

Abstract
The signaling pathways that lead to the localization of cellular protein to the area of interaction between T cell and antigen-presenting cell and the mechanism by which these molecules are further sorted to the peripheral supramolecular activation cluster or central supramolecular activation cluster regions of the immunologic synapse are poorly understood. In this study, we investigated the functional involvement of CD28 costimulation in the T cell receptor (TCR)-mediated immunologic synapse formation with respect to protein kinase C (PKC)θ localization. We showed that CD3 crosslinking alone was sufficient to induce PKCθ capping in naïve CD4+ T cells. Studies with pharmacologic inhibitors and knockout mice showed that the TCR-derived signaling that drives PKCθ membrane translocation requires the Src family kinase, Lck, but not Fyn. In addition, a time course study of the persistence of T cell molecules to the immunologic synapse indicated that PKCθ, unlike TCR, persisted in the synapse for at least 4 h, a time that is sufficient for commitment of a T cell to cell division. Finally, by using TCR-transgenic T cells from either wild-type or CD28-deficient mice, we showed that CD28 expression was required for the formation of the mature immunologic synapse, because antigen stimulation of CD28− T cells led to a diffuse pattern of localization of PKCθ and lymphocyte function-associated antigen-1 in the immunologic synapse, in contrast to the central supramolecular activation cluster localization of PKCθ in CD28+ T cells.
T cell activation involves a complex series of molecular interactions between T cells and antigen-presenting cells that include ligand/receptor pairs such as antigen-MHC/T cell receptor, B7/CD28 and intercellular adhesion molecule/lymphocyte function-associated antigen-1 (ICAM/LFA-1). Effective signaling through these and other ligand/receptor pairs requires a stable coupling of T cell and antigen-presenting cell (APC) for a prolonged period (1–3). This coupling is accomplished by the remodeling of the distribution of these receptors and ligands to the area of contact between APC and T cell, thus creating the so-called immunologic synapse (4). Not only are the interacting surface molecules concentrated at the immunologic synapse, but also localized to this region are a variety of intracellular molecules such as Lck, ZAP-70, and protein kinase C (PKC)θ involved in T cell receptor (TCR) signaling and molecules involved in the formation of a stable adherent complex between T cell and APC (e.g., filamentous actin and talin) (5, 6). In addition, the morphology of the immunologic synapse is complex with respect to its molecular organization in that some ligand/receptor pairs, such as ICAM/LFA-1, are located at the periphery of the synapse [referred to as peripheral supramolecular activation cluster (p-SMAC)], whereas others, such as Ag-MHC/TCR, are concentrated in a small central region [central supramolecular activation cluster (c-SMAC)] (6).
The signaling pathways that lead to the localization of cellular proteins to the area of interaction between T cell and APC and the mechanism by which these molecules are sorted to the p- and c-SMAC regions of the synapse are poorly understood and may involve multiple signaling pathways. The possibility of multiple signaling pathways is suggested by the finding that certain methods of T cell stimulation result in some aspects of the synapse being formed and not others. For instance, TCR-mediated signaling by low-affinity ligands (TCR antagonists) induces conjugates between T cells and APCs with the localization of some of the molecules found in a mature synapse to the region of interaction between APC and T cell (e.g., TCR, LFA-1), but not others (e.g., PKCθ, CD28) (7). Also, the formation of c- and p-SMAC is not observed after stimulation with low-affinity ligands of the TCR (6).
In this study, we have analyzed the requirements for localization of PKCθ to the immunologic synapse. This PKC isoform is a critical participant in the NF-κB pathway leading to IL-2 transcription in T cells and has been shown to localize to the c-SMAC region of the immunologic synapse (8–10). Our studies demonstrate that, whereas TCR-mediated signaling alone is sufficient for the localization of PKCθ to the immunologic synapse, CD28-mediated signals are required for the localization of PKCθ to the c-SMAC region of the synapse.
Materials and Methods
Animals.
The pigeon cytochrome c 88–104-specific TCR-transgenic mice AD10 (B10.A) and AND (B10 Br) were originally obtained from S. Hedrick (University of California, San Diego) (11, 12). Fyn−/− mice were purchased from The Jackson Laboratory. To generate mice that undergo normal thymic development but have no Lck in peripheral T cells, Lck-deficient mice were bred with mice that expressed Lck specifically in thymocytes (LGF+) because of having an Lck gene under control of the Lck proximal promoter. These mice, LGF+Lck−/−, have been described (13). CD28−/− AND mice (obtained from M. Croft, La Jolla Institute for Allergy and Immunology) were produced by breeding CD28−/− mice with AND TCR transgenic mice (14).
Antibodies and Reagents.
The following reagents were used in this study: polyclonal antibodies against PKCθ (Santa Cruz Biotechnology); biotinylated anti-mouse TCR Vβ3 (KJ25) and FITC-labeled anti-mouse LFA-1 (M17/4) (PharMingen); FITC-Phalloidin (Sigma–Aldrich), Cy5 conjugated AffiniPure donkey anti-rabbit IgG or Rhodamine Red-x-conjugated AffiniPure goat anti-hamster IgG (Jackson ImmunoResearch); CD11b, CD45R (B220), mouse CD8a(Ly-2), and streptavidin-coated MicroBeads (MACS, Miltenyi Biotec, Auburn, CA); 2C11-producing hybridoma cells (American Type Culture Collection); FluoroGuard Antifade Reagent (Bio-Rad); PP2 (Calbiochem).
Peptide Synthesis.
Peptides were synthesized on a Rainin symphony synthesizer and purified as described (15). Purity was routinely >95% after high-pressure liquid chromatography.
Cells.
The AD10 T cell clone and CH27 B lymphoma cells were cultured as described (15). Naïve CD4+ T cells were purified from lymph nodes and/or spleens of 8- to 12-week-old mice by using LS Separation Columns (MACS, Miltenyi Biotec) according to the manufacturer's instructions. Purity was assessed by immunofluorescence flow cytometry and was >95% for naive CD4+cells (CD45RB+, CD62L+, CD44low). Viability was >98% as determined by trypan blue exclusion.
T Cell Stimulation and Immunofluorescence Microscopy.
For the study of TCR-mediated capping, 2C11 was conjugated to Dynal M450 beads (Dynal, Lake Success, NY) per manufacturer's instructions. Cells (4 × 105) and beads (4 × 105) were coincubated for 45 min at 37°C and allowed to settle on poly-L-lysine-coated glass coverslips. Cells were fixed with 3% paraformaldehyde for 12 min, washed, and blocked with 3% BSA/PBS for 30 min. Samples were stained with FITC-anti-LFA-1. Phalloidin and PKCθ staining was performed on fixed and permeabilized cells (0.2% Triton-X-100 for 2 min). After washing, the cells were mounted in FluoroGuard antifade reagent. Capping of proteins was analyzed visually by using a confocal microscope (Bio-Rad). At least 100 T cell-bead conjugates were analyzed for cap formation in each experiment. In some experiments, T cells were pretreated at 37°C with 20 μM PP2 for 1 h.
For the study of the formation of the immunologic synapse induced by peptide-pulsed APCs, CH27 cells were preincubated with PCC88–104 (1 μg/ml) for 2 h at 37°C. T cells (3 × 105) and CH27 cells were mixed at 2:1 ratio and incubated at 37°C for the indicated times and allowed to settle on poly-L-lysine-coated glass coverslips. Cells were fixed and permeabilized as described above. Samples were processed for immunofluorescence staining with antibodies to LFA-1, TCR, talin, and/or PKCθ. In some experiments, images were obtained by using the DeltaVision system (Applied Precision, Issaquah, WA) with an Olympus 1 × 70 microscope, equipped with a 100-W mercury lamp and a KAF1400 chip-based cooled charge-coupled device camera. Exposure times were 0.1–0.5 s with 2-binning, 100 × 1.35 oil objectives. The three-dimensional reconstruction of the T-APC contact area was generated with 100-nm serial sections of x–y images alone with the z axis and subsequent analysis with softworx Ver. 2.5 (16). The size of the T-APC contact area was quantitated with the nih image Ver. 1.61 software system.
Results
TCR Signaling Is Sufficient to Mediate Membrane Localization of PKCθ and Requires Lck.
To investigate the requirement for the localization of PKCθ to the immunologic synapse, initial experiments were conducted by using anti-CD3-coated beads and naïve CD4+ T cells to determine the effects of TCR-mediated signaling on the intracellular distribution of PKCθ. The results, shown in Fig. 1, indicated that as a result of the anti-CD3 coated bead stimulation, PKCθ localized to the cell membrane in the area of contact between the T cells and the beads. This localization was similar to that of F-actin (Fig. 1B), LFA-1, and TCR (data not shown). In contrast the distribution in unstimulated cells of PKCθ was homogeneous throughout the cytoplasm.
Figure 1.

Stimulation through the TCR is sufficient for induction of PCKθ capping. Naive AD10 TCR transgenic CD4+ T cells were stimulated at 37°C for 45 min with beads coated with anti-CD3 antibodies. As controls, T cells were also incubated with uncoated beads. Cells were stained with anti-PKCθ (A) or phalloidin (B) and analyzed by confocal microscopy. Both PKCθ and phalloidin were localized to the contact region between T cells and the anti-CD3-coated beads. Data are from one representative experiment of five performed.
To gain some insight into the TCR-mediated signals required for the membrane localization and capping of PKCθ, the effect of PP2, an inhibitor of Src family kinases (17, 18), on PKC capping was studied. As shown in Fig. 2A, pretreatment of T cells with PP2 completely abrogated the capping of PKCθ after stimulation with anti-CD3-coated beads. In contrast, pretreatment of cells with an inhibitor of ZAP-70, piceatannol (19), had no effect on PKCθ capping (data not shown). To determine which of the two major Src family kinases expressed in T cells, Fyn or Lck, was critical for TCR-induced PKCθ capping, we analyzed the distribution of PKCθ after anti-CD3 stimulation of Fyn- or Lck-deficient T cells. The data illustrated in Fig. 2B indicated that, whereas capping of PKCθ proceeded normally in Fyn-deficient T cells, it was not observed in Lck-deficient T cells, thus indicating that Lck, but not Fyn, is required for the cocapping of PKCθ after cross-linking of the TCR with anti-CD3-coated beads.
Figure 2.

TCR-mediated PCKθ capping is Lck-dependent. (A) Naive AD10 CD4+ T cells were pretreated at 37°C for 1 h with vehicle alone (0.1% DMSO) (a) or 20 μM PP2 (b). The T cells were then incubated with anti-CD3-coated beads for 45 min, after which time T cells were stained for PCKθ. The asterisk indicates the position of the CD3-coated beads. (B) Naïve CD4+ T cells were purified from Fyn−/− and LGF+ Lck−/− mice, stimulated with anti-CD3-coated beads, fixed, and stained for PKCθ localization. Results are representative of three similar experiments.
The data presented above with anti-CD3-coated beads indicated that TCR-mediated signals were sufficient to induce membrane localization and capping of PKCθ. However, published data indicate that CD28 plays an important role in the activation and lipid raft localization of PKCθ (20, 21). Because the use of anti-CD3-coated beads is an artificial means of TCR engagement, we thought it important to evaluate the role of CD28 under more physiologic conditions. For this purpose, we used AND TCR transgenic T cells derived from CD28+/+ or CD28−/− mice. These cells were incubated with antigen-pulsed APCs for 45 min, fixed, permeabilized, and stained for PKCθ localization. As shown in Fig. 3, both CD28+ and CD28− T cells localized PKCθ to the immunologic synapse after encounter with antigen-pulsed APCs, confirming the results obtained with anti-CD3-coated beads that stimulation through the TCR was sufficient to cause localization of PKCθ to the immunologic synapse.
Figure 3.
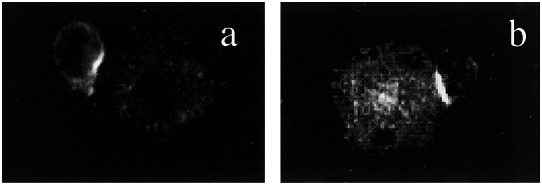
Localization of PKCθ to the immunologic synapse does not require CD28. APC-T cell conjugates were formed between antigen-pulsed CH27 cells and naïve CD4+ T cells purified from CD28+ AND TCR transgenic mice (a) or CD28− AND TCR transgenic mice (b). Cells were incubated at 37°C for 45 min, fixed, permeabilized, and stained with anti-PKCθ antibodies.
PCKθ Localization to the Immunologic Synapse Is Long-Lived.
A recent study by Lee et al. (22) demonstrated that the localization of some of the critical signaling molecules to the immunologic synapse is quite transient (30–60 min) despite a requirement for T-APC conjugation to last for a longer period (≥2 h) to commit T cells to undergo cell division. Because of this observation, it was of interest to study the persistence of PKCθ in the immunologic synapse and to compare it with the TCR and other synapse-localized molecules such as talin and LFA-1. For this purpose, naïve AD10 TCR transgenic CD4+ T cells were incubated with antigen-pulsed APCs (CH27 cells) for varying times before fixation, permeabilization, and staining with the appropriate fluorescent antibodies. The distribution of TCR and PKCθ across a 4-h time span is shown in Fig. 4. The bright field appearance of the conjugates is shown in the left-hand column (DIC). The smaller of the two cells in each frame represents the T cell. Staining for PKCθ (middle column) showed localization of the enzyme to the immunologic synapse at the first time point analyzed (30′), and this localization was stable throughout the 4-h period of observation. In contrast, the TCR (right-hand column) was localized to the synapse for the first hour, but this localization decreased thereafter and was no longer evident by the end of the observation period. Fig. 5 quantitatively summarizes these results as well as the results obtained for LFA-1 and talin. Approximately 75–80% of the conjugates analyzed showed localization to the synapse of talin, LFA-1, and PKCθ at the early time points and, for PKCθ, this remained unchanged for the 4-h observation period. LFA-1 and talin localization declined slightly between the 2- and 4-h time points, such that at 4 h, about 60–65% of conjugates still had these two molecules localized to the synapse. In contrast, only about 50% of conjugates had detectable localization of the TCR to the synapse at the early time points, and this localization was rapidly lost after the first hour with only 5–10% of conjugates having TCR localized to the synapse at 2 h and 0% at 4 h.
Figure 4.

Persistence of localization of PKCθ and TCR to the immunologic synapse. Naïve AD10 CD4+ T cells were incubated with CH27 cells that had been previously pulsed with PCC88–104 (1 μg/ml). At the indicated time points, cells were fixed, permeabilized, and stained for TCR (anti-Vβ3) and PKCθ. Conjugates were examined by confocal microscopy. The left column shows differential interference contrast (DIC) images. The middle column shows the distribution of PKCθ (green), and the right column shows TCR distribution (red).
Figure 5.

PKCθ persists in the immunologic synapse for prolonged periods of time. Antigen-pulsed APCs and naïve AD10 CD4+ T cells were allowed to form conjugates as described in Fig. 4, and at different time points cells were fixed, permeabilized, and stained for PKCθ (○), TCR (▵), talin (◊), and LFA-1 (□). A minimum of 100 conjugates were analyzed for the localization of these molecules to the immunologic synapse over time and the quantitative results are shown as a percentage of conjugates that had detectable localization of the antigen to the region of the synapse.
In summary, unlike TCR, PKCθ and the adhesion- and cytoskeleton-associated molecules LFA-1 and talin persist within the synapse for a long period, a time that has been shown to be sufficient for full commitment to cell division.
CD28 Expression Is Required for the Formation of the Mature Immunologic Synapse.
To explore further the possible role of CD28 in formation of the immunologic synapse, deconvolution microscopy was used so that the formation of the mature synapse containing p-SMAC and c-SMAC regions could be evaluated (6). For this purpose, naïve CD28+ AND T cells and CD28− AND T cells were incubated with antigen-pulsed APCs for 45 min to allow conjugate and synapse formation. The cells were then fixed, permeabilized, and stained for LFA-1 and PKCθ. Fig. 6A shows the distribution of LFA-1 (green) and PKCθ (red) in the immunologic synapse formed at the interface between APCs with CD28+ (Fig. 6A a–c) and CD28− (Fig. 6A d–f) AND T cells. Strikingly different patterns were observed. Whereas CD28+ cells localized PKCθ to the central region of the synapse (b), no such localization was seen with CD28− T cells (e). In CD28+ cells, LFA-1 appeared as multiple clusters distributed throughout the synapse. In some cells, a central area devoid of LFA-1 was discernable (a, c). In CD28− T cells, LFA-1 was diffusely localized throughout the synapse, which seemed to encompass a larger surface area than in CD28+ T cells. To verify this impression, the areas encompassed by LFA-1 and PKCθ were measured in several cells. The data obtained are summarized in Fig. 6B. The area occupied by PKCθ was 1.3 μm2 in CD28+ cells and 11.8 μm2 in CD28− cells, and the area occupied by LFA-1 was 7.1 μm2 in CD28+ and 13.1 μm2 in CD28− T cells.
Figure 6.

CD28 is required for the formation of a mature immunologic synapse. (A) Conjugates were formed between antigen-pulsed CH27 cells and naïve CD4+ T cells that were purified from CD28+ TCR transgenic mice (a–c) and CD28− TCR transgenic mice (d–f). Cells were stained for LFA-1 (a and d) or PKCθ (b and e) and analyzed by deconvolution microscopy. The merged images are shown in c and f. (B) The areas occupied by LFA-1 and PKCθ were determined for several cells and the mean areas calculated for both PKCθ and LFA-1 in CD28+ and CD28− T cell-APC conjugates.
Discussion
In this study, we have analyzed the signaling requirements for the localization of PKCθ to the immunologic synapse and the role of CD28 in this process. We found that for naïve CD4+ T cells, TCR-mediated signals were sufficient to induce the membrane localization and capping of PKCθ to the region of TCR engagement by ligand, which was shown by crosslinking of the TCR with anti-CD3-coated beads. The lack of CD28 requirement in the localization of PKCθ to the immunologic synapse was confirmed by using naïve CD4+ T cells from TCR transgenic, CD28− T cells that formed conjugates with antigen-pulsed APCs. However, further examination of the morphology of the immunologic synapse by deconvolution microscopy revealed that in the absence of CD28, PKCθ was present diffusely throughout the synapse in a pattern similar to that of LFA-1, whereas in the presence of CD28, PKCθ was concentrated to a small central area of the synapse (c-SMAC) with LFA-1 appearing as multiple clusters throughout most of the synaptic region. Also, in CD28−/− T cells the area encompassed by the synapse was almost 2-fold greater than that in CD28+ cells. In contrast to the TCR, the concentration of which in the synapse decreased rapidly such that it was at background levels 2 h after the initial contact with APCs, LFA-1, talin, and PKCθ remained localized to the synaptic region for at least 4 h, a time that has been shown to be sufficient to commit T cells to divide.
The observation that incubation with anti-CD3-coated beads was a sufficient stimulus to induce translocation of PKCθ from the cytoplasm to the cell membrane is consistent with biochemical data that showed an increase in PKCθ localization to membrane fractions after stimulation with anti-CD3 (20, 23). In contrast, Monks et al. (8), with the use of fluorescent microscopy, failed to demonstrate membrane localization of PKCθ after stimulation with anti-CD3 antibodies. The reason for this discrepancy with our data is not known but may be related to technical differences such as their use of a TH2 clone, D10, rather than naïve CD4 cells and the use of soluble rather than bead-coated anti-CD3 antibodies. With respect to the intracellular signaling events involved in the membrane localization of PKCθ, Villalba et al. (24) showed that PP2, an inhibitor of Src family kinases, inhibited the localization of PKCθ to membrane fractions after anti-CD3 stimulation of Jurkat cells. We confirmed these results by showing that PP2 inhibited the capping of PKCθ to the area of contact between naïve CD4+ T cells and anti-CD3-coated beads. Furthermore, by using Lck- or Fyn-deficient T cells, we demonstrated that the capping of PKCθ required Lck but not Fyn. Data from Villalba et al. further suggest that PI3K (but not PLCγ) may also be important for the membrane localization of PKCθ (24). Whether the binding of PKCθ to the membrane involves direct interaction with membrane lipids or whether PKCθ binds indirectly to the membrane through interactions with a membrane-localized adapter protein remains to be elucidated (25–28).
Our study demonstrated that CD28 expression was specifically required for PKCθ localization to the c-SMAC region of the immunologic synapse. Whether this effect is specific for PKCθ or whether CD28 is important in the overall formation of p-SMAC and c-SMAC is not clear. The observation that in CD28− cells LFA-1 was diffusely distributed over a 2-fold greater area than in CD28+ cells suggests a more general role for CD28 in SMAC formation. This possibility is further supported by the findings of Davis and coworkers (29), who used APCs that expressed green fluorescent protein-tagged MHCII to study the formation of the immunologic synapse. They showed that preventing the interaction between CD28 and B7 with anti-B7 antibodies inhibited the localization of MHCII into a small concentrated area on the APC side of the immunologic synapse; i.e., the APC equivalent of c-SMAC. Instead, in the absence of CD28/B7 interaction MHC localization was diffuse and/or unstable. However, the conclusion that CD28 is critical for p-SMAC and c-SMAC formation conflicts with the experiments of Dustin and colleagues (30, 31), who used fluorescent-tagged MHC peptide and ICAM-1 incorporated into planar lipid bilayers to explore the APC side of the immunologic synapse. Their work showed MHC localization to the central area and ICAM-1 to a peripheral area within the contact zone between the planar membrane and T cell, which would suggest that CD28 is not necessary for the formation of p- and c-SMAC. Furthermore, in their experiments, inclusion of B7 into the lipid bilayer had no discernable effect on the kinetics or extent of localization of MHC and ICAM to central and peripheral regions of the contact zone. How the T cell counterparts of these ligands were arranged could only be inferred from their studies. One possible explanation for the discrepancy in the results with planar membranes and APCs is to postulate that the interaction between other APC and T cell ligand-receptor pairs may interfere with the formation of p- and c-SMACs and that CD28 signaling is necessary to overcome this inhibition. The absence of these ligands in the artificial membrane system would render the formation of the mature synapse to be independent of CD28.
The physiologic relevance of the localization of T cell molecules into p-SMAC and c-SMAC regions is still unclear (32, 33). The kinetics of formation of the mature synapse is relatively slow, taking several minutes. As recently emphasized (22), much of the early signaling through the TCR precedes its formation. Furthermore, in this same study, TCR and activated Lck could be shown to be localized to the c-SMAC for only a relatively short period (30–60′), a time that was insufficient for a full commitment to cell division. Our observation that, in contrast to TCR and activated Lck, PKCθ remains localized to the immunologic synapse for at least 4 h suggests the possibility that the function of the c-SMAC may be to allow the late costimulatory signals required for IL-2 synthesis to be generated, a process in which CD28 and PKCθ are critically involved (9, 34, 35).
Acknowledgments
We thank Dr. Katsuji Sugie for helpful discussions and Nancy Martorana for assistance in preparation of the manuscript. This article is publication 494 from the La Jolla Institute for Allergy and Immunology. This work was supported in part by National Institutes of Health Grants AI18634 (to H.M.G.) and GM65230 (to N.R.J.G.).
Abbreviations
- p-SMAC
peripheral supramolecular activation cluster
- c-SMAC
central supramolecular activation cluster
- TCR
T cell receptor
- APC
antigen-presenting cell
- PKC
protein kinase C
- ICAM
intercellular adhesion molecule
- LFA-1
lymphocyte function-associated antigen-1
References
- 1.Iezzi G, Karjalainen K, Lanzavecchia A. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 2.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. J Exp Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldsmith M A, Weiss A. Science. 1988;240:1029–1031. doi: 10.1126/science.3259335. [DOI] [PubMed] [Google Scholar]
- 4.Paul W E, Seder R A. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 5.Kupfer A, Singer S J, Janeway C A, Jr, Swain S L. Proc Natl Acad Sci USA. 1987;84:5888–5892. doi: 10.1073/pnas.84.16.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monks C R, Freiberg B A, Kupfer H, Sciaky N, Kupfer A. Nature (London) 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Sugie K, La Face D M, Altman A, Grey H M. Eur J Immunol. 2000;30:50–58. doi: 10.1002/1521-4141(200001)30:1<50::AID-IMMU50>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 8.Monks C R, Kupfer H, Tamir I, Barlow A, Kupfer A. Nature (London) 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- 9.Sun Z, Arendt C W, Ellmeier W, Schaeffer E M, Sunshine M J, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg P L, Littman D R. Nature (London) 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- 10.Isakov N, Altman A. Annu Rev Immunol. 2002;20:761–794. doi: 10.1146/annurev.immunol.20.100301.064807. [DOI] [PubMed] [Google Scholar]
- 11.Kaye J, Vasquez N J, Hedrick S M. J Immunol. 1992;148:3342–3353. [PubMed] [Google Scholar]
- 12.Kimachi K, Croft M, Grey H M. Eur J Immunol. 1997;27:3310–3317. doi: 10.1002/eji.1830271230. [DOI] [PubMed] [Google Scholar]
- 13.Trobridge P A, Levin S D. Eur J Immunol. 2001;31:3567–3579. doi: 10.1002/1521-4141(200112)31:12<3567::aid-immu3567>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Rogers P R, Song J, Gramaglia I, Killeen N, Croft M. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 15.La Face D M, Couture C, Anderson K, Shih G, Alexander J, Sette A, Mustelin T, Altman A, Grey H M. J Immunol. 1997;158:2057–2064. [PubMed] [Google Scholar]
- 16.Zal T, Zal M A, Gascoigne N R. Immunity. 2002;16:521–534. doi: 10.1016/s1074-7613(02)00301-1. [DOI] [PubMed] [Google Scholar]
- 17.Hanke J H, Gardner J P, Dow R L, Changelian P S, Brissette W H, Weringer E J, Pollok B A, Connelly P A. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Wong N, Jabali M D, Johnson P. J Biol Chem. 2001;276:28767–28773. doi: 10.1074/jbc.M100158200. [DOI] [PubMed] [Google Scholar]
- 19.Soede R D, Wijnands Y M, Van Kouteren-Cobzaru I, Roos E. J Cell Biol. 1998;142:1371–1379. doi: 10.1083/jcb.142.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coudronniere N, Villalba M, Englund N, Altman A. Proc Natl Acad Sci USA. 2000;97:3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk M J, Altman A. Nat Immunol. 2001;2:556–563. doi: 10.1038/88765. [DOI] [PubMed] [Google Scholar]
- 22.Lee K H, Holdorf A D, Dustin M L, Chan A C, Allen P M, Shaw A S. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 23.Szamel M, Appel A, Schwinzer R, Resch K. J Immunol. 1998;160:2207–2214. [PubMed] [Google Scholar]
- 24.Villalba M, Bi K, Hu J, Altman Y, Bushway P, Reits E, Neefjes J, Baier G, Abraham R T, Altman A. J Cell Biol. 2002;157:253–263. doi: 10.1083/jcb.200201097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valensin S, Paccani S R, Ulivieri C, Mercati D, Pacini S, Patrussi L, Hirst T, Lupetti P, Baldari C T. Eur J Immunol. 2002;32:435–446. doi: 10.1002/1521-4141(200202)32:2<435::AID-IMMU435>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. Science. 1999;283:680–682. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- 27.Huang J, Tilly D, Altman A, Sugie K, Grey H M. Proc Natl Acad Sci USA. 2000;97:10923–10929. doi: 10.1073/pnas.97.20.10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villalba M, Coudronniere N, Deckert M, Teixeiro E, Mas P, Altman A. Immunity. 2000;12:151–160. doi: 10.1016/s1074-7613(00)80168-5. [DOI] [PubMed] [Google Scholar]
- 29.Wulfing C, Sumen C, Sjaastad M D, Wu L C, Dustin M L, Davis M M. Nat Immunol. 2002;3:42–47. doi: 10.1038/ni741. [DOI] [PubMed] [Google Scholar]
- 30.Grakoui A, Bromley S K, Sumen C, Davis M M, Shaw A S, Allen P M, Dustin M L. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 31.Bromley S K, Iaboni A, Davis S J, Whitty A, Green J M, Shaw A S, Weiss A, Dustin M L. Nat Immunol. 2001;2:1159–1166. doi: 10.1038/ni737. [DOI] [PubMed] [Google Scholar]
- 32.van Der Merwe P A, Davis S J. Science. 2002;295:1479–1480. doi: 10.1126/science.1069896. [DOI] [PubMed] [Google Scholar]
- 33.Dustin M L, Chan A C. Cell. 2000;103:283–394. doi: 10.1016/s0092-8674(00)00120-3. [DOI] [PubMed] [Google Scholar]
- 34.Linsley P S, Brady W, Grosmaire L, Aruffo A, Damle N K, Ledbetter J A. J Exp Med. 1991;173:721–730. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraser J D, Irving B A, Crabtree G R, Weiss A. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]