

Abstract
Erk5 is a mitogen-activated protein kinase, the biological role of which is largely undefined. Therefore, we deleted the erk5 gene in mice to assess its function in vivo. Inactivation of the erk5 gene resulted in defective blood-vessel and cardiac development leading to embryonic lethality around embryonic days 9.5–10.5. Cardiac development was retarded largely, and the heart failed to undergo normal looping. Endothelial cells that line the developing myocardium of erk5−/− embryos displayed a disorganized, rounded morphology. Vasculogenesis occurred, but extraembryonic and embryonic blood vessels were disorganized and failed to mature. Furthermore, the investment of embryonic blood vessels with smooth muscle cells was attenuated. Together, these data define an essential role for Erk5 in cardiovascular development. Moreover, the inability of Erk5-deficient mice to form a complex vasculature suggests that Erk5 may play an important role in controlling angiogenesis.
Mitogen-activated protein kinases (MAPKs) represent key signaling molecules through which cells integrate a variety of extracellular stimuli and transduce intracellular signals (1). To date, there are at least four recognized MAPK families: the extracellular signal-related kinases (Erks) 1/2, c-Jun N-terminal kinases, p38 MAPKs, and the recently described big mitogen-activated kinase, Erk5. Each MAPK is activated by specific MAPK kinases (Mkks or Meks), that in turn are regulated by Mek kinases (Mekks), thus producing the core MAPK module. Although significant research efforts have elucidated many aspects of the regulation of the Erk, c-Jun N-terminal kinase, and p38 MAPK signaling cascades in vitro and in vivo, the regulation of the Erk5 MAPK pathway is less well understood. However, recent biochemical studies have demonstrated a direct link between the activation of Mekk3 and subsequent activation of Erk5 via Mek5 (2). Furthermore, myocyte enhancer factor 2C (Mef2C) was shown to be a downstream substrate of activated Erk5 (3, 4). Taken together, these data suggest a linear MAPK cascade (Mekk3–Mek5–Erk5) for the activation of Erk5 in vitro and demonstrate that Mef2C is a substrate for activated Erk5.
Recently, many MAPKs, their upstream activators, and downstream targets have been shown to play important roles in cardiovascular pathophysiology (5) and the development of the cardiovascular system. For example, mice with a homozygous deletion of Mek1, the upstream activator of Erk1/2, displayed defects in placental and yolk-sac vascular development (6). Additionally, homozygous deletion of the p38α allele resulted in multiple embryonic cardiovascular defects (7). Furthermore, studies from Yang et al. (8) showed that deletion of an upstream activator of MAPKs, Mekk3, in mice resulted in defects in placental and embryonic vascular development as well as cardiac development. Interestingly, in addition to Erk5 activation, Mekk3 activation has been shown also to lead to p38α activation (8, 9), and p38α has been shown to phosphorylate Mef2C (3, 10). Moreover, mice deficient in Mef2C displayed multiple defects in cardiovascular development (11–13) that were similar to mekk3−/− (8) and p38α−/− (7) mice. Taken together, these data suggest one mechanism whereby the transduction of Mekk3 signals in the developing cardiovascular system may occur via p38α-dependent phosphorylation of downstream targets such as Mef2C. However, because activated Mekk3 also can lead to Erk5-dependent Mef2C phosphorylation (2, 3), the importance of this pathway and the specific role of Erk5 in vivo have yet to be elucidated fully.
To begin to address the specific role of Erk5 and its signaling pathway in vivo, we generated mice deficient in Erk5. Heterozygous mice were viable and appeared normal. However, erk5−/− embryos died as early as embryonic day (E)9.5 and demonstrated abnormal placental vascular, yolk-sac vascular, and embryonic cardiac and vascular development. Interestingly, the phenotype of erk5−/− embryos was highly analogous to the knockout phenotypes of the Erk5 upstream activator, Mekk3 (8), and the Erk5 substrate, Mef2C (11–13). As such, it is interesting to speculate that Erk5 may be a critical MAPK for transducing signals from Mekk3 to downstream factors that are required for early cardiovascular development. Moreover, because our data suggest a role for Erk5 in angiogenesis, these data may indicate that Erk5 may represent a novel kinase target for the development of antiangiogenic therapies.
Materials and Methods
Northern Blot and in Situ Hybridization Analysis.
Mouse multiple tissue and embryo Northern blot membranes were purchased from CLONTECH. An expressed sequence tag clone (W98507) was used to PCR-amplify a 530-bp fragment of Erk5 corresponding to amino acid sequence 678–855. Hybridization was conducted following manufacturer instructions. Membranes then were stripped and rehybridized with a β-actin probe provided by the manufacturer to verify equal loading of each lane. Detection of erk5 and SM22α mRNA by in situ hybridization was carried out according to previously published methods (14). The source of erk5 cDNA for the preparation of the in situ probes was the same as the one used for Northern blot analysis, and the SM22α probe has been described previously (14).
Generation of erk5+/− Embryonic Stem Cells and erk5−/− Mice.
erk5-specific primers were used to amplify a 1.4-kb fragment of erk5 genomic DNA that then was used to screen a mouse genomic DNA library (strain 129/sv, Stratagene). Screening yielded a clone containing a 16-kb erk5 insert. Restriction mapping identified a 4.6-kb NheI–EcoRI and a 4.5-kb SacI–AvrII fragment within the 16-kb insert that contained part of exons 2 and 3 of erk5, which were used for targeted gene disruption (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). Subcloned fragments were ligated with FseI and AscI linkers and inserted into a targeting vector. TC1 embryonic stem cells (a gift from Phillip Leder, Harvard Medical School, Boston, MA) then were transfected with the linearized targeting vector and screened by using a 2-kb fragment of the erk5 gene 3′ to the targeting vector (see Fig. 6). Two clones that underwent proper homologous recombination were injected into C57BL/6 blastocysts to generate two heterozygous mouse lines. The Vertex Pharmaceutical animal care and use committee approved all animal protocols.
Reverse Transcription (RT)-PCR/Western Analysis.
mRNA was isolated from wild-type and mutant embryos or embryonic hearts by using Oligotex Direct mRNA Mini kit (Qiagen, Chatsworth, CA). First-strand cDNA synthesis was performed by using commercially available kits. Specific primers (primer sequences have been published previously; refs. 11 and 15) were added to equal amounts of cDNA, and PCR cycles were determined under conditions of linearity by sampling the reaction every 5 cycles up to 35 PCR cycles for each primer set. Conditions were as follows: 94°C for 2 min, 20–35 cycles of 94°C for 30 sec, 56°C for 45 sec, and 72°C for 1 min. Control RT-PCRs also were conducted in the absence of reverse transcriptase to verify the absence of genomic DNA. PCR products were separated by electrophoresis, and gels were ethidium bromide-stained for image analysis. To confirm deletion of Erk5 protein in mutant embryos, mouse embryo fibroblasts (MEFs) were cultured from +/+ and −/− E9.5 embryos. Cells were placed in serum-free medium overnight and treated with epidermal growth factor (EGF, 100 ng/ml) or vehicle for 15 min. Cells lysates were separated by SDS/PAGE, and Western analysis (1:1,000 rabbit anti-human BMK1, Upstate Biotechnology, Lake Placid, NY) was performed as described previously (16).
Histology and Immunohistochemistry.
For histologic and immunohistologic analysis, embryos were fixed in 4% paraformaldehyde, processed for routine paraffin embedding, and sectioned at 4 μm. Sections were either stained with hematoxylin/eosin for morphologic evaluation or used for immunohistochemistry. Immunohistochemical staining with a mouse anti-human smooth muscle actin (SmαA) antibody (1:2,000, clone 1A4, Sigma) was done as described previously (17). Whole-mount embryos were stained with rat anti-mouse platelet endothelial cell adhesion molecule (PECAM)-1 (10 μg/ml, PharMingen) as described previously (7, 11).
Results
erk5 Transcript Was Differentially Expressed Throughout Embryonic Development.
Because the expression of erk5 has been studied primarily in vitro, we first performed Northern blot analysis on mouse embryos and adult tissues and in situ hybridization on embryos to assess the level and distribution of erk5 mRNA throughout development. Specificity of the antisense erk5 probe was confirmed by hybridization of both blots and embryo sections with a sense erk5 probe, which produced no signal (data not shown). The expression of erk5 was detectable at various developmental stages (Fig. 1A). Of note, there was a significant increase in the expression of erk5 between E7 and E11. Northern blot analysis of various adult mouse tissues showed low but ubiquitous expression of a 3.2-kb transcript corresponding to erk5 mRNA in tissues such as heart, lung, testis, liver, and brain (Fig. 1B).
Figure 1.
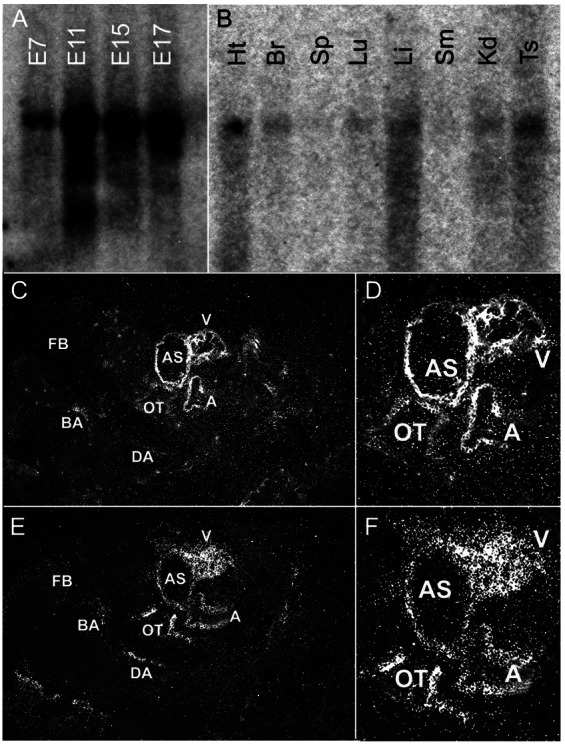
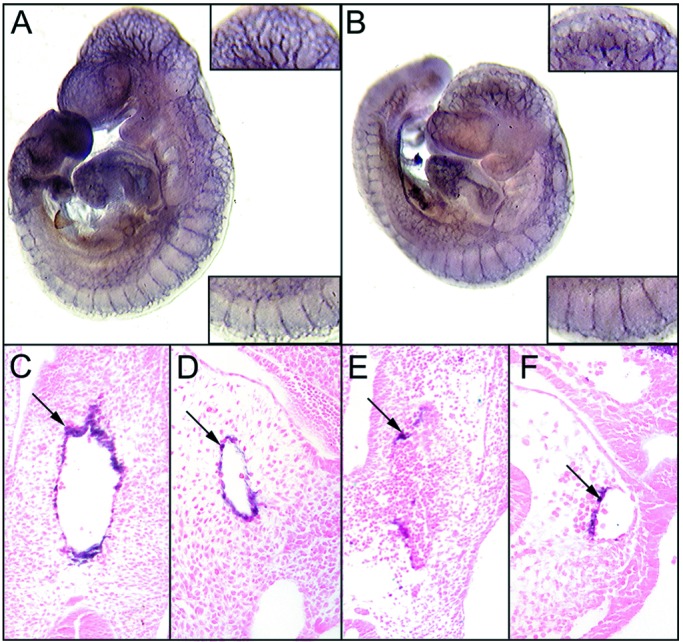
Expression of erk5 during development and in adult tissues. Multitissue blots from CLONTECH were hybridized with erk5 cDNA probes as described in Materials and Methods. (A) A high level of erk5 mRNA was present at various embryonic stages, with a significant increase between E7 and E11 of development. (B) Ubiquitous but low-level erk5 expression was observed in various adult tissues: Ht, heart; Br, brain; Sp, spleen; Lu, lung; Sm, skeletal muscle; Kd, kidney; Ts, testis. (C and D) In situ hybridization of E9.5 was performed to localize erk5 expression. High levels of erk5 expression were localized to the developing heart and vascular structures. (E and F) Serial sections also were hybridized with an SM22α probe to highlight the developing cardiovascular system. Of note, regions that showed an SM22α signal were similar to those that showed an erk5 signal. FB, forebrain; BA, branchial artery; DA, dorsal aorta; OT, outflow tract; AS, aortic sac; A, atrium, V, ventricle.
Because there was a significant increase in erk5 expression between E7 and E11, we examined the distribution of erk5 transcript in E9.5 embryos by in situ hybridization. erk5 transcripts were expressed in the heart and developing vasculature (Fig. 1 C and D). The expression of erk5 was apparent in the outflow tract and some arteries such as the branchial arch arteries (Fig. 1C), and it was strongly detected in the developing myocardium (Fig. 1D). Serial sections of embryos also were hybridized with a specific SM22α probe to highlight the developing cardiovascular system (Fig. 1 E and F). erk5 expression colocalized with areas showing strong SM22α expression, providing further support that the highest levels of erk5 expression were localized to the developing cardiovascular system.
Loss of Erk5 Resulted in Embryonic Lethality at E9.5–10.5.
To investigate the role of Erk5 in development, we disrupted the erk5 gene by homologous recombination in embryonic stem cells (see Fig. 6). Because the data were similar from two independently generated mouse lines for all parameters studied, the data were combined. Mice heterozygous for the erk5 mutant allele grew to adulthood, were fertile, and appeared normal phenotypically. However, genotyping of 258 adult offspring from heterozygous intercrosses detected no viable homozygous null mice (erk5−/−, see Table 1). Subsequently, staged embryos from heterozygous crosses were analyzed. The distribution of erk5−/− embryos was normal at E8.5, and +/+ and −/− embryos were of similar size. Thus, wild-type and mutant embryos did not appear to have significantly different rates of growth up to this time, although Erk5 has been shown to play a key role in the proliferative response to certain growth factors such as EGF (16). However, it is likely that any Erk5-specific effect on EGF-dependent proliferation in vivo could be masked by the up-regulation or increased activation of other signaling molecules/growth factors. At E9.5, mutant embryos appeared smaller than +/+ embryos (likely because of multiple vascular and cardiac developmental defects; see below), and abnormal or necrotic erk5−/− embryos appeared with increasing frequency from E10.5 to E11.5. In fact, although small numbers of −/− embryos could be detected at E10.5 and E11.5, all mutant embryos recovered at these stages were found to be nonviable (Table 1). Taken together, these data indicate that the erk5−/− embryos died at E9.5–E10.5.
Table 1.
Mouse genotypes for various embryonic developmental stages and adult mice
| E | +/+ | +/− | −/− | Total | |
|---|---|---|---|---|---|
| Adults | — | 86 | 172 | 0 | 258 |
| Embryos | 8.5 | 18 | 37 | 14 | 69 |
| 9.5 | 17 | 48 | 13 | 78 | |
| 10.5 | 5 | 7 | 6* | 18 | |
| 11.5 | 10 | 16 | 4* | 30 | |
| 13.5 | 2 | 5 | 0 | 7 |
Two independent embryonic stem cell-derived erk5+/− lines were generated and bred, and the data were combined. No viable erk5−/− adult mice were observed after genotyping 258 offspring of erk5+/− mating. Timed matings revealed that −/− embryos appeared with decreasing frequency after E9.5, and all −/− embryos that were identifiable from E10.5 to E11.5 were nonviable.
Embryos not viable.
erk5−/− Embryos Displayed Defects in Cardiac Development.
The embryonic cardiovascular system is the first organ system to develop and is critical for fetal survival beyond E9.5 (18). At the linear heart tube stage (E8.0), the hearts of erk5−/− appeared normal both phenotypically and histologically (data not shown). At E9.5, however, erk5−/− hearts failed to undergo normal rightward looping, and there was excessive pericardial fluid accumulation (compare Fig. 2A, wild type, to 2B, erk5−/−), suggesting hemodynamic insufficiency. Histological analysis confirmed that further development of the right ventricle in erk5−/− hearts was retarded compared with wild type (Fig. 2 C and D). In addition, a large space between the heart and pericardium in erk5−/− embryos was evident, consistent with pericardial fluid accumulation (Fig. 2D, asterisk). Histological analysis also revealed that the well organized trabeculae in the wild-type hearts (Fig. 2C, arrowhead) were disorganized in erk5−/− hearts (Fig. 2D, arrowhead). Although the endocardial lining in wild-type embryos was populated with thin, elongated endothelial cells (Fig. 2E), the endothelial cells lining the myocardium of erk5−/− embryos appeared disorganized and had a rounded morphology (Fig. 2F).
Figure 2.
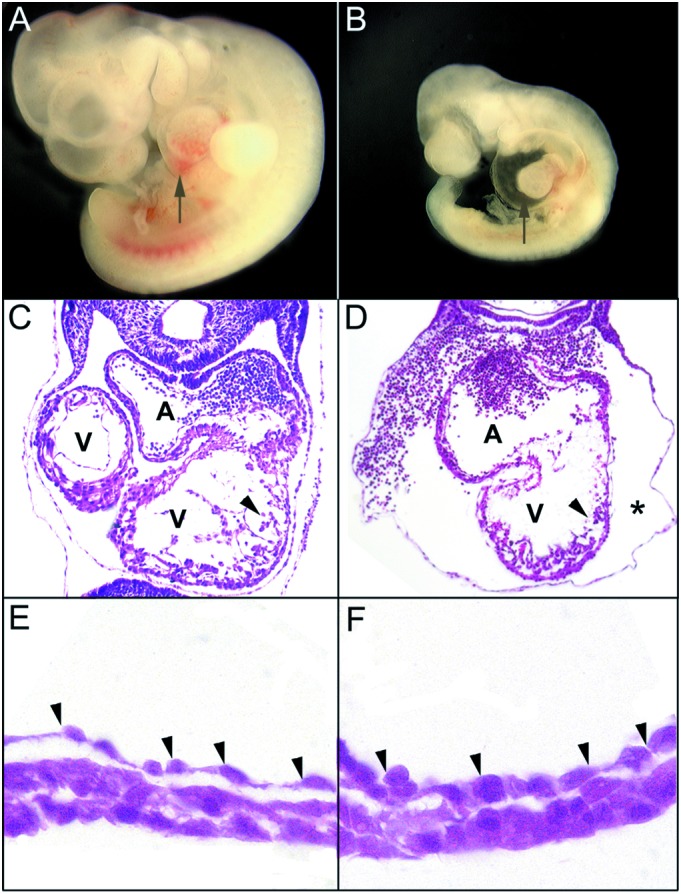
erk5−/− embryos displayed multiple defects in cardiac development. Lateral view of the wild-type (A) and mutant (B) E9.5 embryos demonstrate retarded cardiac development in erk5−/− embryos. Arrows point to the heart region. Histological examination of +/+ (C) and −/− (D) hearts revealed that erk5−/− hearts failed to undergo normal looping. The common atrial chamber of the mutant hearts also appeared enlarged, the ventricular trabeculations (arrowheads) were attenuated and disorganized, and there was notable pericardial edema (asterisk). A, atrium; V, ventricle. High-power magnification of the hearts of wild-type (E) and erk5−/− (F) embryos revealed that the endothelial cells (arrowheads) of mutant hearts were rounded as compared with the thin, elongated cells present in the wild type.
Effect of erk5 Deletion on Embryonic Gene Expression.
To verify that the gene-targeting strategy resulted in a loss of functional Erk5, we cultured MEFs from E9.5 embryos and treated them with either EGF or vehicle. Unphosphorylated and phosphorylated Erk5 could be readily detected in +/+ MEFs, and EGF caused an increase in the amount of phospho-Erk5 detected (Fig. 3A, lanes 1 and 2). In contrast, no Erk5 could be detected in either vehicle or EGF-treated MEFs derived from −/− embryos (Fig. 3A, lanes 3 and 4). Because the erk5−/− phenotype was similar to the mef2c−/− phenotype (12), we examined the expression of Mef2C in E9.5 hearts by Taqman PCR and in MEFs by Western blot. Both Taqman and Western analysis revealed no change in the level of Mef2C in +/+ vs. −/− embryos (data not shown).
Figure 3.
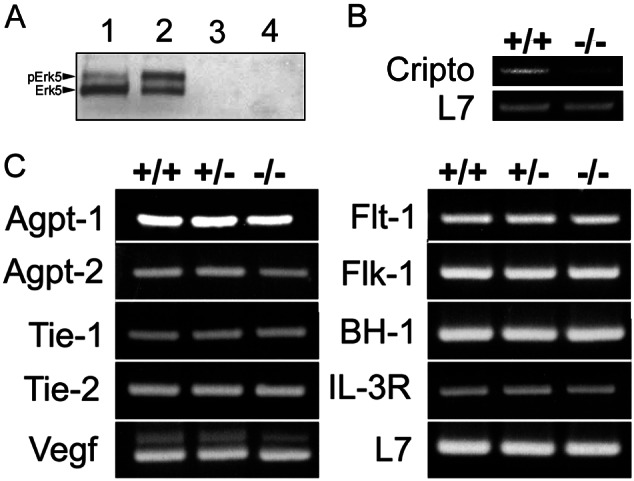
Analysis of gene expression in E9.5 erk5 embryos. (A) Western blot of erk5+/+ and erk5−/− MEFs shows absence of functional Erk5 in knockout mice. Lane 1, +/+, unstimulated; lane 2, +/+, 100 ng/ml EGF; lane 3, −/−, unstimulated; lane 4, −/−, 100 ng/ml EGF. (B) RT-PCR of RNA isolated from erk5+/+ and erk5−/− E9.5 hearts showed decreased expression of a potential Mef2C-dependent gene, cripto, in mutant embryos. L7 was used as an internal control. (C) RNA extracted from +/+, +/−, and −/− embryos was used to amplify a panel of genes known to play important roles for vascular development (flt-1 and flt-4, tie-1 and tie-2, agpt-1 and agpt-2, flk-1, and vegf) and hematopoiesis (IL-3R and βH-1). L7 was used as an internal control. No obvious expression differences among genotypes were noted.
To obtain some initial evidence of the proposed Erk5–Mef2C pathway in vivo, we examined the expression of two genes that were determined recently to be differentially expressed in wild-type vs. Mef2C mutant mice, cripto and CHAMP (15), in erk5+/+ and −/− E9.5 hearts. Both are highly expressed in the embryonic heart early in development, and although the function of CHAMP is yet to be elucidated, evidence suggests that cripto is required for cardiac development (19). Additionally, they are not contractile proteins or metabolic pathway genes, either of which could be altered secondarily to the cardiovascular development defect in erk5−/− embryos. Preliminary RT-PCR analysis revealed decreased CHAMP (data not shown) and cripto (Fig. 3B) expression in erk5−/− hearts compared with control hearts. As such, these data provide initial evidence for a functional Erk5–Mef2C signaling pathway in vivo.
Because many of the extraembryonic and embryonic vascular defects were reminiscent of angiogenic growth factor/receptor-deficient mice (20), we also used RT-PCR to determine whether deletion of erk5 could potentially affect expression of one or more of these factors. As shown in Fig. 3C, similar expression patterns were observed among +/+, +/−, and erk5−/− embryos at E9.5 for a variety of angiogenic signaling molecules/receptors. Additionally, RT-PCR analysis did not reveal altered expression of flt-4 (data not shown) or genes relevant to hematopoiesis such as βH-1 and IL-3R. We also performed Taqman PCR on several genes that were examined originally by RT-PCR and found that the Taqman data agreed closely with that of the original RT-PCR results. These results suggest that Erk5 does not regulate the expression of important vasculogenic/angiogenic molecules. However, given the similar phenotype of erk5−/− embryos to other knockouts such as agpt-1 and tie-2, Erk5 may be involved in the signaling cascade of specific angiogenic molecules.
erk5−/− Embryos Displayed Yolk-Sac, Placental, and Embryonic Vascular Defects.
The yolk sacs of both the wild-type and homozygous mutants at E8.5 were expanded and had identifiable blood islands, and embryos were of similar size and appearance (data not shown). Because blood islands are formed de novo from the condensation of mesoderm, their presence in erk5−/− yolk sacs suggested that vasculogenesis was largely unaffected by deletion of the erk5 gene. At E9.5, the wild-type yolk sac developed a well formed, hierarchical vascular network with large collecting vessels (Fig. 4A). In contrast, erk5−/− yolk sacs at E9.5 generally were pale, with blood cells scattered in some regions and no distinct blood vessels evident (Fig. 4B). Histologic analysis confirmed that +/+ yolk sacs had discrete, well defined vessels, whereas the vessels of mutant yolk sacs were thinner and contained fewer red blood cells (Fig. 4 C and D). PECAM staining revealed that endothelial cells were found in both wild-type and mutant yolk sacs (data not shown). Taken together, these data suggest that the failure to form blood vessels in the mutant yolk sacs was not caused by a lack of endothelial cells but rather a failure of further vascular maturation.
Figure 4.
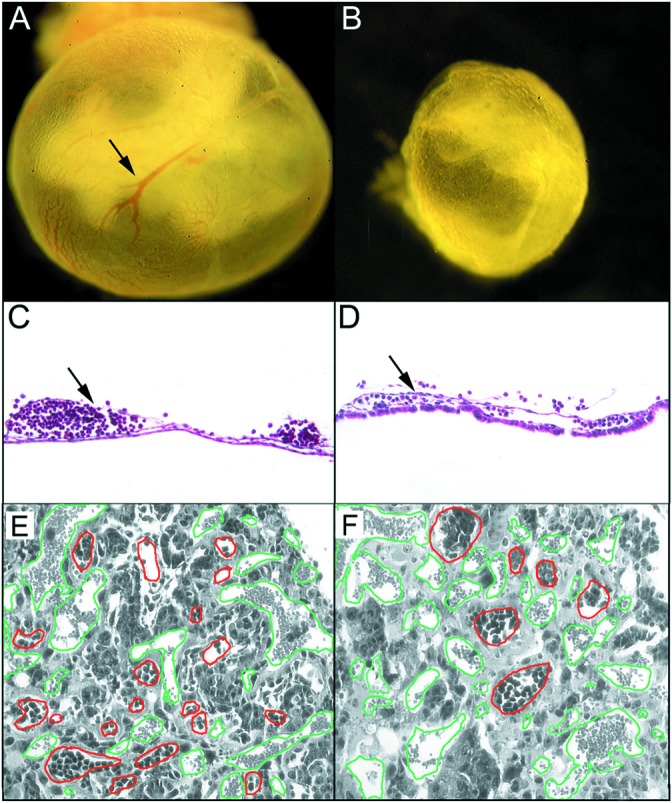
erk5−/− embryos displayed defects in yolk-sac and placental angiogenesis. Whole-mount visualization of wild-type (A) and mutant (B) E9.5 yolk sacs demonstrated that the large collecting vessels present in wild-type yolk sacs (arrow) were nearly absent in erk5−/− mice. Further histological analysis revealed the well defined collecting vessels in wild-type yolk sacs (arrow in C) were collapsed and contained fewer blood cells in erk5−/− yolk sacs (arrow in D). Placental vasculature was analyzed in hematoxylin/eosin-stained sections of wild-type (E) and mutant (F) placentas. Embryonic vessels, as defined by the presence of nucleated red cells, are outlined in red and maternal vessels are outlined in green to better highlight differences. (F) erk5−/− placentas had fewer embryonic vessels, and the vessels failed to invade deep into the labyrinth layer as compared with wild type.
We further examined the development of the placenta and placental vasculature in wild-type and erk5−/− mice. As compared with wild-type placentas (Fig. 4E), the labyrinthine region of erk5−/− placentas had few embryonic blood vessels (Fig. 4F, outlined in red). Furthermore, there was much less intermingling of maternal (outlined in green) and embryonic blood vessels in the erk5−/− placenta compared with wild type (Fig. 4 E and F). In general, vascular endothelial cells were present in erk5−/− placentas but seemed unable to invade the labyrinthine region efficiently, suggesting a defect in angiogenesis.
In addition to yolk-sac and placental vascular defects, whole-mount PECAM staining at E9.5 revealed significant alterations in the embryonic vasculature. Staining revealed that blood vessels in the intersomitic regions of wild-type and erk5−/− embryos were similar (Fig. 5 A and B Lower Right Insets), suggesting that the absence of Erk5 did not significantly prevent vasculogenesis. However, vascular complexity, as demonstrated particularly in capillaries in the head region, was reduced in erk5−/− embryos compared with wild-type embryos (Fig. 5 A and B). Specifically, wild-type embryos had undergone vascular pruning/maturation resulting in the formation of a complex vascular network (Fig. 5A Upper Right Inset). On the other hand, vessels in this region of erk5−/− embryos were all of similar size and lacked the complex branching pattern indicative of vascular maturation (Fig. 5B Upper Right Inset). Further examination of the embryonic vasculature of erk5−/− embryos revealed attenuation in the degree of investment of SmαA-positive cells surrounding multiple embryonic vessels. At this stage of development the caudal dorsal aorta as well as branchial arch arteries of wild-type embryos were invested with a thin layer of SmαA-positive cells (Fig. 5 C and D, respectively). However, the dorsal aorta and branchial arteries of erk5−/− embryos were invested with comparably fewer SmαA-positive cells (Fig. 5 E and F). The observed defects in yolk-sac and placental vascular development, the failure of the vascular plexus in erk5−/− embryos to remodel into a mature vascular network, and the evidence of decreased smooth muscle cell investment of embryonic vessels are consistent with a role for Erk5 in the angiogenic process.
Figure 5.
Embryonic vascular defects were present in erk5−/− embryos. Whole-mount PECAM staining of +/+ (A) and −/− (B) E9.5 embryos revealed defects in vascular maturation/angiogenesis in erk5−/− mice. Intersomitic vessels (wild-type, Lower Right Inset in A; mutant, Lower Right Inset in B) appeared normal in erk5−/− embryos. However, the angiogenic pruning and remodeling that was present in the head region of wild-type embryos (Upper Right Inset in A) was absent in erk5−/− embryos (Upper Right Inset in B). Note the disorganized vasculature and lack of complex branching in the erk5−/− embryos. Further analysis of the embryonic vasculature revealed that SmαA-positive cells did not invest the vessels of mutant embryos efficiently. (C) Dorsal aorta. (D) Branchial artery, wild type. (E) Dorsal aorta. (F) Branchial artery, erk5−/−.
Discussion
Although the intracellular kinase signaling pathways required for normal cardiovascular development have yet to be elucidated fully, the generation of kinase-deficient mouse strains has enabled researchers to begin to dissect several key elements (6–8). When these data are examined together with in vitro studies, where the upstream activators and downstream effectors within these pathways have been demonstrated, putative MAPK modules begin to emerge that may play a role in cardiovascular development. Previous in vitro studies have shown that Mekk3 can activate Mek5 (2), and Mek5 has been shown specifically to activate Erk5 (21). Erk5, in turn, can translocate to the nucleus and affect gene expression through its transcriptional activation domain (22) or through phosphorylation of transcription factors such as Mef2C (3). Taken together, these studies provide evidence for one possible MAPK module: Mekk3 activation–Mek5 activation–Erk5 activation–Mef2C phosphorylation.
Consistent with the above-mentioned proposed Erk5 signaling pathway, the phenotype of erk5−/− embryos closely resembles that of mekk3−/− (8) and mef2c−/− (11–13) embryos. Mekk3 also has been shown to induce p38-dependent Mef2C phosphorylation (3, 10). In fact, p38α−/− embryos (7) display a similar phenotype as mekk3−/− (8), mef2c−/− (11–13), and erk5−/− embryos. Taken together, these data suggest a Mekk3-dependent pathway that may activate p38α- and/or Erk5-dependent phosphorylation of downstream targets such as Mef2C that are critical for normal cardiovascular development. Of note, although hearts from erk5−/− embryos expressed similar levels of Mef2C compared with +/+ embryos, RT-PCR analysis suggested that erk5−/− hearts expressed lower levels of the Mef2C-dependent genes, CHAMP and cripto, providing preliminary evidence for a functional Erk5–Mef2C pathway in vivo. Although these data provide initial in vivo evidence of Erk5-dependent Mef2C activation during cardiovascular development, future studies that use a combination of in vivo/in vitro model systems together with expression and mutation analysis will define further the role of Erk5/Mef2C-dependent signaling.
Previous studies have shown that both p38α and Erk5 can phosphorylate Mef2C, but at distinct sites (3). Given that Mef2C, a MADS-box transcription factor, associates with other proteins to alter gene transcription (23), Erk5 phosphorylation likely would result in different interactions from those that depend on p38α phosphorylation. Indeed, although the phenotypes of erk5−/− and p38α−/− embryos are somewhat similar, they also are distinct in some aspects (7, 24, 25). For example, erk5−/− embryos displayed a more severe defect in looping morphogenesis than p38α−/− embryos. Also, although both erk5−/− and p38α−/− embryos formed a disorganized vascular network, deletion of erk5 caused a further defect in smooth muscle cell investment of the vasculature. It is possible that the distinct posttranslational modifications of Mef2C by p38α or Erk5 would allow for unique protein–protein interactions that then would differentially affect gene expression. It is possible also that Erk5 may phosphorylate other as-yet-unidentified factors distinct from those that can be phosphorylated by p38α. Furthermore, Erk5 may affect gene transcription directly through its transcriptional activation domain (22). To distinguish among these possibilities in vivo, further studies that use mice containing more subtle mutations of genes within the proposed signaling pathway or that use more restricted gene-deletion techniques will be necessary.
Although the extracellular stimuli that lead to activation of Erk5 in vivo have not been elucidated fully, Erk5 has been shown to be activated by receptor tyrosine kinases (16, 26, 27) as well as other stimuli (3, 26, 28). Interestingly, deletion of the receptor tyrosine kinase, Tie-2 (29), and its ligand, Angiopoietin 1 (Agpt-1; ref. 30), lead to a similar phenotype as that of erk5−/− mice. Indeed, mice deficient in both Tie-2 and its ligand, Agpt-1, displayed defects in the formation of a complex branching network of vessels, the investment of vessels with pericytes/smooth muscle cells, and the morphology of the endocardial lining (31), all of which were observed in erk5−/− embryos. Although there is no direct evidence linking the Tie-2 kinase to Erk5 activation in vivo, it is interesting to speculate that Erk5 may represent one downstream kinase target of Agpt-1/Tie-2 signaling given the similar phenotype among the knockout mice. Additionally, shear stress may represent another potential extracellular stimulant that lies upstream of Erk5 activation, because it has been shown to lead to Erk5 phosphorylation in endothelial cells in a tyrosine kinase-dependent manner (26). It is well known that changes in flow and shear stress are major stimuli for vascular maturation/remodeling during embryonic development. Of note, the vascular developmental defects in erk5−/− embryos were observed shortly after the time when the embryonic cardiovascular system would be exposed to increases in vessel flow/shear stress (30). As such, these data suggest an interesting model whereby changes in mechanical forces may up-regulate signaling cascades that lead to subsequent activation of Erk5. Erk5 then may transduce signals required for further vascular maturation/remodeling either directly, through its transcriptional activation domain (22), or indirectly, through phosphorylation of downstream targets such as Mef2C (3). Further experiments aimed at examining the in vivo relationship among receptor tyrosine kinase signaling and/or mechanical forces and MAPK activation in embryonic angiogenesis may define these pathways further.
In this study, we generated erk5-deficient mice to assess the function of this MAPK and found that deletion of the erk5 gene resulted in embryonic lethality at E9.5–E10.5. Histological and immunohistological analysis of mutant embryos demonstrated severe defects in cardiac development and vascular maturation/angiogenesis. In contrast to cardiac and smooth muscle, we did not detect any overt morphologic changes in skeletal muscle that might have been anticipated on the basis of recent in vitro studies (32). However, because deletion of erk5 caused embryonic lethality at E9.5, mutants may have died before any overt change in skeletal muscle development could be examined. Defects in heart development were marked by failure to undergo normal looping and morphologic changes in the endocardial lining. Extraembryonic angiogenesis in the placenta and yolk sac also were affected severely. Furthermore, the formation of a complex branching network of vessels in the embryo, through further vascular pruning/maturation, was attenuated. Consistent with defects in vascular maturation/angiogenesis, there was a marked deficiency in investment of embryonic vessels with smooth muscle cells. Together, these data suggest a role for Erk5 in multiple aspects of cardiovascular development. The embryonic defects in maturation of the vascular network may further implicate Erk5 in angiogenic signaling. However, interpretation of data from mice that display complex embryonic cardiovascular phenotypes must be approached cautiously. Because the gene of interest has been deleted in all cells of the embryo, it is difficult to determine whether vascular and cardiac defects occur independently or whether defects in cardiac development have caused changes in early hemodynamic parameters that then, secondarily, affect vascular development. Concomitant changes in extraembryonic vasculature may complicate analysis further. However, we did localize high levels of erk5 expression to both the developing heart and vessels. Furthermore, previous studies have provided evidence that Erk5 is expressed in cultured endothelial cells (26), smooth muscle cells (28), and adult blood vessels and heart (21, 33). Taken together, these data provide correlative evidence for a potential role of Erk5 in both embryonic and adult cardiac and vascular biology. However, to address such complex issues in vivo, future studies that use tissue-restricted knockouts, conditionally regulated knockouts, and/or chimeric techniques to define the role of Erk5 in the embryonic and adult cardiovascular system further are needed. Nevertheless, the data obtained from the erk5−/− mice provide evidence for an in vivo role of Erk5 in embryonic development. As such, these data provide an important first step in elucidating the role of Erk5 signaling in cardiac and vascular biology.
Supplementary Material
Acknowledgments
We thank Ms. Lina Du for blastocyst injection, Ms. Brinley Furey for technical assistance, Ms. Heidi Cyr for animal facility maintenance, and Dr. Gary Owens for critically reviewing the manuscript.
Abbreviations
- MAPK
mitogen-activated protein kinase
- Erk
extracellular signal-related kinase
- Mek
MAPK kinase
- Mekk
Mek kinase
- Mef2C
myocyte enhancer factor 2C
- E
embryonic day
- RT
reverse transcription
- MEF
mouse embryonic fibroblast
- EGF
epidermal growth factor
- PECAM
platelet endothelial cell adhesion molecule
- Agpt-1
angiopoietin 1
References
- 1.Chang L, Karin M. Nature (London) 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Chao T H, Hayashi M, Tapping R I, Kato Y, Lee J D. J Biol Chem. 1999;274:36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 3.Kato Y, Kravchenko V V, Tapping R I, Han J, Ulevitch R J, Lee J D. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan C, Luo H, Lee J D, Abe J, Berk B C. J Biol Chem. 2001;276:10870–10878. doi: 10.1074/jbc.M009286200. [DOI] [PubMed] [Google Scholar]
- 5.Feuerstein G Z, Young P R. Cardiovasc Res. 2000;45:560–569. doi: 10.1016/s0008-6363(99)00372-7. [DOI] [PubMed] [Google Scholar]
- 6.Giroux S, Tremblay M, Bernard D, Cardin-Girard J F, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, Charron J. Curr Biol. 1999;9:369–372. doi: 10.1016/s0960-9822(99)80164-x. [DOI] [PubMed] [Google Scholar]
- 7.Mudgett J S, Ding J, Guh-Siesel L, Chartrain N A, Yang L, Gopal S, Shen M M. Proc Natl Acad Sci USA. 2000;97:10454–10459. doi: 10.1073/pnas.180316397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J, Boerm M, McCarty M, Bucana C, Fidler I J, Zhuang Y, Su B. Nat Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 9.Deacon K, Blank J L. J Biol Chem. 1999;274:16604–16610. doi: 10.1074/jbc.274.23.16604. [DOI] [PubMed] [Google Scholar]
- 10.Han J, Jiang Y, Li Z, Kravchenko V V, Ulevitch R J. Nature (London) 1997;386:296–299. doi: 10.1038/386296a0. [DOI] [PubMed] [Google Scholar]
- 11.Bi W, Drake C J, Schwarz J J. Dev Biol. 1999;211:255–267. doi: 10.1006/dbio.1999.9307. [DOI] [PubMed] [Google Scholar]
- 12.Lin Q, Schwarz J, Bucana C, Olson E N. Science. 1997;276:1404–1407. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Q, Lu J, Yanagisawa H, Webb R, Lyons G E, Richardson J A, Olson E N. Development (Cambridge, UK) 1998;125:4565–4574. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- 14.Shanahan C M, Cary N R, Metcalfe J C, Weissberg P L. J Clin Invest. 1994;93:2393–2402. doi: 10.1172/JCI117246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z P, Nakagawa O, Nakagawa M, Yanagisawa H, Passier R, Richardson J A, Srivastava D, Olson E N. Dev Biol. 2001;234:497–509. doi: 10.1006/dbio.2001.0277. [DOI] [PubMed] [Google Scholar]
- 16.Kato Y, Tapping R I, Huang S, Watson M H, Ulevitch R J, Lee J D. Nature (London) 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- 17.Regan C P, Adam P J, Madsen C S, Owens G K. J Clin Invest. 2000;106:1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava D, Olson E N. Nature (London) 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 19.Adamson E A, Minchiotti G, Salomon D S. J Cell Physiol. 2002;190:267–278. doi: 10.1002/jcp.10072. [DOI] [PubMed] [Google Scholar]
- 20.Carmeliet P. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 21.Zhou G, Bao Z Q, Dixon J E. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 22.Kasler H G, Victoria J, Duramad O, Winoto A. Mol Cell Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molkentin J D, Black B L, Martin J F, Olson E N. Mol Cell Biol. 1996;16:2627–2636. doi: 10.1128/mcb.16.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Sudo T, Senftleben U, Dadak A M, Johnson R, Karin M. Cell. 2000;102:221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 25.Adams R H, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda A R. Mol Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- 26.Yan C, Takahashi M, Okuda M, Lee J D, Berk B C. J Biol Chem. 1999;274:143–150. doi: 10.1074/jbc.274.1.143. [DOI] [PubMed] [Google Scholar]
- 27.Kamakura S, Moriguchi T, Nishida E. J Biol Chem. 1999;274:26563–26571. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 28.Abe J, Kusuhara M, Ulevitch R J, Berk B C, Lee J D. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- 29.Sato T N, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Nature (London) 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 30.Suri C, Jones P F, Patan S, Bartunkova S, Maisonpierre P C, Davis S, Sato T N, Yancopoulos G D. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- 32.Dinev D, Jordan B W, Neufeld B, Lee J D, Lindemann D, Rapp U R, Ludwig S. EMBO Rep. 2001;2:829–834. doi: 10.1093/embo-reports/kve177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang C C, Ornatsky O I, McDermott J C, Cruz T F, Prody C A. Nucleic Acids Res. 1998;26:4771–4777. doi: 10.1093/nar/26.20.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.