

Abstract
The function of GATA transcription factors in diverse developmental contexts depends in part on physical interaction with cofactors of the Friend of GATA (FOG) family. However, previous studies indicate that FOG-1 may play a GATA-1-independent role in early megakaryopoiesis, suggesting that FOG proteins might act in a GATA factor-independent manner. Here, we have generated mouse knock-in (KI) mutants harboring a critical valine-to-glycine substitution in the amino-terminal zinc fingers of GATA-1 and GATA-2 to ablate FOG interaction. In contrast to male GATA-1KI (GATA-1 is located on the X-chromosome) or GATA-2KI/KI mice, compound GATA-1KI GATA-2KI/KI mutant mice display complete megakaryopoietic failure, a phenocopy of FOG-1−/− mice. We conclude that FOG-1 requires an interaction with either GATA-1 or -2 as part of its essential role in early megakaryopoiesis. On the basis of these and previous reports, we infer that GATA factor dependence is a critical aspect of FOG protein function.
Tissue-specific transcription factors play essential roles throughout development by regulation of gene expression. In addition to binding DNA, these factors participate in critical protein–protein interactions that affect their function. The Friend of GATA (FOG) family of proteins are a novel class of large multitype zinc-finger polypeptides that interact with GATA transcription factors and modulate their activity. Like GATA factors, FOG proteins play essential roles in the development of diverse tissue types.
FOG-1, the founding member of this family, was identified in a yeast two-hybrid screen for GATA-1-interacting proteins (1). It is a 998-aa nuclear protein that contains nine predicted zinc fingers. FOG-1 is expressed in erythroid and megakaryocytic cells, liver, and testis. FOG-1−/− murine embryos die by embryonic day (E) 11.5 of gestation because of arrested erythroid cell maturation and complete failure of megakaryopoiesis (2). FOG-2, a 1,151-aa protein containing eight zinc fingers, was cloned by homology to FOG-1 (3–5). It is expressed in cardiac, neural, and gonadal tissues. FOG-2−/− mice die during embryogenesis because of a complex congenital heart defect (6, 7). FOG orthologues are present in divergent species, including humans and flies where they have also been shown to play critical roles in blood, neural, cardiac, and eye development (8–11).
Six members of the GATA transcription factor family are known to exist in vertebrates (reviewed in ref. 13). GATA-1, -2, and -3 play roles within the hematopoietic system, whereas GATA-4, -5, and -6 are involved in the development of nonhematopoietic tissues including neuronal, cardiac, endocrine, and gastrointestinal cell types (14–17). All six members can bind to FOG proteins through highly conserved amino acid sequences present in their amino-terminal zinc finger (3). This interaction occurs on a surface of the GATA zinc finger opposite to its DNA contact sites (18).
The molecular mechanism of FOG protein function is incompletely understood. Several lines of evidence suggest that FOG proteins require physical interaction with GATA factors to fulfill their essential roles in development. First, a mutant version of GATA-1 with markedly reduced binding affinity for FOG-1 (substitution of valine by glycine at codon 205 in its amino zinc finger; GATA-1V205G) is unable to rescue erythroid maturation of a GATA-1− cell line, whereas the wild-type molecule is highly active (18). Coexpression of an interaction-restoring FOG-1 mutant overcomes this block, which indicates that the defect is directly attributable to loss of interaction between GATA-1 and FOG-1. A similar, naturally occurring mutation in humans (GATA-1V205M) results in severe congenital dyserythropoietic anemia and thrombocytopenia (8). Second, mutant mice carrying a homologous substitution in their GATA-4 gene (GATA-4V217G), one of the cardiac-expressed GATA factors, die during embryogenesis of a constellation of cardiac defects similar to FOG-2−/− embryos (19). Third, a FOG-1 mutant molecule with impaired binding to GATA factors is unable to rescue erythroid or megakaryocytic maturation of a FOG-1−/− cell line, whereas the wild-type protein is active (20).
Previous studies also suggest that FOG-1 functions in a GATA-1-independent manner during early megakaryopoiesis (2). FOG-1−/− mice fail to produce any megakaryocytes, whereas mice with megakaryocytic-selective loss of GATA-1 expression generate abundant megakaryocytes, although these fail to undergo late stages of maturation (21, 22).
If interaction of FOG proteins with GATA factors is a prerequisite for FOG's biological activity, then one explanation for megakaryocytic failure in the absence of FOG-1, but not GATA-1, is that FOG-1 might function in concert with a different GATA factor in early megakaryocyte development. A candidate is GATA-2, because it is expressed in immature hematopoietic progenitors and also interacts physically with FOG-1 (1). However, in vitro differentiated GATA-2−/− murine embryonic stem (ES) cells produce normal megakaryocytes indicating that GATA-2, by itself, is not required for megakaryopoiesis (23).
In this study, we test the hypothesis that FOG-1 requires an interaction with either GATA-1 or GATA-2 as the basis for its essential role in early megakaryopoiesis. We have used a gene knockin (KI) approach to generate mice containing point mutations in their GATA-1 and GATA-2 genes which disrupt binding to FOG proteins. We report that, in contrast to either GATA-1KI males (GATA-1 is located on the X-chromosome) or GATA-2KI/KI single mutants, compound GATA-1KI GATA-2KI/KI mutants fail to produce any yolk-sac-derived megakaryocytes, a phenocopy of FOG-1−/− mice. Our results demonstrate that FOG-1 functions with either GATA-1 or GATA-2 in controlling an early stage of megakaryopoiesis. Taken together with previous reports, we propose that GATA factor binding is a general requirement for FOG function.
Materials and Methods
Targeted KI Mutagenesis of GATA-1.
A 4-kb EagI GATA-1 genomic fragment from a 129Sv mouse strain library (Stratagene), spanning exons 2–6, was cloned into the NotI site of pBS (Stratagene). The amino-finger valine-to-glycine substitution mutation at residue 205, as well as a silent mutation in the adjacent codon generating a unique MunI site, was incorporated by using the GeneEditor kit (Promega). We subsequently inserted a blunt-ended DNA fragment containing the floxed neomycin (neo) selection cassette into a BstZ171 site within the intron downstream of exon 4. Finally, we added a thymidine kinase negative selection cassette into the SalI site of pBS, distal to the GATA-1 genomic sequences. The targeting construct was linearized by digestion with PvuII. Constructs were electroporated into the TL1 ES cell line (gift of B. Hogan, Vanderbilt University) and selected for homologous recombination by using neomycin resistance for positive selection and thymidine kinase sensitivity for negative selection. Properly targeted ES clones were screened by Southern blot of XbaI/MunI-digested genomic DNA and a 5′ probe external to the targeting construct (probe A).
Targeted KI Mutagenesis of GATA-2.
A 6.3-kb spanning genomic region of mGATA-2 DNA from a 129Sv mouse strain library (Stratagene) was cloned into the pLNTK vector containing neo (floxed) and thymidine kinase selectable markers. Site-directed mutagenesis by using the QuikChange Kit (Stratagene) was performed on exon 4 (amino finger) to mutate valine 296 to glycine. The modified 3-kb BamHI-SalI exon 4 fragment was flanked by a 2.3-kb NcoI-BamHI fragment encoding exons 2 and 3, and by a 1-kb SalI-BamHI fragment containing exon 5 in the final targeting construct. A loxP site was introduced upstream of exon 4 to facilitate conditional targeting of this locus in future experiments. The final 13.5-kb construct was linearized with SalI. A 3′ probe external to the construct (probe B) was used in Southern blots to check for targeting on HindIII-digested genomic DNA.
In Vitro Cre Excision of Neo Cassette, Blastocyst Injection, and Breeding.
To excise the neor cassette, targeted ES clones were transfected with the Cre recombinase expressing plasmid pMC-Cre and subjected to a second round of neomycin selection (24). Sensitive clones were analyzed further by PCR or Southern blot analysis to confirm proper excision of the neor cassette. For the GATA-1KI clones, PCR was performed with primers flanking the neor cassette (see below). For GATA-2KI clones, Southern blot analysis was performed by using an internal probe (probe C) on BamHI-digested genomic DNA. Properly excised clones containing a normal karyotype were then injected into C57BL/6 blastocysts to generate male chimeras. These male chimeras were bred to homozygosity with C57BL/6 females. Crossbreeding of GATA-1KI and GATA-2KI alleles occurred after several generations of inbreeding these lines.
Genotyping PCR Primer Sequences and Conditions.
PCR primers flanking the remaining 5′ loxP were used for animal genotyping. GATA-1KI: GATA1-KI-F: 5′-TATATAGCCCTGGGTGGCCT-3′; GATA1-KI-R: 5′-CCTGCCTCTGTCTCCAGTTC-3′. GATA-2KI: GATA2-KI-F: 5′-GGAGCGAGGGCTTAGTAGCT-3′; GATA2-KI-R: 5′-CACTGCAGAGATCTAGGCCC-3′. PCR reactions were subjected to 1 cycle of 94°C for 4 min, then 35 cycles of 94°C for 1 min, 60°C for 1 min, 72°C for 1 min. GATA-1KI = 321 bp; GATA-1+ = 241 bp; GATA-2KI = 502 bp; GATA-2+ = 366 bp. PCR products were resolved on a 2% agarose gel.
Hematological Assays.
For embryonic blood evaluation, blood cells were collected from E10.5 days postcoitum (dpc) embryo yolk sacs, cytospun, and stained with May–Grunwald–Giemsa with standard techniques. For evaluation of adult blood, retro-orbital phlebotomy was performed on anesthetized adult animals and collected into EDTA-containing tubes. Blood smears were stained with May–Grunwald–Giemsa and hematologic indices measured on an automated clinical hematology system blood analyzer (ADVI A120B) (Bayer, Tarrytown, NY). All procedures were in compliance with institutional and federal animal care requirements.
Yolk Sac/Fetal Liver Differentiation Assays.
Embryonic yolk sacs dissected at E10.5 dpc were digested in 0.1% collagenase/20% FCS (GIBCO) for 1.5 h at 37°C. The cells were dissociated by using an 18-gauge needle, washed once in Iscove's modified Dulbecco's medium (IMDM)/2% FCS, and seeded into methylcellulose (StemCell Technologies, Vancouver) or IMDM/15% FCS directly (growth media). Fetal livers were collected at E14.5 dpc, dispersed by using a 20-gauge needle, washed, and seeded into methylcellulose as above. Both yolk sac and fetal liver hematopoietic cells were cultured for 7 days in growth medium containing any combination of recombinant murine thrombopoietin (Tpo; 5 ng/ml), rat Kit ligand (50 ng/ml), erythropoietin (2 units/ml), IL-3 (10 ng/ml), or IL-11 (5 ng/ml). Acetylcholinesterase staining of megakaryocytes was performed as described (2).
Semiquantitative Reverse Transcription (RT)-PCR.
Yolk sac cells from E10.5 dpc embryos (one embryo each for wild type, GATA-1KI, GATA-2KI/KI, and five embryos for compound GATA-1KI GATA-2KI/KI) were obtained after collagenase treatment and cultured in growth media containing Tpo (5 ng/ml) for 7 days at 5% CO2, 37°C. Total RNA was extracted by using Trizol (GIBCO) and treated with RNase-free RQ1 DNase (Promega). cDNA synthesis and semiquantitative PCR was performed on 1.75 μg of total RNA for each sample as described (22). [α-32P]dCTP-labeled PCR products were collected at 20–28 cycles and separated on 6% acrylamide gels. PCR products were quantified on a PhosphorImager (Molecular Dynamics). The amount of cDNA in the samples was normalized by using primers for hypoxanthine phosphoryibosyltransferase. Primers: AChE-F: 5′-TAGTGGTCGAACTGGTTCTTCCAG-3′ and AChE-R: 5′-GAGGATCTTTGCTCAGCGACTTATG-3′ (314-bp product); P-selectin-F: 5′-CTCGCTCCGCTGCAGGGC-3′ and P-selectin-R: 5′-GGGGTTGGGTCATATGCAGCG-3′ (443-bp product). Primers for von Willebrand factor, GPIbα, and hypoxanthine phosphoryibosyltransferase have been described (12, 22, 29).
Results and Discussion
KI of FOG-1-Binding Mutation into the GATA-1 and -2 Loci.
To study GATA–FOG interactions in vivo, we introduced a point mutation separately into homologous positions of a critical FOG-binding region of GATA-1 (substitution of valine by glycine at codon 205) and GATA-2 (substitution of valine by glycine at codon 296) by gene targeting of murine ES cells (Fig. 1 A, B, and D). Previous studies have shown that this mutation markedly impairs GATA-1's ability to interact with FOG-1 without affecting its DNA-binding activity (18). The homologous mutation in GATA-4 disrupts its interaction with FOG-2 in vivo (19). We chose a gene KI approach to preserve endogenous expression levels throughout development. In addition, we excised the neomycin resistance cassette after selection of targeted cells to prevent possible deleterious effects on expression (24).
Figure 1.

Homologous recombination of targeting constructs and subsequent neo cassette excision in ES cells. (A) Amino acid sequence alignment of the amino-terminal zinc fingers of GATA-1 and GATA-2. Mutated positions in GATA-1 and GATA-2 N-terminal zinc fingers (N-f) are indicated by the red “V” and residue number. Identical sequences are shown in black, and nonidentical residues are shown in blue. (B) Schematic representation of V205G KI mutation gene-targeting strategy into the GATA-1 locus on the X-chromosome. Subsequent Cre-mediated excision of neomycin resistance gene (neo) cassette is shown below. Solid triangles represent loxP sites. Location of probe used for Southern analysis is indicated by solid bar (probe A). PCR primers are shown as horizontally opposed arrows. (C) Southern blot and PCR analyses depicting correctly recombined and Cre-excised GATA-1KI loci. (D) Schematic representation of V296G KI targeting strategy in the GATA-2 locus and subsequent Cre-mediated excision of neo cassette. Locations of probes used for Southern analysis are indicated by solid bars (probes B and C). (E) Southern blot analysis depicting correctly recombined and Cre-excised GATA-2KI loci.
Correctly targeted and excised clones were identified by Southern blot and PCR analyses (Fig. 1 C and E). The resulting ES cells were injected into host C57/BL6 blastocysts and transferred to recipient mice. Chimeric mice containing germ-line transmission of the mutant alleles were bred to generate individual mutant KI lines.
Megakaryocyte Production in Hemizygous GATA-1KI Male Mutants.
Most male GATA-1KI mutants were severely anemic and died at about E11.5 dpc. A few survived to birth but perished in the neonatal period. Mutant embryos were then examined at E10.5 dpc. Yolk sac erythrocytes displayed arrested maturation at an early precursor stage similar to that observed for FOG-1−/− and GATA-1− murine erythrocytes and GATA-1V205M human erythrocytes (Fig. 2) (2, 8, 32), which provides further evidence that an interaction between FOG and GATA-1 is required for normal erythroid development in vivo.
Figure 2.
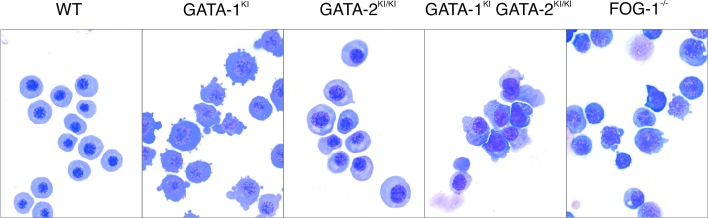
May–Grunwald–Giemsa stains of cytospun erythrocytes collected from E10.5 yolk sacs (original magnification ×1,000).
To assess the loss of FOG–GATA-1 interaction on megakaryopoiesis, yolk sac cells from E10.5 dpc animals were cultured in methylcellulose in the presence of Tpo for 7 days (25). As shown in Fig. 3, megakaryocytic colonies were readily generated from male GATA-1KI embryos. Although slightly fewer colonies existed compared with wild type, their presence contrasted with the absence of megakaryocytic colonies from FOG-1−/− yolk sac cells.
Figure 3.

In vitro megakaryocyte differentiation from E10.5 yolk sac. (A) Phase-contrast appearance and AChE stains of representative megakaryocyte colonies at day 7 of culture. Arrows indicate proplatelet processes. AchE-positive cells stain orange [original magnification, ×320 (phase-contrast); ×1,000 (AchE)]. (B) Quantitation of megakaryocyte colony formation. Colonies were counted on day 7 of culture and expressed as the mean number of megakaryocyte colonies (MK) per 1 × 105 cells plated. Error bars represent the SEM. n = number of embryos analyzed. (C) RT-PCR analysis of the murine megakaryocytic markers AChE, von Willebrand factor (vWF), GPIbα and P-selectin, from yolk sac cells cultured for 7 days in liquid culture in the presence of recombinant Tpo. Hypoxanthine ribosyltransferase (HPRT) represents “housekeeping” control gene for normalization of cDNA content. Fold decrease of mRNA levels relative to wild type (WT) (after normalization to HPRT signal) is indicated below each panel. “−RT” indicates no reverse transcriptase was added to sample.
Morphologically, GATA-1KI megakaryocyte colonies seemed abnormal compared with wild type. They grew more compactly and exhibited fewer proplatelet processes (Fig. 3A). RT-PCR analysis indicated a markedly reduced number of mRNA transcripts for P-selectin, a platelet-specific adhesion molecule expressed during late stages of megakaryopoiesis (Fig. 3C) (33). Electron microscopy revealed reduced numbers of platelet-specific granules and disorganized platelet demarcation membranes, features also observed in murine GATA-1− megakaryocytes and human GATA-1V205M megakaryoctyes (data not shown) (8, 21). Taken together, these observations support previous findings that an interaction between FOG-1 and GATA-1 is specifically required for late stages of megakaryocyte maturation, but not early megakaryocyte development (8).
Normal Steady-State Hematopoiesis of GATA-2KI/KI Mutants.
GATA-2KI/KI animals were born at the expected Mendelian frequency and seemed normal. No significant differences were observed in the morphology or quantitation of embryonic and adult erythrocytes compared with wild-type littermates (Fig. 2 and data not shown). Likewise, no differences were observed in the number, morphology, or marker gene expression levels of megakaryocyte colonies cultured from yolk sac (Fig. 3).
GATA-2 plays a role in the proliferation of multipotential progenitor cells (26). To test whether FOG binding is required for this function, we performed multipotential colony assays from E14.5 dpc GATA-2KI/KI fetal livers. No differences were observed compared with wild-type littermates (data not shown). We conclude that an interaction between FOG-1 and GATA-2, by itself, is dispensable for steady-state hematopoiesis.
Complete Failure of Megakaryopoiesis in GATA-1KI GATA-2KI/KI Compound Mutants.
To test whether FOG-1 requires an interaction with either GATA-1 or -2 for its roles in hematopoiesis, we interbred GATA-1KI and -2KI mice to obtain compound GATA-1KI GATA-2KI/KI mutants. All compound mutant embryos began dying by late E10.5 dpc. Examination of yolk sac erythrocytes demonstrated an arrest in maturation at a precursor stage (Fig. 2), which is similarly observed in GATA-1KI, GATA-1−, and FOG-1−/− mutants, suggesting this phenotype reflects the presence of the GATA-1KI allele specifically (Fig. 2) (2, 32).
We next examined the ability of compound mutant animals to generate megakaryocytes. In contrast to either GATA-1KI or -2KI/KI mice by themselves, yolk sac cells from compound mutant embryos failed to produce any identifiable megakaryocyte colonies in semisolid culture. This failure phenocopies FOG-1−/− embryos (Fig. 3). Only macrophage and granulocytic cells were detected. RT-PCR analysis of cells cultured in liquid media also failed to detect significant mRNA levels for the murine megakaryocytic marker genes acetylcholinesterase (AChE), von Willebrand factor, GPIbα, or P-selectin, whereas these were all present in GATA-1KI- or -2KI/KI-derived cultures.
All genotypes tested produced a similar number and morphology of granulocytes and macrophages (Fig. 4). Therefore, the failure of megakaryopoiesis in the compound mutant animals is not attributable to the loss of early multipotential progenitor cells.
Figure 4.
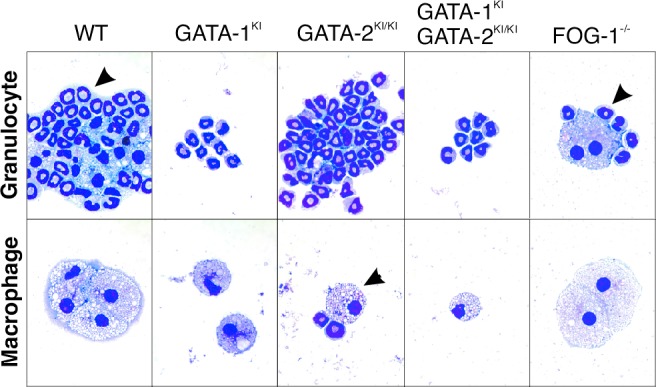
Myeloid colony formation from E10.5 yolk sacs. May–Grunwald–Giemsa stains of cytospun preparation from E10.5 yolk sac cells cultured in methylcellulose in the presence of erythropoietin, Tpo, and Kit ligand for 7 days.
If GATA factors serve to stabilize FOG protein, then loss of FOG expression could explain the failure of megakaryopoiesis in the compound mutant animals. However, we have previously shown that a mutant FOG-1 molecule incapable of binding to GATA family proteins is stably expressed in hematopoietic cells, arguing against this possibility (20). Likewise, the megakaryocytic failure could be explained if the mutations in the GATA-1 and -2 proteins resulted in their instability. The finding of normal numbers of multipotential fetal liver progenitors in the GATA-2 KI/KI animals argues against this possibility, because GATA-2+/− mice have a reduced number of multipotential progenitors (F.-Y. Tsai and S.H.O., unpublished data). It is not feasible to measure directly expression levels of FOG-1, GATA-1, or GATA-2 proteins in the compound mutant progenitor cells.
Requirement of GATA Factor Interaction for the Role of FOG-1 in Early Megakaryopoiesis.
Our results here with compound, altered-specificity mutants of GATA-1 and -2 demonstrate that FOG-1 indeed requires an interaction with GATA factors for its essential role in early megakaryopoiesis. However, unlike its role in late megakaryopoiesis, where GATA-1 is specifically required, either GATA-1 or -2 can serve this function (Fig. 5).
Figure 5.
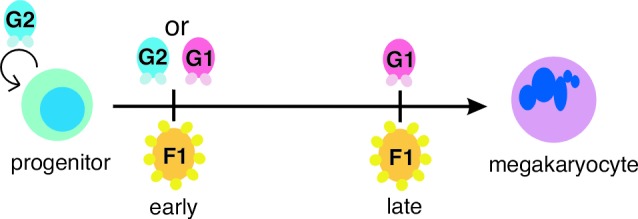
Model of FOG-1–GATA factor interaction requirements during megakaryocyte development. During an early stage of megakaryocyte commitment, FOG-1 requires an interaction with either GATA-1 or GATA-2. During a later stage, FOG-1 specifically requires an interaction with GATA-1. GATA-2 also plays a role in proliferation of progenitor cells (indicated by looping arrow). F1, FOG-1; G1, GATA-1; G2, GATA-2.
FOG family members play essential roles in multiple developmental settings, yet the molecular mechanism of FOG protein function remains incompletely understood. Our findings here, in conjunction with previous studies, indicate that interaction with a GATA factor is critical to the biological function of FOG. Given that neither murine FOG-1 nor FOG-2 exhibit sequence-specific, high-affinity DNA binding (A. Tsang, S. Tevosian, and S.H.O., unpublished data), GATA factors may serve to tether FOG proteins to specific cis-regulatory sites where they influence gene expression through unknown mechanisms. An alternate possibility is that FOG proteins mediate a step in the activation of GATA factors, perhaps by altering their conformation or disrupting interactions with repressor molecules. Further studies are necessary to test these possibilities.
Although our results demonstrate that FOG proteins require interactions with GATA factors for their function, the converse is not true. Within the hematopoietic system, GATA proteins also participate in mast and eosinophil cell differentiation (23, 27). However, FOG expression is absent in mature cells from these lineages (1, 28). Consistent with this finding, we have found that the compound KI animals produce abundant yolk sac-derived mast cells (A.B.C., A.N.C., and S.H.O., unpublished data). In addition, Querfurth et al. have shown that down-regulation of FOG expression is a prerequisite for differentiation of eosinophils from myb-ets transformed multipotent avian cells (28). Whether GATA factors interact with alternate cofactors in these lineages is as yet unknown.
Functional Compensation Between GATA-1 and -2 in Early Megakaryopoiesis.
Our results indicate that GATA-1 and -2 are functionally interchangeable at an early stage in megakaryocytic development. Functional redundancy between GATA-1 and -2 has been suggested in mouse studies of erythroid development (29). Transcription factor compensation has also been described in other protein families, such as the basic helix–loop–helix transcription factors MyoD and Myf-5, which are required for skeletal myocyte differentiation (30).
GATA-1− megakaryocytes express GATA-2 yet fail to progress through late stages in their maturation (P. Vyas and S.H.O., unpublished data) (22). Thus, GATA-2 seems able to compensate for loss of GATA-1 during early but not late megakaryopoiesis. What accounts for this disparity? A possibility is that structural differences between GATA-1 and -2 proteins allow only GATA-1 to activate late target genes. Indeed, GATA-1 and -2 share little sequence homology outside of their two zinc fingers (31). Alternatively, the levels of expression of each factor may be critical to their functions.
Implications for Functions of FOG Proteins.
Our findings demonstrate that FOG-1 function is strictly GATA-factor-dependent with respect to transcriptional events necessary to program megakaryocytic differentiation. Similarly, the function of FOG-2 in gonad development seems to rely on interaction with GATA-4 (S. G. Tevosian, K. H. Albrecht, J.D.C., Y.F., E. M. Eicher, and S.H.O., unpublished data). Hence, the dependence of FOG function on physical interaction with GATA factors seems to be quite general. No data are yet available to indicate that FOG factors function independent of a direct interaction with GATA factors.
Acknowledgments
We thank C. Browne, A. Williams, S. Galusha, and M. Hamblen for technical assistance with ES cell culture and mouse generation, and S. Katz for FOG-1−/− mice. Special thanks to F. Alt for targeting vectors and B. Hogan for the TL1 ES cell line. A.B.C. is supported by a National Cancer Institute KO8 Mentored Clinical Scientist Award (CA 82175–03). J.D.C. is a recipient of a Burroughs Wellcome Fund Career Award in the Biomedical Sciences. S.H.O. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- En
embryonic day n
- dpc
days postcoitum
- FOG
friend of GATA
- Tpo
thrombopoietin
- RT
reverse transcription
- ES
embryonic stem
- KI
knockin
- AChE
acetylcholinesterase
References
- 1.Tsang A P, Visvader J E, Turner C A, Fujiwara Y, Yu C, Weiss M J, Crossley M, Orkin S H. Cell. 1997;90:109–119. doi: 10.1016/s0092-8674(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 2.Tsang A P, Fujiwara Y, Hom D B, Orkin S H. Genes Dev. 1998;12:1176–1188. doi: 10.1101/gad.12.8.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tevosian S G, Deconinck A E, Cantor A B, Rieff H I, Fujiwara Y, Corfas G, Orkin S H. Proc Natl Acad Sci USA. 1999;96:950–955. doi: 10.1073/pnas.96.3.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Svensson E C, Tufts R L, Polk C E, Leiden J M. Proc Natl Acad Sci USA. 1999;96:956–961. doi: 10.1073/pnas.96.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J R, McKinsey T A, Xu H, Wang D Z, Richardson J A, Olson E N. Mol Cell Biol. 1999;19:4495–4502. doi: 10.1128/mcb.19.6.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tevosian S G, Deconinck A E, Tanaka M, Schinke M, Litovsky S H, Izumo S, Fujiwara Y, Orkin S H. Cell. 2000;101:729–739. doi: 10.1016/s0092-8674(00)80885-5. [DOI] [PubMed] [Google Scholar]
- 7.Svensson E C, Huggins G S, Lin H, Clendenin C, Jiang F, Tufts R, Dardik F B, Leiden J M. Nat Genet. 2000;25:353–356. doi: 10.1038/77146. [DOI] [PubMed] [Google Scholar]
- 8.Nichols K E, Crispino J D, Poncz M, White J G, Orkin S H, Maris J M, Weiss M J. Nat Genet. 2000;24:266–270. doi: 10.1038/73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fossett N, Tevosian S G, Gajewski K, Zhang Q, Orkin S H, Schulz R A. Proc Natl Acad Sci USA. 2001;98:7342–7347. doi: 10.1073/pnas.131215798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes M, Turner J, Fox A, Chisholm O, Crossley M, Chong B. J Biol Chem. 1999;274:23491–23498. doi: 10.1074/jbc.274.33.23491. [DOI] [PubMed] [Google Scholar]
- 11.Cubadda Y, Heitzler P, Ray R P, Bourouis M, Ramain P, Gelbart W, Simpson P, Haenlin M. Genes Dev. 1997;11:3083–3095. doi: 10.1101/gad.11.22.3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols W C, Cooney K A, Mohike K L, Ballew J D, Yang A, Bruck M E, Reddington M, Novak E K, Swank R T, Ginsburg D. Blood. 1994;83:3225–3231. [PubMed] [Google Scholar]
- 13.Orkin S H. Blood. 1992;80:575–581. [PubMed] [Google Scholar]
- 14.Nardelli J, Thiesson D, Fujiwara Y, Tsai F Y, Orkin S H. Dev Biol. 1999;210:305–321. doi: 10.1006/dbio.1999.9278. [DOI] [PubMed] [Google Scholar]
- 15.Molkentin J D, Lin Q, Duncan S A, Olson E N. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 16.Dasen J S, O'Connell S M, Flynn S E, Treier M, Gleiberman A S, Szeto D P, Hooshmand F, Aggarwal A K, Rosenfeld M G. Cell. 1999;97:587–598. doi: 10.1016/s0092-8674(00)80770-9. [DOI] [PubMed] [Google Scholar]
- 17.Laverriere A C, MacNeill C, Mueller C, Poelmann R E, Burch J B, Evans T. J Biol Chem. 1994;269:23177–23184. [PubMed] [Google Scholar]
- 18.Crispino J D, Lodish M B, MacKay J P, Orkin S H. Mol Cell. 1999;3:219–228. doi: 10.1016/s1097-2765(00)80312-3. [DOI] [PubMed] [Google Scholar]
- 19.Crispino J D, Lodish M B, Thurberg B L, Litovsky S H, Collins T, Molkentin J D, Orkin S H. Genes Dev. 2001;15:839–844. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cantor A B, Katz S G, Orkin S H. Mol Cell Biol. 2002;12:4268–4279. doi: 10.1128/MCB.22.12.4268-4279.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shivdasani R A, Fujiwara Y, McDevitt M A, Orkin S H. EMBO J. 1997;16:3965–3973. doi: 10.1093/emboj/16.13.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vyas P, Ault K, Jackson C J, Orkin S H, Shivadasani R. Blood. 1999;93:2867–2875. [PubMed] [Google Scholar]
- 23.Tsai F-Y, Orkin S H. Blood. 1997;89:3636–3643. [PubMed] [Google Scholar]
- 24.Torres R M, Kuhn R. Laboratory Protocols for Conditional Gene Targeting. New York: Oxford Univ. Press; 1997. [Google Scholar]
- 25.Wong P M C, Chung S W, Chui D H K, Eaves C J. Proc Natl Acad Sci USA. 1986;83:3851–3854. doi: 10.1073/pnas.83.11.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai F-Y, Keller G, Kuo F C, Weiss M, Chen J, Rosenblatt M, Alt F W, Orkin S H. Nature (London) 1994;371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 27.Kulessa H, Frampton J, Graf T. Genes Dev. 1995;9:1250–1262. doi: 10.1101/gad.9.10.1250. [DOI] [PubMed] [Google Scholar]
- 28.Querfurth E, Schuster M, Kulessa H, Crispino J D, Doderlein G, Orkin S H, Graf T, Nerlov C. Genes Dev. 2000;14:2515–2525. doi: 10.1101/gad.177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss M J, Keller G, Orkin S H. Genes Dev. 1994;8:1184–1197. doi: 10.1101/gad.8.10.1184. [DOI] [PubMed] [Google Scholar]
- 30.Rudnicki M A, Braun T, Hinuma S, Jaenisch R. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- 31.Zon L I, Mather C, Burgess S, Bolce M E, Harland R M, Orkin S H. Proc Natl Acad Sci USA. 1991;88:10642–10646. doi: 10.1073/pnas.88.23.10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujiwara Y, Browne C P, Cunniff K, Goff S C, Orkin S H. Proc Natl Acad Sci USA. 1996;93:12355–12358. doi: 10.1073/pnas.93.22.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schick P K, Barbara K A, He X, Thornton R D. J Lab Clin Med. 1993;121:714–721. [PubMed] [Google Scholar]