDa each provided maximal stimulation of DNA supercoiling.
Factors That Influence Transcription-Coupled Supercoiling of DNA.
To this point, the DNA templates used in the T-S reactions all contained two or more recognition sequences for the specific DNA-binding proteins being tested. Therefore, we constructed a plasmid DNA, pRLM411 (Fig. 7, which is published as supporting information on the PNAS web site), derived from pRLM375, which contains just a single O iteron and a single EcoRI site. T-S reaction mixtures containing pRLM411 as the DNA template were supplemented with either λ O or with an inactive form of EcoRI, EcoRI-Gln-111, which remains capable of binding tightly to its recognition sequence (26). Addition of each protein alone to the T-S reaction mixture yielded a moderate stimulation of transcription-coupled DNA supercoiling (Fig. 5D), indicating that formation of a single nucleoprotein complex is sufficient to observe an effect on supercoiling. However, when both proteins were included simultaneously in the reaction mixture, production of (− −) SC DNA was strongly enhanced (Fig. 5D, lanes 4 and 8). This result indicates that these two proteins act independently to stimulate supercoiling in the in vitro transcription system.
A number of additional combinations of templates and specific DNA-binding proteins was tested in the transcription-supercoiling reaction. In each case, the capacity of the added binding protein to stimulate the production of (− −) SC DNA was measured. The results for transcription reactions that used T7 RNA polymerase are tabulated in Table 1, which is published as supporting information on the PNAS web site, whereas those for reactions with E. coli RNA polymerase are listed in Table 2, which is published as supporting information on the PNAS web site. Several observations can be made. It is apparent that an increase in the number of binding sites in the DNA template for a specific DNA-binding protein enhances the rate of DNA supercoiling during T7 transcription. This effect was true for both λ O and for the λ cI repressor. Also, the nature of the interaction of the specific DNA-binding protein with its recognition sequence, rather than the mass of the nucleoprotein complex, seems to be paramount. Finally, the absolute effect of a DNA-binding protein on DNA supercoiling during transcription can depend on the type of RNA polymerase used. A dimer of O bound to a single iteron provided only a weak enhancement of DNA supercoiling during transcription by T7 RNA polymerase but greatly boosted DNA supercoiling when E. coli RNA polymerase was used (Tables 1 and 2). It may be relevant here that simply binding E. coli RNA polymerase to a promoter is sufficient to boost supercoiling (Table 1).
Discussion
In this article, we have demonstrated that the binding of a protein to its recognition site or sites on a plasmid DNA molecule can produce a striking stimulation of transcription-coupled DNA supercoiling. This supercoiling process was found to be especially potent when proteins such as the λ O replication initiator or the Gal or Lac repressors were present during transcription. We also have confirmed earlier reports (13, 22) that there exists another mechanism for producing (− −) SC DNA. This second mechanism apparently involves the formation of R loops during transcription. Our studies clearly indicate, however, that DNA supercoiling by the “R-loop” mechanism is quite modest in comparison to that generated by the binding of proteins to the transcription template.
Our characterization of the transcription–supercoiling reaction suggests that a twin-supercoiled-domain mechanism (9) is involved. Extensive (− −) SC was obtained only under conditions where transcription produced long RNA chains (Fig. 2 A and C), precisely as hypothesized by Liu and Wang (9) in their original proposal. In this regard, control experiments demonstrated that O has no effect on the length or amount of RNA synthesized and that both T7 and E. coli RNA polymerases readily transcribe through a bound O-some (S.-H. Chung, F.L, and R.M., unpublished data). It is also notable that the presence of a specific DNA-binding protein in the transcription mixture stimulated production of positively supercoiled, rather than negatively supercoiled, DNA when a DNA Topo that relaxes (− −) SC DNA (Topo I) was substituted for DNA gyrase (Fig. 3). This finding, too, is fully consistent with a twin-domain mechanism. Taken together, our results suggest that the site-specific DNA-binding proteins tested here either assist in the generation or promote the stabilization of negatively and positively supercoiled domains during transcription.
We favor the model depicted in Fig. 6 to explain how DNA-binding proteins stimulate transcription-coupled DNA supercoiling. In the early stages of transcription, the RNA polymerase (green oval) rotates around the helical axis as it translocates along the template DNA (Fig. 6b). However, the growing RNA chain soon obtains a length where it and the RNA polymerase can no longer be rotated freely around the template. Once this point is reached, continued movement of the transcription complex instead forces rotation of the template DNA, producing positive supercoils ahead of the polymerase and negative ones behind it (Fig. 6c). The specific binding of a protein (blue hourglass) to a recognition sequence (red circle) is proposed to form a barrier that slows or prevents diffusion of supercoils past the nucleoprotein complex (Fig. 6d). Such a nucleoprotein barrier would lessen supercoil annihilation, a process that occurs when supercoils of opposite sign merge by diffusion, and would be expected to increase the average lifetime of transient DNA supercoils. In turn, this should present DNA gyrase with a greater opportunity to convert transient positive supercoils into “permanent” negative supercoils (Fig. 6e). The net result is a more negatively supercoiled DNA template (Fig. 6f).
Figure 6.

Proposed scheme for the stimulation of transcription-coupled DNA supercoiling by a sequence-specific DNA-binding protein. Green oval, RNA polymerase; blue hourglass, a site-specific DNA-binding protein; red circle, DNA recognition sequence for the site-specific DNA-binding protein. See text for details.
It is apparent that a subset of specific DNA-binding proteins is particularly effective at stimulating transcription-coupled DNA supercoiling (Table 1). What underlies the potency of such proteins? We considered the possibility that two or more bound protein complexes interact in cis to form DNA loops or pair in trans to join two DNA molecules together. Both events would presumably create an effective barrier to diffusion of supercoils generated by transcription (10, 27). Although O, GalR, and LacI are each capable of forming DNA loops (28–32), it is unlikely that either DNA-looping or DNA-pairing events explain the stimulation of DNA supercoiling mediated by open complexes of E. coli RNA polymerase (Table 1) or by the combination of λ O and EcoRI-Gln-111 proteins (Fig. 5D). On the other hand, it is striking that, of the DNA-binding proteins we tested, the ones that bend DNA most strongly are generally those most effective at promoting transcription-coupled DNA supercoiling (Table 1). In this regard, there is evidence that strongly bent DNA sequences preferentially are localized at the apices of interwound supercoiled DNA molecules (33, 34). We suggest that the localization of a protein–DNA complex at the end of a supercoiled domain may enhance the capacity of the protein to function as a barrier to the rotational diffusion of nascent toroidal supercoils that arise during transcription. Barriers of this sort need not be highly efficient to facilitate localized DNA supercoiling. Even the relatively slow-moving E. coli RNA polymerase is capable of producing 120 transient positive supercoils in a single minute. If only 10% of the positive supercoils are captured, before annihilation, by DNA gyrase and converted into negative supercoils (resulting in a net gain of 24 negative supercoils), the superhelical density of the starting plasmid template would quickly double.
Previously, it had generally been assumed that DNA-bound proteins absolutely needed to be attached directly or indirectly to some large cellular entity before they could act as a barrier to supercoil diffusion. Our demonstration that many typical DNA-binding proteins apparently have the capacity to form such barriers in vitro greatly expands the spectrum of proteins that potentially could generate localized DNA supercoiling effects in vivo. The double-helical structure of DNA makes it possible to produce localized DNA supercoiling through the action of any molecular motor (e.g., RNA polymerases, DNA helicases, branch-migration enzymes, certain ATP-dependent restriction enzymes, etc.) that translocates unidirectionally along a DNA strand. All that is needed is (i) a mechanism to fix the DNA translocase to prevent it from rotating around the DNA axis as it moves and (ii) a nucleoprotein structure that is positioned to block or slow passage of transient DNA supercoils generated by the DNA translocase. Of course, the final level of localized DNA supercoiling will be modulated by the action of cellular DNA Topos.
Changes in local DNA superhelicity can have a powerful impact on the conformation and function of critical DNA sequence elements (35–37). With the recent discovery that chromatin remodeling activities can generate superhelical torsion (38), it would not be surprising to find that mechanisms similar to those postulated here influence transcriptional activation or repression of eukaryotic genes.
Supplementary Material
Acknowledgments
We are particularly grateful to the many colleagues, mentioned in the text, who supplied us with preparations of purified proteins or with bacterial strains and plasmids. We also thank Brian Learn and C. C. Victor Fok for assistance with some of the experiments. F.L. especially thanks Drs. P. C. Huang and James C. Wang for helpful discussions. This work was supported by National Institutes of Health Grant GM32253.
Abbreviations
- T-S
transcription/supercoiling
- GalR
E. coli gal repressor
- LacI
E. coli lac repressor
- Topo I
E. coli DNA topoisomerase I
- (−−) SC DNA
hypernegatively supercoiled DNA
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wang J C. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg A, Baker T A. DNA Replication. New York: Freeman; 1992. [Google Scholar]
- 3.Pruss G J, Drlica K. Cell. 1989;56:521–523. doi: 10.1016/0092-8674(89)90574-6. [DOI] [PubMed] [Google Scholar]
- 4.Snoep J L, Der Weijden C C, Andersen H W, Westerhoff H V, Jensen P R. Eur J Biochem. 2002;269:1662–1669. doi: 10.1046/j.1432-1327.2002.02803.x. [DOI] [PubMed] [Google Scholar]
- 5.Zechiedrich E L, Khodursky A B, Bachellier S, Schneider R, Chen D, Lilley D M, Cozzarelli N R. J Biol Chem. 2000;275:8103–8113. doi: 10.1074/jbc.275.11.8103. [DOI] [PubMed] [Google Scholar]
- 6.Pruss G J, Drlica K. Proc Natl Acad Sci USA. 1986;83:8952–8956. doi: 10.1073/pnas.83.23.8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figueroa N, Bossi L. Proc Natl Acad Sci USA. 1988;85:9416–9420. doi: 10.1073/pnas.85.24.9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lockshon D, Morris D R. Nucleic Acids Res. 1983;11:2999–3017. doi: 10.1093/nar/11.10.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu L F, Wang J C. Proc Natl Acad Sci USA. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J C, Lynch A S. Curr Opin Genet Dev. 1993;3:764–768. doi: 10.1016/s0959-437x(05)80096-6. [DOI] [PubMed] [Google Scholar]
- 11.Droge P. BioEssays. 1994;16:91–99. doi: 10.1002/bies.950160205. [DOI] [PubMed] [Google Scholar]
- 12.Wu H Y, Shyy S H, Wang J C, Liu L F. Cell. 1988;53:433–440. doi: 10.1016/0092-8674(88)90163-8. [DOI] [PubMed] [Google Scholar]
- 13.Drolet M, Bi X, Liu L F. J Biol Chem. 1994;269:2068–2074. [PubMed] [Google Scholar]
- 14.Hager D A, Jin D J, Burgess R R. Biochemistry. 1990;29:7890–7894. doi: 10.1021/bi00486a016. [DOI] [PubMed] [Google Scholar]
- 15.Zawadzki V, Gross H J. Nucleic Acids Res. 1991;19:1948. doi: 10.1093/nar/19.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mensa-Wilmot K, Seaby R, Alfano C, Wold M S, Gomes B, McMacken R. J Biol Chem. 1989;264:2853–2861. [PubMed] [Google Scholar]
- 17.Mensa-Wilmot K, Carroll K, McMacken R. EMBO J. 1989;8:2393–2402. doi: 10.1002/j.1460-2075.1989.tb08369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norrander J, Kempe T, Messing J. Gene. 1983;26:101–106. doi: 10.1016/0378-1119(83)90040-9. [DOI] [PubMed] [Google Scholar]
- 19.Vieira J, Messing J. Gene. 1982;19:259–268. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 20.Russell R, Jordan R, McMacken R. Biochemistry. 1998;37:596–607. doi: 10.1021/bi972025p. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 22.Masse E, Drolet M. J Biol Chem. 1999;274:16659–16664. doi: 10.1074/jbc.274.23.16659. [DOI] [PubMed] [Google Scholar]
- 23.Rouviere-Yaniv J, Yaniv M, Germond J E. Cell. 1979;17:265–274. doi: 10.1016/0092-8674(79)90152-1. [DOI] [PubMed] [Google Scholar]
- 24.Lewis D E, Geanacopoulos M, Adhya S. Mol Microbiol. 1999;31:451–461. doi: 10.1046/j.1365-2958.1999.01186.x. [DOI] [PubMed] [Google Scholar]
- 25.Alfano C, McMacken R. J Biol Chem. 1989;264:10699–10708. [PubMed] [Google Scholar]
- 26.Wright D J, King K, Modrich P. J Biol Chem. 1989;264:11816–11821. [PubMed] [Google Scholar]
- 27.Wu H Y, Liu L F. J Mol Biol. 1991;219:615–622. doi: 10.1016/0022-2836(91)90658-s. [DOI] [PubMed] [Google Scholar]
- 28.Schnos M, Zahn K, Blattner F R, Inman R B. Virology. 1989;168:370–377. doi: 10.1016/0042-6822(89)90278-x. [DOI] [PubMed] [Google Scholar]
- 29.Schnos M, Inman R B. Virology. 1991;183:753–756. doi: 10.1016/0042-6822(91)91005-2. [DOI] [PubMed] [Google Scholar]
- 30.Kramer H, Niemoller M, Amouyal M, Revet B, Wilcken-Bergmann B, Muller-Hill B. EMBO J. 1987;6:1481–1491. doi: 10.1002/j.1460-2075.1987.tb02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haber R, Adhya S. Proc Natl Acad Sci USA. 1988;85:9683–9687. doi: 10.1073/pnas.85.24.9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandal N, Su W, Haber R, Adhya S, Echols H. Genes Dev. 1990;4:410–418. doi: 10.1101/gad.4.3.410. [DOI] [PubMed] [Google Scholar]
- 33.Laundon C H, Griffith J D. Cell. 1988;52:545–549. doi: 10.1016/0092-8674(88)90467-9. [DOI] [PubMed] [Google Scholar]
- 34.Heggeler-Bordier B, Wahli W, Adrian M, Stasiak A, Dubochet J. EMBO J. 1992;11:667–672. doi: 10.1002/j.1460-2075.1992.tb05098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahmouni A R, Wells R D. J Mol Biol. 1992;223:131–144. doi: 10.1016/0022-2836(92)90721-u. [DOI] [PubMed] [Google Scholar]
- 36.Lilley D M, Chen D, Bowater R P. Q Rev Biophys. 1996;29:203–225. doi: 10.1017/s0033583500005825. [DOI] [PubMed] [Google Scholar]
- 37.Sheridan S D, Opel M L, Hatfield G W. Mol Microbiol. 2001;40:684–690. doi: 10.1046/j.1365-2958.2001.02416.x. [DOI] [PubMed] [Google Scholar]
- 38.Havas K, Flaus A, Phelan M, Kingston R, Wade P A, Lilley D M, Owen-Hughes T. Cell. 2000;103:1133–1142. doi: 10.1016/s0092-8674(00)00215-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}