

Abstract
The human protein kinase X gene (PRKX) is a member of an ancient family of cAMP-dependent serine/threonine kinases here shown to be phylogenetically distinct from the classical PKA, PKB/Akt, PKC, SGK, and PKG gene families. Renal expression of the PRKX gene is developmentally regulated and restricted to the ureteric bud epithelium of the fetal metanephric kidney. Aberrant adult kidney expression of PRKX was found in autosomal dominant polycystic kidney disease. PRKX kinase expression markedly activated migration of cultured renal epithelial cells in the presence of cAMP; this effect was blocked by cell treatment with the PKA inhibitor H89 and was not observed in PKA-transfected cells. In addition, expression of PRKX kinase activated branching morphogenesis of Madin–Darby canine kidney cells in collagen gels even in the absence of cAMP and/or hepatocyte growth factor, an effect not seen with either PKA expression or expression of a mutant, kinase-inactivated PRKX. These results suggest that the PRKX kinase may regulate epithelial morphogenesis during mammalian kidney development. Because another member of the PRKX gene family (the Dictyostelium discoideum gene KAPC-DICDI) also plays a role in cellular migration, these studies suggest that regulation of morphogenesis may be a distinctive property of these genes that has been conserved in evolution that is not shared with PKA family genes.
The organization of cells into defined structures during development depends on morphogenetic programs of gene expression. During mammalian kidney organogenesis, reciprocal signaling interactions between the ureteric bud and metanephric blastema initiate the branching growth of the collecting duct system that result in the radially organized, highly intricate structure of the adult kidney (1, 2). Tubular morphogenesis of epithelial cells depends partly on the directional outgrowth of individual cell processes in response to bone-morphogenetic protein 7 (BMP-7; ref. 3) and signaling molecules that activate receptor tyrosine kinases including hepatocyte growth factor (HGF; ref. 4), epidermal growth factor, and other epithelial growth factor receptor ligands (5). Importantly, BMP regulation of epithelial branching morphogenesis in model systems and lower organisms has been shown to depend on cAMP-dependent protein kinases (3, 6, 7), although the individual roles played by different genes encoding these kinases in vertebrate epithelial organ development has yet to be systematically investigated.
In studies designed to elucidate signaling pathways in renal epithelial morphogenesis, we identified PRKX, a serine/threonine kinase gene on the X chromosome at Xp22.3 (8, 9) as an interesting candidate regulatory gene, because it was activated transcriptionally in fetal kidneys during kidney organogenesis but not expressed in adult kidneys. Previous studies by others have demonstrated functional differences between protein kinase X (PRKX) and protein kinase A (PKA) kinases (10) and implicated PRKX in granulocyte/macrophage lineage differentiation (11), also suggesting that PRKX may have important developmental functions. Because the catalytic domain of PRKX shares greater amino acid homology with the Dictyostelium and Drosophila kinase genes KAPC-DICDI and DC2 than with mammalian PKA genes (12–15), it is plausible that PRKX and KAPC-DICDI might share unique functions not conserved in PKA genes. KAPC-DICDI plays an important role in the development of Dictyostelium discoideum, notably in morphogenetic cell migration (16, 17) as well as in transcriptional regulation of cellular differentiation (16–19), implying that PRKX also might regulate morphogenesis in higher eukaryotes in addition to its proposed role in regulating cellular differentiation in hematopoietic lineages.
In studies reported here, we show by phylogenetic analysis that PRKX, KAPC-DICDI, DC2, Ascaris suum KAPC ASCU, and Caenorhabditis elegans kin-1/CAB41352 convincingly comprise an ancient gene family distinct from the PKA, PKB, PKC, SGK, or PKG kinase gene families. We also show that expression of the PRKX kinase, but not the PKA kinase, strongly activates cellular morphogenesis and drives the formation of epithelial tubular structures in vitro associated with a stimulation of cellular migration. Taken together, our results suggest that the PRKX kinase might regulate tubulogenesis during kidney development and supports the hypothesis that the PRKX gene family plays an important function in cellular morphogenesis in multicellular eukaryotes.
Materials and Methods
Reverse Transcription–PCR Cloning of PRKX.
Total RNA was isolated from human normal adult and autosomal dominant polycystic kidney disease (ADPKD) kidneys (20), and reverse transcription–PCR was performed by using degenerate primers (a gift of R. Reed, Johns Hopkins University School of Medicine, Baltimore) designed against the conserved protein kinase C (PKC)/PKA catalytic domains sequences FYAAE/QI/V (coding strand: GGCCGGATCCTTT/CTAT/CGCXGCXG/CAA/GA/GT, 512× degenerate) and YI/M/LAPEI (noncoding strand: GGCCGAATTCATT/CTCXGGXGCXAXA/GTA, 1,024× degenerate). A 215-bp PCR product amplified from ADPKD RNA was subcloned into pBluescript II KS(−) (pBSIIKS−) and used to screen a 19–23-week human fetal kidney λgt10 cDNA library (CLONTECH). Positive plaques were used for PCR amplification of the full-length PRKX (GenBank accession no. X85545; ref. 8) ORF, which was subcloned into PT-7 blue LIC (Novagen).
In Situ Hybridization.
Deparaffinized, dehydrated kidney sections were treated with proteinase K, prehybridized with triethanolamine/acetic anhydride, and hybridized overnight with digoxigenin-substituted PRKX antisense or sense probes. After washing in 0.1× SSC (0.15 M sodium chloride/0.015 M sodium citrate, pH 7.0), sections were incubated in antidigoxigenin antibody/alkaline phosphatase and color-developed with NBT reagent (Roche Molecular Biochemicals).
Construction of PRKX Expression Vectors.
Recombinant pEGFP/PRKX was made by PCR amplification of the 1,074-bp ORF of PRKX from the pT-7 blue/PRKX plasmid, which was subcloned in-frame to the C terminus of the enhanced green fluorescent protein (EGFP) coding sequence of the pEGFP-C3 expression vector (CLONTECH). Recombinant pFLAG/PRKX was made by adding an N-terminal Met codon followed by the FLAG epitope tag (MDYKDDDDK) coding sequence in-frame to the PRKX ORF by PCR subcloning into a modified pEGFP-C3 vector lacking EGFP. A K78R kinase-dead mutation was introduced into pFLAG/PRKX to generate the construct pFLAG/PRKX/K78R.
PRKX Immunoprecipitation Kinase Assay.
HEK293 cells were transfected by using Effectene (Qiagen, Chatsworth, CA) with pFLAG/PRKX washed after 16 h, grown for 48 h, washed, and lysed in 10 mM Tris⋅HCl, pH 7.2/150 mM NaCl/0.5% Triton X-100/0.5% Tween-20 + protease inhibitor mixture. Two milligrams of lysate protein was immunoprecipitated with anti-Flag M2 monoclonal antibody covalently coupled to agarose beads (Sigma), and after four washes PRKX kinase activity was measured by using kemptide (LRRASLG) substrate in the presence of ATP (100 μM), magnesium (10 mM), and 1 μCi (1 Ci = 37 GBq) of [32P]ATP (specific activity 3,000 Ci/mmol) by subtracting background kinase activity observed in vector-transfected cells. The v0 reaction rates and Km values were determined in immunoprecipitates of PKA-deficient FIB4 cells (21) transfected with pFLAG/PRKX or pFC-PKA over a range of kemptide concentrations (0.25–10 μM; 1 μM 8-Br-cAMP; 2-min, 30°C incubation).
PRKX cAMP-Responsive Element (CRE) Luciferase Promoter-Reporter Assays.
JAR and GH3 cells (22, 23) were transfected with pCRE-Luc reporter (Stratagene) alone, pEGFP-C3 alone, pFLAG/PRKX, or pFC-PKA (Stratagene). After washing and 48-h culture, cell lysates were used for luciferase analysis (Stratagene).
PRKX Nuclear Translocation Studies in Cell Culture.
PKA-deficient FIB4 cells (21) cultured on cover slips were transfected with 1 μg of pEGFP/PRKX by using Effectene, washed, grown for 48 h, treated with 100 μM 8-Br-cAMP at 0, 1, 3, 5, 10, 15, 20, 30, and 120 min, fixed with 4% paraformaldehyde in PBS, washed twice with 0.2% PBS/Tween, mounted on slides, and examined by confocal microscopy (MSSM Microscopy Core). For detection of the PKA regulatory subunit I (RI) by immunofluorescence, cells were incubated also with anti-RI antibody (1:500, Transduction Laboratories, Lexington, KY), washed three times in PBS, and incubated in Texas red/goat anti-mouse IgM (1:100) before washing, mounting, and viewing.
Coimmunoprecipitation of PRKX and RI Subunits.
FIB4 lysates prepared from pFLAG/PRKX-transfected cells were immunoprecipitated with anti-Flag M2 antibody beads (4°C overnight). After extensive washing, aliquots were fractionated by 10% SDS/PAGE and immunoblotted with anti-FLAG (1:500), anti-RIα (1:500, Transduction Laboratories), anti RII (1:500, Upstate Biotechnology, Lake Placid, NY), and anti-PKACα (Transduction Laboratories).
Epithelial Cell Migration Assay.
FIB4 cells were transfected with pEGFP-C3 vector, pFLAG/PRKX, or pFC-PKA as described above and incubated with calcein acetoxymethyl ester (Molecular Probes). For each experiment, 1,000 cells were plated in 0.8 ml of DMEM + 1% FBS in triplicate 24-well chambers adapted for the Fluoroblock apparatus (Becton Dickinson), a modified Boyden chamber allowing labeled cells to attach in the upper chamber, and detecting fluorescent cells that migrate through the 8-μm pores into the lower chamber containing medium + 5% FBS. Measurements were taken after 4, 8, 12, 18, and 24 h in an HTS 7000 microfluorimeter (Perkin–Elmer) in the presence or absence of 100 μM 8-Br-cAMP and 10 μM H89.
Branching Tubulogenesis in Three-Dimensional Collagen Gels.
Madin–Darby canine kidney (MDCK) cells were transfected with pEGFP-C3 vector alone, pFLAG/PRKX, pEGFP/PRKX, the kinase-dead pFLAG/PRKX/K78R, or pFC-PKA by using VectorStat (1.25×; Genespan, Bothell, WA) to extend expression for 10–14 days. Washed cells were plated in 12-well chambers (500 cells per chamber) and cultured in type I collagen gels (1 vol of 10× MEM/1 vol of NaHCO3/942 mg/ml/1/2 vol of FBS/3 vol of collagen/4.5 vol of sterile water) with either no treatment or 100 μM 8-Br-cAMP, 10 μM H89 inhibitor, or 25 ng/ml HGF. After 10 days, gels were fixed with ice-cold methanol and stained with rhodamine-phalloidin for 8 h and examined on a Zeiss IM35 inverted microscope.
Results
Identification of PRKX as a Developmentally Regulated Serine/Threonine Kinase.
Because our previous studies implicated protein kinase abnormalities in ADPKD (24, 25), a degenerate reverse transcription–PCR strategy was used to identify novel serine/threonine kinases. A 215-bp reverse transcription–PCR product obtained from RNA prepared from an ADPKD sample was subcloned and sequenced and found to contain an ORF that fulfilled sequence criteria for serine/threonine kinases.
Phylogenetic Analysis of the PRKX Family.
A human 19–23-week fetal kidney cDNA library was screened by using the above kinase probe, and three cDNA clones were obtained and sequenced. One clone contained a full-length ORF for PRKX (8). Although the catalytic domain of PRKX has significant homology (56.6% identity) to human PKA-α (Table 1), it is more closely homologous to mouse PRKX (85.9%) Drosophila melanogaster DC2 (65.5%), D. discoideum PKA-C (59.6%), and A. suum PKA-C (57.8%) kinases. By phylogenetic analysis, the PRKX kinase genes can be distinguished from the PKA, PKB, SGK, PKC, and PKG kinase gene families (Fig. 1) and have been conserved broadly throughout eukaryote evolution.
Table 1.
Catalytic domain sequence comparisons of PRKX relation cAMP-dependent protein kinases
| Hs PRKX | Mm PRKX | Dm DC2 | Dd PKA-C | AsPKA-C | Hs PKAα | Ce PRKX | |
|---|---|---|---|---|---|---|---|
| Hs PRKX | 100 | 85.9 | 65.5 | 59.6 | 57.8 | 56.6 | 54.9 |
| Mm PRKX | 85.9 | 100 | 65.9 | 58.8 | 56.1 | 56.1 | 55.3 |
| Dm DC2 | 65.5 | 65.9 | 100 | 60.8 | 57.3 | 58.8 | 54.1 |
| Dd PKA C | 59.6 | 58.8 | 60.8 | 100 | 58 | 58.4 | 52.5 |
| As PRKX | 57.8 | 56.1 | 57.3 | 58 | 100 | 54.9 | 66.3 |
| Hs PKAα | 56.6 | 56.1 | 58.8 | 58.4 | 54.9 | 100 | 54.1 |
| Ce PRKX | 54.9 | 55.3 | 54.1 | 52.5 | 66.3 | 54.1 | 100 |
Figure 1.
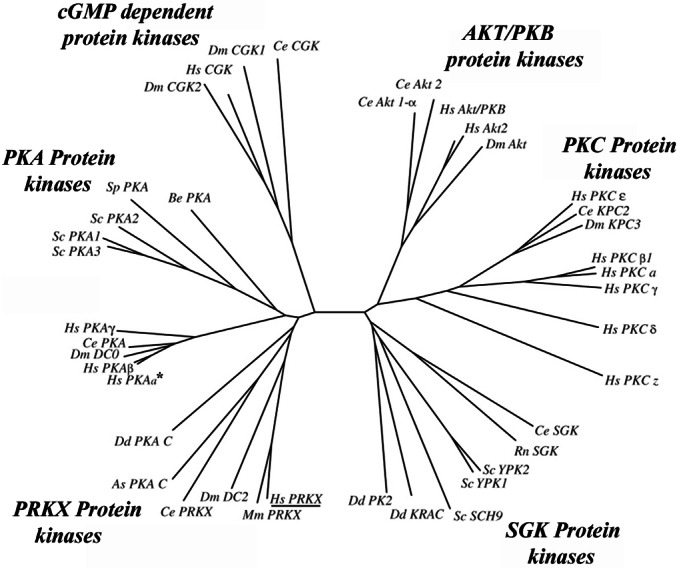
Phylogenetic analysis of the PRKX, PKA, PKC, protein kinase B (PKB), cGMP protein kinases, and serum- and glucocorticoid-inducible kinase (SGK)-related kinase families. The catalytic domains of the most highly PRKX homologous human, nematode, yeast, Dictyostelium, and D. melanogaster protein kinases identified by BLAST search were aligned with CLUSTALW (http://workbench.sdsc.edu/CGI/BW.cgi) to generate an unrooted phylogenetic tree with PHYLIP 3.5C (46). Protein kinase Y (PRKY, GenBank accession no. CAA75792) was excluded, because it lacks 81 C-terminal amino acid residues essential for kinase activity (47). The PKC family was added to the phylogenetic analysis as an outgroup (48). Except for the SGK kinase (from Rattos norvegicus) and mouse PRKX, all mammalian kinases used in this analysis were the human orthologs (see Table 2, which is published as supporting information on the PNAS web site, www.pnas.org). The PKA-α kinase is indicated by an asterisk, and human PRKX is underlined.
PRKX and PKA (26–29) family member amino acid sequences were compared and aligned (Fig. 2). The PRKX sequence (10, 30, 31) contains all essential conserved residues for serine/threonine kinase catalytic activity (32, 33). The aligned PRKX and PKA catalytic domains (Fig. 2A) showed extensive conservation of sequence in multiple regions including (i) the Mg-ATP binding domain (the β2–β3 glycine-rich loop starting with G50 in PKA and G56 in PRKX); (ii) the adjacent functionally critical kinase residues (PKA K72 and E91; PRKX K78 and E97); (iii) the PKA D166LKPEN catalytic loop (PRKX D172LKPEN); (iv) the PKA D184FG β8–β9 loop (PRKX D190FG); (v) the PKA P+1 peptide recognition residues L198, P202, and L205 (PRKX L204, P208, and L211); and (vi) the PKA T197 autophosphorylation site (T203 in PRKX). However, several residues (indicated by arrows in Fig. 2A) crucial for specific binding of the regulatory (R) subunit to the catalytic subunit (34) were not conserved in the PRKX family (PRKX Q92 and PKA E86; PRKX N140 and PKA R134; PRKX D199 and PKA G193), which might contribute to the reduced binding affinity of the RI regulatory subunit to PRKX (10) in comparison to PKA.
Figure 2.

Sequence alignment of PKA and PRKX family members. (A) Conserved residues of kinase domains I, II, III, VIb, VII, and VIII (horizontal lines) of the catalytic core domain (10), the PKA β-strands β2, β3, β7, β8, and β9 (filled arrows), the α-helices B, C, and D (double lines), and the PKA T197 autophosphorylation site are shown. The PRKX residues (G92, N140, and D199), which differ from PKA residues involved in RI subunit binding, are indicated by asterisks; PKA substrate P+1 recognition residues L198, P202, and L205 (domain VIII) are designated by small, filled pentagons. (B) C-terminal region alignments for PRKX and PKA families are shown with the large and small lobe anchor and C-terminal gate of PKA designated (33). Regions of conservation within the PRKX family shared with the PKA family are designated by the consensus sequences PxxP and GDtsNF.
Alignment of the C-terminal region of PRKX and PKA family sequences revealed previously unrecognized sequence conservation (PRKX G328DtsNF) (Fig. 2B), although many of the functionally important PKA residues in this region are not found in the PRKX family (of the PKA D329, Y330, E332, S338, and E346 residues, only one is conserved, Y336 in PRKX). In PKA, functional interactions of this C terminus with the large and small lobes of the catalytic domain have been found in the closed ternary PKA complex (29). Intriguingly, there is also a previously unrecognized conserved PxxP motif in the large lobe anchor region of both PKA and PRKX proteins conforming to the SH3 domain consensus binding-site sequence ψPxψP (35).
Developmental Regulation of PRKX Expression in Human Kidney.
PRKX Northern blot analysis showed expression of the 6-kb PRKX mRNA in human fetal kidneys (Fig. 3A, lanes 1 and 2) and fetal brain and lung (data not shown) but not in adult kidneys (Fig. 3A, lanes 3 and 4). In situ hybridization (Fig. 3B) detected PRKX mRNA in fetal kidney ureteric bud epithelial (arrowhead) and ADPKD epithelial cells but not in normal adult kidneys.
Figure 3.

Northern blot and in situ hybridization analysis of PRKX. (A) Lanes 1 and 2, 16-week normal fetal human kidney (FHK); lanes 3 and 4, normal adult human kidney (NHK). Arrow, 6-kb PRKX mRNA; arrowhead, 14S rRNA control. (B) Digoxigenin-substituted PRKX antisense riboprobe was hybridized to human fetal kidney sections and visualized by antidigoxigenin-alkaline phosphatase immunohistochemical staining. (A) Fetal human kidney at 12 weeks gestation showing ureteric bud PRKX expression (arrowhead). (B) Normal human adult kidney cortex (female). (C) Early-stage ADPKD kidney showing PRKX expression in cyst epithelial cells.
PRKX cAMP-Dependent Kinase Activity: Function as an Activator of CRE Promoter Elements and Nuclear Translocation.
Immunoprecipitation kinase assays of FLAG epitope-tagged PRKX expressed in HEK293 cells demonstrated PRKX kinase activity that was stimulated significantly by 8-Br-cAMP (P < 0.01) and inhibited completely by protein kinase inhibitor [PRKX kinase assay results, basal activity 39.7 ± 4.5; 8Br-cAMP (1 μM) stimulation, 278 ± 26.3; 8Br-cAMP (1 μM) plus protein kinase inhibitor (1 μM), 30.7 ± 2.9; units are pmol of 32P incorporated per min per mg of protein (mean ± SEM)]. By using anti-FLAG immunoprecipitates from pFLAG/PRKX or pFC-PKA-transfected PKA-deficient FIB4 (21) renal epithelial cells, calculated kemptide Km values for PRKX and PKA were found to be comparable for both kinases (6.7 ± 1.6 μM for PRKX and 4.4 ± 0.8 μM for PKA, mean ± SEM).
Cotransfection of JAR choriocarcinoma (data not shown) or GH3 pituitary tumor cell lines (22, 23) with either pFLAG/PRKX or pFC-PKA and the pCRE-Luc reporter led to a significant increase in luciferase-reporter activity (P < 0.01) and was stimulated 3–5-fold by treatment with 10 μM 8-Br-cAMP and completely inhibited by 10 μM H89 (Fig. 4A). The level of activation of the CRE luciferase reporter by PRKX was equivalent to that observed with cotransfection of the PKA expression construct pFC-PKA in JAR and GH3 cell lines.
Figure 4.
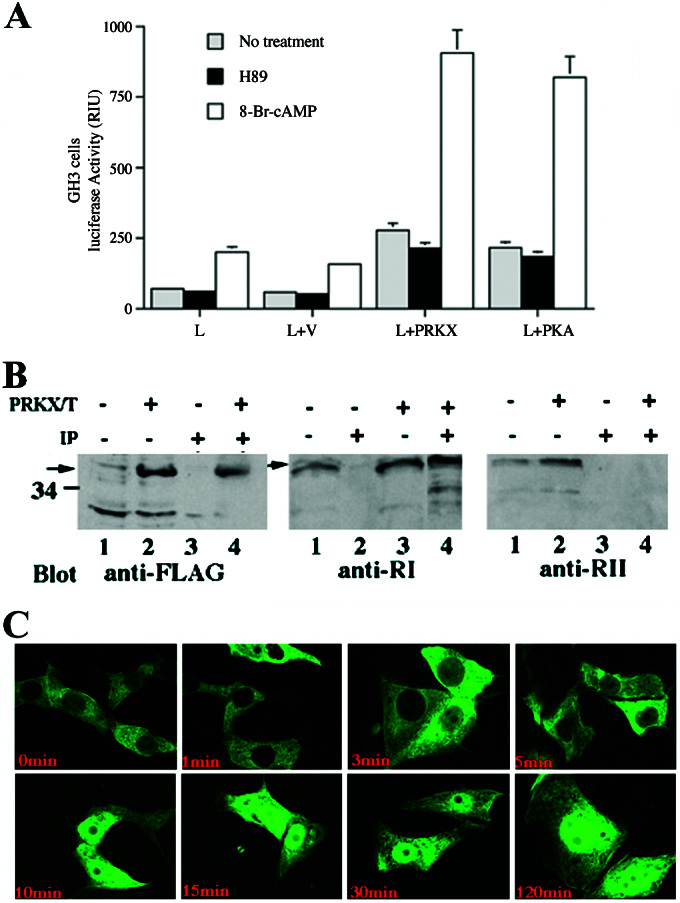
cAMP stimulation of PRKX-dependent CRE promoter elements, detections of PRKX–RI holoenzyme complexes, and nuclear translocation of EGFP/PRKX fusion protein. (A) Five micrograms of PRKX expression construct cotransfected with 3 μg of pCRE-luciferase into JAR cells incubated in the presence or absence of 1 μM 8-Br-cAMP or 1 μM H89 followed by luciferase activity assay 24 h after transfection. The letters on the abscissa refer to constructs used for cell transfection (L, CRE-luciferase promoter-reporter; V, empty expression vector; PRKX, pFLAG/PRKX; PKA, pFC-PKA). All values are means ± SEM for two separate transfection experiments carried out in triplicate. (B) PKRX–RI complexes were detected by immunoprecipitation experiments of PRKX-transfected cells. PRKX/T (+, −) indicates cell transfection with pFLAG/PRKX. IP (+, −) indicates immunoprecipitation with anti-FLAG antibody beads. Lanes 1 and 2 (Left and Right) and lanes 1 and 3 (Middle) represent immunoblots performed on whole-cell lysates. Lanes 3 and 4 (Left and Right) and lanes 2 and 4 (Middle) are immunoblots performed on anti-FLAG immunoprecipitated proteins. (C) cAMP-dependent PRKX nuclear translocation demonstrated in FIB4 cells transfected with pEGFP/PRKX and treated with 100 μM 8-Br-cAMP for 0, 1, 3, 5, 10, 15, 30, and 120 min.
PRKX subcellular localization to the cytoplasm in FIB4 cells (21) after transfection with pEGFP/PRKX was demonstrated by confocal microscopy (data not shown). Double immunolabeling of FIB4 cells after 48 h of transfection with pEGFP/PRKX showed partly overlapping diffuse cytoplasmic colocalization of EGFP/PRKX with the RI subunit. PRKX–RI complexes were detected in PRKX-transfected cells (Fig. 4B) by coimmunoprecipitation, although PRKX–RII complexes were not found. Treatment with 8-Br-cAMP resulted in nuclear translocation of the EGFP/PRKX within 10 min (Fig. 4C). These results are comparable to studies of hemagglutinin-tagged PRKX in a fibroblast cell line (10) and confirm that the GFP/PRKX fusion protein used in our experiments is comparable to native PRKX in this respect.
PRKX Stimulation of Epithelial Cell Migration.
Fluorescence microscopy of FIB4 cells transfected with pEGFP/PRKX showed elongation of cell shape after 15 min of treatment with 8-Br-cAMP, which was not seen in untransfected cells. To test whether PRKX might have an effect on cell migration, modified Boyden-chamber assays were carried out by using FIB4 cells. A significant increase in cell migration was measured in cells transfected with pFLAG/PRKX (Fig. 5) after treatment with 8-Br-cAMP that was inhibited by the PKA inhibitor H89. By contrast, no significant increase in migration was seen in untransfected cells, in cells transfected with empty vector alone, or in cells transfected with pFC-PKA. We also observed significant increases in epithelial cell migration in pFLAG/PRKX but not pFC-PKA-transfected MDCK and LLC-PK1 cell lines after treatment with 8-Br-cAMP (data not shown), demonstrating that the effect of PRKX on cell migration is not restricted to the PKA-deficient FIB4 cell line. To confirm that the observed differences on cell migration were not caused by deficient PKA expression in these experiments, we demonstrated comparable increases in cell lysate kinase activities in pFC-PKA- and pFLAG/PRKX-transfected cells.
Figure 5.
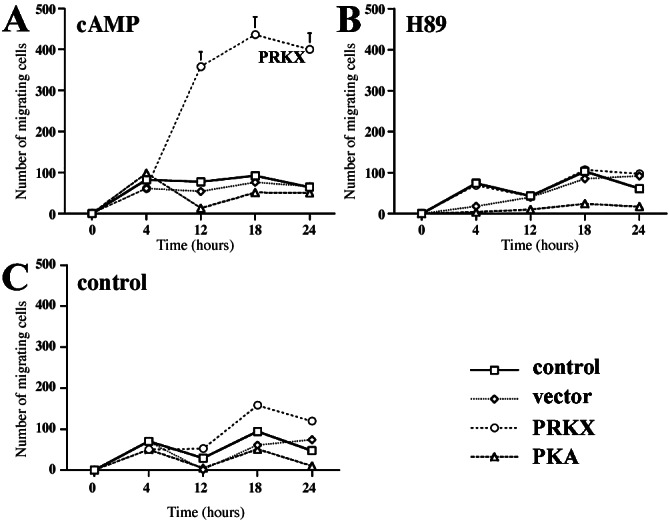
PRKX activates renal epithelial cell migration. pFLAG/PRKX-, pEGFP-C3-, pFC-PKA-transfected, and control FIB4 cells were labeled with calcein acetoxymethyl ester and plated 24 h posttransfection in Fluoroblock cell migration chamber wells. Four hours postattachment, cells cultured in 1% FBS were either untreated (C) or treated with 8-Br-cAMP (A) or the PKA inhibitor H89 (B) and cultured in wells containing 5% FBS in the lower chamber. The number of migrated cells was determined by fluorescence reading after 0, 4, 8, 12, 18, and 24 h. The values are mean ± SEM from three independent experiments in triplicate.
PRKX Stimulation of Branching Morphogenesis.
Cultured MDCK cells form small cysts in type I collagen gels that can be stimulated to undergo branching morphogenesis by addition of HGF (5, 36). In previously published work and our own experiments (Fig. 6), no branched structures are formed even after 10 days of culture in the absence of HGF in untransfected cells (Fig. 6A). MDCK cells transfected either with vector or pFC-PKA showed a complete absence of branched epithelial structures no different from untransfected control cultures (Fig. 6 D and G). However, MDCK cell transfection with pEGFP/PRKX alone induced branching morphogenesis in the absence of HGF (Fig. 6, J versus G, D, and A). Quantitative analysis of the effect of PRKX expression showed that pEGFP/PRKX transfection resulted in 3.0 ± 0.25 (mean ± SE) branched epithelial structures observed per 10 epithelial structures scored (n = 4 sets of observations) versus 0 ± 0 (mean ± SE) branched structures observed per 10 epithelial structures scored (n = 4 sets of observations) for vector-transfected or untransfected cultures. This activation of branching morphogenesis was shown to completely depend on the kinase activity of PRKX, because pFLAG/PRKX/K78R transfection expressing a kinase-dead PRKX (Fig. 6M) did not result in the formation of any branched epithelial structures. In the presence of 8-Br-cAMP, we observed modest cyst deformations in cultures transfected with pFC-PKA (Fig. 6H), suggestive of abortive branching morphogenesis not seen in untransfected or vector-transfected control cultures (Fig. 6 B and E). With 8Br-cAMP treatment of pEGFP/PRKX-transfected MDCK cells, large dilated branched cystic structures were observed (Fig. 6K) that were absent from 8-Br-cAMP-treated cultures after transfection with the kinase-dead pFLAG/PRKX/K78R expression vector (Fig. 6N). The addition of H89 to pEGFP/PRKX-transfected cells cultured with 8-Br-cAMP (Fig. 6L) completely abolished epithelial branching morphogenesis.
Figure 6.
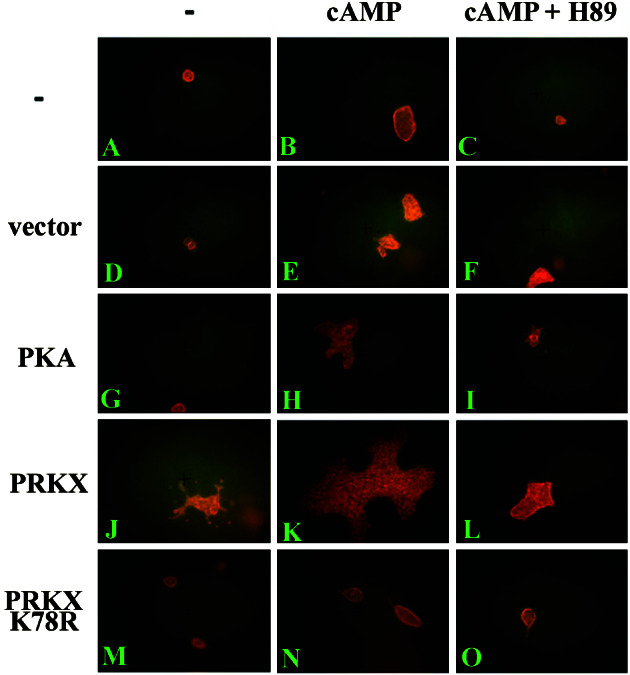
PRKX activates epithelial branching morphogenesis of cultured MDCK cells in collagen gels. MDCK cells (A–C) or MDCK cells transfected with an empty expression vector pEGFP-C3 (D–F), PKA expression vector pFC-PKA (G–I), pFLAG/PRKX (J–L), or the kinase-dead pFLAG/PRKX/K78R (M–O) were cultured in type I collagen gels for 10 days. The gels were untreated (A, D, G, J, and M) or treated with 100 μM 8-Br-cAMP (B, E, H, K, and N) or 100 μM 8-Br-cAMP and 10 μM H89 inhibitor (C, F, I, L, and O) and stained with rhodamine-phalloidin for 8 h for fluorescence microscopy visualization.
Discussion
Our studies show that PRKX, a recently discovered human serine/threonine kinase gene (8), is expressed normally during human metanephric kidney development, not expressed in the normal adult kidney, and expressed aberrantly in ADPKD (37, 38). By immunoprecipitation kinase assays in PKA-deficient FIB4 renal epithelial cells, we showed that PRKX is a protein kinase inhibitor-inhibitable cAMP-dependent protein kinase with a similar Km for the kemptide substrate to that observed for PKA-α kinase, a result that parallels the similar efficacy of these two kinases in the activation of a CRE-promoter reporter minigene in cotransfection assays.
Phylogenetic analysis based on sequence alignments of the core catalytic domains showed that PRKX belongs to a previously unrecognized unique and ancient family of related protein kinase genes rather than to the PKA, PKG, PKC, PKB/Akt, or SGK kinase gene families (12, 14, 15, 39–42). The PRKX kinases share significant amino acid sequence homology with PKA kinases and show conservation of residues that function in nucleotide binding, catalysis of phosphotransfer, substrate binding, and protein kinase inhibitor interactions. An SH3 domain ligand site conforming to the consensus ψPxψP (35) was found in PRKX and PKA kinase C termini that might mediate regulatory protein–protein interactions. The PRKX kinases are more divergent in this region of the protein from each other and PKA than in the more highly conserved regions involved in phosphotransfer and substrate binding. However, the protein alignments of PRKX family members showed conserved blocks of amino acids in this region, likely signifying that this portion of the protein plays an important regulatory role or is essential for kinase activity. Nonconservative sequence differences between these two kinase families identified in key PKA amino acids (E86, R134, and G193) required for RI interactions may contribute to reduced RI binding by PRKX. The N terminus of PKA also plays an important role in RI binding (43), and the lack of conservation here also may significantly alter RI as well as RII interactions with PRKX. Taken together, these results support the notion that PRKX and PKA kinases will be found to have multiple differences in substrate specificity and regulatory protein interactions, which may result in different biological functions that account for the independent evolutionary conservation of these two distinct cAMP-dependent kinase families.
The biological functions of PRKX family kinases has been studied most intensively in D. discoideum, in which the PKA-C kinase has been shown to play an essential role in the cell-shape changes and cell migration necessary for cell sorting during morphogenesis as well as in the transcriptional regulation of later cell fate differentiation during development (18, 19, 44, 45). The sole function thus far proposed for PRKX is in the regulation of granulocyte/macrophage differentiation (11) during hematopoiesis. The developmentally regulated expression of PRKX in the ureteric bud of the fetal kidney suggested that this kinase, similar to the Dictyostelium PKA-C kinase, also might regulate cell migration and morphogenesis earlier in development. To test this hypothesis, we developed a GFP/PRKX fusion protein that showed similar subcellular localization and kinetics of nuclear translocation as found previously with an hemagglutinin-tagged PRKX (10). Expression of this GFP/PRKX fusion protein in renal epithelial cells induced rapid elongation of cell shape, a dramatic increase in growth factor-mediated migration, and activation of branching morphogenesis in three-dimensional collagen gels. Strikingly, transfection of PKA or a kinase-dead PRKX had no effect on cell shape, migration, or branching morphogenesis in control experiments performed in parallel. These results, pointing to a specific role for the PRKX kinase in the control of epithelial motility and migration necessary for morphogenesis in the developing kidney, support the notion that the PRKX gene family may play an important role in the regulation of cellular migration in a broad range of eukaryotic organisms. Because migration and tubulogenesis depend on alterations in cell-matrix interactions, mediated via focal adhesions, as well as on alterations of the actin cytoskeleton, there are many potential molecular targets that might be phosphorylated by PRKX, but not PKA, in the developing kidney. The persistent and abnormal expression of PRKX in ADPKD further suggests that elucidating these molecular targets may be relevant to dissecting the molecular mechanisms associated with cystic diseases of the kidney.
Supplementary Material
Acknowledgments
We thank Barbara Bloswick for her expert technical assistance and Dr. Marius Sudol for his helpful comments and discussion. This work was supported by National Institutes of Health Grants RO1 DK44833 (to P.D.W.), RO1 DK 40698 (to P.D.W. and C.R.B.), and 1F32 DK10130 (to X.L.) and a New York/New Jersey National Kidney Foundation Fellow award (to X.L.). Confocal scanning microscopy was performed at the Mount Sinai School of Medicine core facility supported by National Institutes of Health Shared Instrumentation Grant ISIORRO 9145-01 and National Science Foundation Major Instrumentation Grant DBI-9724504.
Abbreviations
- HGF
hepatocyte growth factor
- ADPKD
autosomal dominant polycystic kidney disease
- PRKX
protein kinase X
- PKA
protein kinase A
- PKC
protein kinase C
- GFP
green fluorescent protein
- EGFP
enhanced GFP
- CRE
cAMP-responsive element
- RI
regulatory subunit I of PKA
- MDCK
Madin–Darby canine kidney
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Saxen L. Organogenesis of the Kidney. Cambridge, U.K.: Cambridge Univ. Press; 1987. [Google Scholar]
- 2.Ekblom P. In: The Kidney: Physiology and Pathophysiology. Seldin D W, Giebisch G, editors. New York: Raven; 1992. pp. 475–501. [Google Scholar]
- 3.Gupta I R, Piscione T D, Grisaru S, Phan T, Macias-Silva M, Zhou X, Whiteside C, Wrana J L, Rosenblum N D. J Biol Chem. 1999;274:26305–26314. doi: 10.1074/jbc.274.37.26305. [DOI] [PubMed] [Google Scholar]
- 4.Montesano R, Matsumoto K, Nakamura T, Orci L. Cell. 1991b;67:901–908. doi: 10.1016/0092-8674(91)90363-4. [DOI] [PubMed] [Google Scholar]
- 5.Sakurai H, Tsukamoto T, Kjelsberg C A, Cantley L G, Nigam S K. Am J Physiol. 1997;273:F463–F472. doi: 10.1152/ajprenal.1997.273.3.F463. [DOI] [PubMed] [Google Scholar]
- 6.Piscione T D, Yager T D, Gupta I R, Grinfeld B, Pei Y, Attisano L, Wrana J L, Rosenblum N D. Am J Physiol. 1997;273:F961–F975. doi: 10.1152/ajprenal.1997.273.6.F961. [DOI] [PubMed] [Google Scholar]
- 7.Lee Y S, Chuong C M. J Cell Physiol. 1997;170:153–165. doi: 10.1002/(SICI)1097-4652(199702)170:2<153::AID-JCP7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 8.Klink A, Schiebel K, Winkelmann M, Rao E, Horsthemke B, Ludecke H J, Claussen U, Scherer G, Rappold G. Hum Mol Genet. 1995;4:869–878. doi: 10.1093/hmg/4.5.869. [DOI] [PubMed] [Google Scholar]
- 9.Schiebel K, Winkelmann M, Mertz A, Xu X, Page D C, Weil D, Petit C, Rappold G A. Hum Mol Genet. 1997;6:1985–1989. doi: 10.1093/hmg/6.11.1985. [DOI] [PubMed] [Google Scholar]
- 10.Zimmermann B, Chiorini J A, Ma Y, Kotin R M, Herberg F W. J Biol Chem. 1999;274:5370–5378. doi: 10.1074/jbc.274.9.5370. [DOI] [PubMed] [Google Scholar]
- 11.Semizarov D, Glesne D, Laouar A, Schiebel K, Huberman E. Proc Natl Acad Sci USA. 1998;95:15412–15417. doi: 10.1073/pnas.95.26.15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann S K, Yonemoto W M, Taylor S S, Firtel R A. Proc Natl Acad Sci USA. 1992;89:10701–10705. doi: 10.1073/pnas.89.22.10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anjard C, Etchebehere L, Pinaud S, Veron M, Reymond C D. Biochemistry. 1993;32:9532–9538. doi: 10.1021/bi00088a003. [DOI] [PubMed] [Google Scholar]
- 14.Melendez A, Li W, Kalderon D. Genetics. 1995;141:1507–1520. doi: 10.1093/genetics/141.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalderon D, Rubin G M. Genes Dev. 1988;2:1539–1556. doi: 10.1101/gad.2.12a.1539. [DOI] [PubMed] [Google Scholar]
- 16.Mann S K, Brown J M, Briscoe C, Parent C, Pitt G, Devreotes P N, Firtel R A. Dev Biol. 1997;183:208–221. doi: 10.1006/dbio.1996.8499. [DOI] [PubMed] [Google Scholar]
- 17.Harwood A J, Hopper N A, Simon M N, Bouzid S, Veron M, Williams J G. Dev Biol. 1992;149:90–99. doi: 10.1016/0012-1606(92)90266-j. [DOI] [PubMed] [Google Scholar]
- 18.Primpke G, Iassonidou V, Nellen W, Wetterauer B. Dev Biol. 2000;221:101–111. doi: 10.1006/dbio.2000.9662. [DOI] [PubMed] [Google Scholar]
- 19.Aubry L, Firtel R. Annu Rev Cell Dev Biol. 1999;15:469–517. doi: 10.1146/annurev.cellbio.15.1.469. [DOI] [PubMed] [Google Scholar]
- 20.Lane M E, Kalderon D. Genes Dev. 1993;7:1229–1243. doi: 10.1101/gad.7.7a.1229. [DOI] [PubMed] [Google Scholar]
- 21.Botterell S H, Jans D A, Hemmings B A. Eur J Biochem. 1987;164:39–44. doi: 10.1111/j.1432-1033.1987.tb10989.x. [DOI] [PubMed] [Google Scholar]
- 22.Howard P, Day K H, Kim K E, Richardson J, Thomas J, Abraham I, Fleischmann R D, Gottesman M M, Maurer R A. J Biol Chem. 1991;266:10189–10195. [PubMed] [Google Scholar]
- 23.Maurer R A. J Biol Chem. 1989;264:6870–6873. [PubMed] [Google Scholar]
- 24.Du J, Wilson P D. Am J Physiol. 1995;269:C487–C495. doi: 10.1152/ajpcell.1995.269.2.C487. [DOI] [PubMed] [Google Scholar]
- 25.Kuo N, Norman J T, Wilson P D. Biochem Mol Med. 1997;61:178–191. doi: 10.1006/bmme.1997.2583. [DOI] [PubMed] [Google Scholar]
- 26.Zheng J H, Knighton D R, Parello J, Taylor S S, Sowadski J M. Methods Enzymol. 1991;200:508–521. doi: 10.1016/0076-6879(91)00167-u. [DOI] [PubMed] [Google Scholar]
- 27.Taylor S S, Zheng J, Radzio-Andzelm E, Knighton D R, Ten Eyck L F, Sowadski J M, Herberg F W, Yonemoto W M. Philos Trans R Soc London B. 1993;340:315–324. doi: 10.1098/rstb.1993.0073. [DOI] [PubMed] [Google Scholar]
- 28.Zheng J, Knighton D R, ten Eyck L F, Karlsson R, Xuong N, Taylor S S, Sowadski J M. Biochemistry. 1993;32:2154–2161. doi: 10.1021/bi00060a005. [DOI] [PubMed] [Google Scholar]
- 29.Narayana N, Cox S, Nguyen-huu X, ten Eyck L F, Taylor S S. Structure (London) 1997;5:921–935. doi: 10.1016/s0969-2126(97)00246-3. [DOI] [PubMed] [Google Scholar]
- 30.Chiorini J A, Zimmermann B, Yang L, Smith R H, Ahearn A, Herberg F, Kotin R M. Mol Cell Biol. 1998;18:5921–5929. doi: 10.1128/mcb.18.10.5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Pasquale G, Stacey S N. J Virol. 1998;72:7916–7925. doi: 10.1128/jvi.72.10.7916-7925.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanks S K, Quinn A M. In: Methods in Enzymology. Hunter T, Sefton B M, editors. Vol. 200. New York: Academic; 1991. pp. 38–62. [DOI] [PubMed] [Google Scholar]
- 33.Hanks S K, Quinn A M, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 34.Gibbs C S, Knighton D R, Sowadski J M, Taylor S S, Zoller M J. J Biol Chem. 1992;267:4806–4814. [PubMed] [Google Scholar]
- 35.Sudol M. Oncogene. 1998;17:1469–1474. doi: 10.1038/sj.onc.1202182. [DOI] [PubMed] [Google Scholar]
- 36.Montesano R, Schaller G, Orci L. Cell. 1991;66:697–711. doi: 10.1016/0092-8674(91)90115-f. [DOI] [PubMed] [Google Scholar]
- 37.Burrow C R. Pediatr Nephrol. 2000;14:240–253. doi: 10.1007/s004670050049. [DOI] [PubMed] [Google Scholar]
- 38.Wilson P D. J Am Soc Nephrol. 2001;12:834–845. doi: 10.1681/ASN.V124834. [DOI] [PubMed] [Google Scholar]
- 39.Mann S K, Firtel R A. Mech Dev. 1991;35:89–101. doi: 10.1016/0925-4773(91)90060-j. [DOI] [PubMed] [Google Scholar]
- 40.Mann S K, Firtel R A. Development (Cambridge, UK) 1993;119:135–146. doi: 10.1242/dev.119.1.135. [DOI] [PubMed] [Google Scholar]
- 41.Jung S, Hoffmann R, Rodriguez P H, Mutzel R, Hofer H W. Eur J Biochem. 1995;232:111–117. doi: 10.1111/j.1432-1033.1995.tb20788.x. [DOI] [PubMed] [Google Scholar]
- 42.Blaschke R J, Monaghan A P, Bock D, Rappold G A. Genomics. 2000;64:187–194. doi: 10.1006/geno.2000.6116. [DOI] [PubMed] [Google Scholar]
- 43.Herberg F W, Zimmermann B, McGlone M, Taylor S S. Protein Sci. 1997;6:569–579. doi: 10.1002/pro.5560060306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander S, Sydow L M, Wessels D, Soll D R. Differentiation (Berlin) 1992;51:149–161. doi: 10.1111/j.1432-0436.1992.tb00691.x. [DOI] [PubMed] [Google Scholar]
- 45.Brown J M, Firtel R A. Dev Biol. 1999;216:426–441. doi: 10.1006/dbio.1999.9485. [DOI] [PubMed] [Google Scholar]
- 46.Felsenstein J. Cladistics. 1989;5:164–166. [Google Scholar]
- 47.Orellana S A, Amieux P S, Zhao X, McKnight G S. J Biol Chem. 1993;268:6843–6846. [PubMed] [Google Scholar]
- 48.Paradis S, Ruvkun G. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.