

Abstract
Nearly all photochemical transformations known to date follow Kasha’s rule, implying that reactions occur only from the lowest electronically excited state of a given spin multiplicity due to the fast relaxation of higher-energy states. We challenge this foundational principle by demonstrating with time-resolved laser spectroscopy that the 4,4″-dicyano-p-terphenyl radical anion can undergo photoinduced electron transfer directly from a higher-energy excited state, enabling reactivity inaccessible to the lowest excited state with the same spin multiplicity. Preassociation with the substrate and driving-force optimization are critical for overcoming the kinetic barrier to subnanosecond electron transfer, enabling bimolecular anti-Kasha reactivity. This advance establishes a general and broadly applicable framework for bypassing one of the most fundamental principles of photophysics and photochemistry, Kasha’s rule, and opens new possibilities in photoredox catalysis and solar energy conversion by rethinking the energetic and kinetic landscape.


Introduction
One of the most fundamental principles in photophysics is that light emission usually occurs exclusively from the lowest electronically excited state of a given spin multiplicity, a behavior known as Kasha’s rule. Higher excited states typically undergo internal conversion to the lowest excited state on a (sub)picosecond time scale, a rate too fast to allow radiative decay from these higher excited states. By analogy, photochemical reactions are generally observed to proceed from the lowest excited state of a given spin multiplicity. − The ultrafast relaxation of higher excited states dissipates energy that is no longer available for photochemical reactions, limiting the achievable reactivity. Reactions that would proceed directly from higher excited states, referred as anti-Kasha reactivity, offer an opportunity to unlock new photochemistry, requiring more energy input and higher electrochemical potentials. − Several applications could greatly benefit from predictable and reliable access to high-energy (anti-Kasha) photochemistry, including the photodegradation of per- and polyfluoroalkyl substances (PFAS), the conversion of sunlight to chemical fuels, and synthetic photochemistry targeting fine chemicals.
Any photochemical activation step of substrate molecules must effectively compete with the relaxation processes on a photocatalyst to enable productive chemical reactions. , Most photocatalytic systems rely on bimolecular diffusion to bring the substrate (Sub) to react with the excited photocatalyst (*PC), yielding a maximum reaction rate (k R) of approximately 109 s–1, which is the diffusion-limited rate at typical substrate concentrations (Figure a). This requires natural excited-state lifetimes (τ0) in the nanosecond regime or longer to effectively compete against intrinsic decay processes. Consequently, under such diffusion-based conditions, bimolecular reactivity can usually only originate from the lowest excited state of a given spin multiplicity, as internal conversion rates (k IC) from the second excited states (ES2) to the lowest excited state (ES1) are orders of magnitude faster than the diffusion-limited reaction rate. Diffusion-based photochemistry, therefore, commonly follows Kasha’s rule, making the available redox potentials independent of the excitation energy (Figure c, orange decay pathway).
1.

(a) Classical photoredox reactions rely on bimolecular diffusion for substrate (Sub) activation by the excited photocatalyst (*PC), limiting the electron transfer rate (k ET) to ≤109 s–1 at typical substrate concentrations, requiring at least nanosecond-scale excited state lifetimes. (b) Preassociation between Sub and *PC enables electron transfer rates of up to 1013 s–1, facilitating reactions of much shorter-lived excited states. (c) Simplified energy diagram of classical Kasha reactivity (left), where internal conversion rates (k IC) of up to ∼1013 s–1 to the lowest excited state (ES1) prevent reactivity from higher excited states (ES2), causing significant energy loss. Anti-Kasha reactivity (right): preassociation between PC and Sub enables competitive k ET, allowing ultrafast electron transfer from higher excited states to form redox products (Sub•±) inaccessible from ES1. (d) Molecular structure of 4,4″-dicyano-p-terphenyl (DCT). (e) UV–vis absorption spectrum (solid black line), and normalized luminescence spectrum (dashed black line) of DCT in DMF. Absorption changes following the one-electron reduction of DCT in DMF are shown in orange and blue, representing the absorption bands resulting from excitation into the D1 ([2*DCT•–]D1 ) and D2 ([2*DCT•–]D2 ) excited states, respectively. The dashed straight lines indicate the excitation wavelengths of the two-color pump–pump–probe experiments.
Few compounds, like azulenes and zinc(II) porphyrins, exhibit much slower internal conversion rates due to an unusually large energy gap between ES1 and ES2, allowing competitive radiative decay of ES2 with luminescence lifetimes of around 1 ps up to a few nanoseconds. − These excited state lifetimes enable anti-Kasha reactivity when zinc(II) porphyrins are covalently linked or preassociated with a suitable reaction partner. − This preassociation is essential for enabling static photoinduced electron transfer (ET) with reaction rates reaching up to 1013 s–1 (Figure b). , Consequently, static quenching , offers potential for systematic access to anti-Kasha reactivity, which is of interest, among other areas, in dye-sensitized solar cells (DSSCs) and water-splitting dye systems, where higher excited states can inject an electron into the covalently bound semiconductor. , However, solution based bimolecular anti-Kasha reactivity remains largely unexplored, and is generally considered unattainable for most photocatalysts owing to their ultrafast internal conversion rates requiring femtosecond reactivitya condition viewed as unrealistic even with preassociation. While some synthetic studies have speculated about the possibility of anti-Kasha reactivity in excited organic radicals as photocatalysts , and kinetic analysis suggests the possibility of femtosecond reactivity, spectroscopic evidence has been absent.
In this study, we observe anti-Kasha reactivity from 4,4″-dicyano-p-terphenyl (Figure d) radical anion (DCT•–), a strong photoreductant. By leveraging preassociation between DCT•– and substrate molecules we achieve static ET rates that compete against nonproductive internal conversion. Using two-color pump–pump–probe laser flash photolysis we directly observe ultrafast ET from higher-energy excited states. Our results reveal a threshold ET driving force required to achieve anti-Kasha reactivity, establishing design principles for photocatalytic systems capable of exploiting this phenomenon. This work lays the foundation for broad applications of anti-Kasha reactivity and redefines the energetic landscape of photocatalysis.
Results and Discussion
Direct Observation of Anti-Kasha Reactivity
Based on the UV–vis absorption spectrum of 2DCT•– (Figure e), the electronic transitions from the doublet ground state (D0) to the first (D1) and second (D2) doublet excited states yield [2*DCT•–]D1 and [2*DCT•–]D2 energies (E D) of 1.0 and 2.3 eV, respectively. Using the one-electron reduction potential (E red) of DCT at −1.7 V versus SCE (Figure S1), the reduction potentials of the D1 and D2 excited states (2* E red) are estimated using the Rehm–Weller equation, as approximately −2.7 and −4.0 V versus SCE (Figure S2). While the Rehm–Weller formalism is established for excited organic radicals, , its applicability to higher excited states is not yet established, though it appears plausible to us. Nonetheless, the 1.3 V potential difference between D1 and D2 likely leads to distinct reactivity profiles.
Ultrafast transient absorption spectroscopy initially seems to be the most logical approach to investigate the proposed subpicosecond anti-Kasha reactivity. However, in our system, the anticipated static quenching between [2*DCT•–]D2 and the electron acceptor (Sub) would further reduce the already weak transient absorption signal, as the potentially ultrafast electron transfer to the substrate may occur within the instrument response function, making detection challenging. Furthermore, a two-color pump–pump–probe femtosecond transient absorption experiment (or a combination of spectro-electrochemistry and subpicosecond pump–probe spectroscopy) , would be required to first generate the active photocatalyst DCT•–, further limiting the accessibility of such measurements. To overcome this technical limitation, we employed nanosecond two-color pump–pump–probe spectroscopy (Figure a). , In this experiment, direct excitation of DCT with 355 nm laser pulses in the presence of an electron donor (Dsac), such as N,N-dimethylaniline (DMA), generates DCT•–. After a delay of several microseconds, a second laser pulse at either 1064 or 532 nm selectively excites DCT•–, forming [2*DCT•–]D1 or [2*DCT•–]D2 , respectively. Without an electron acceptor, these excited states decay back to the doublet ground state (DCT•–) within the 10 ns duration of the second laser pulse, resulting in no change in the transient absorption dynamics. When an electron acceptor capable of oxidizing the electronically excited DCT•– is present, the second excitation triggers an ET reaction. This results in an instantaneous bleach of the DCT•– transient absorbance signal, as ET from 2*DCT•– to the acceptor forms charge-neutral DCT and a (neutral) phenyl radical along with halogenide anions, which do not absorb in the visible region. ,
2.

(a) Schematic representation of the two-color pump–pump–probe experiment. (b) Transient absorption kinetics for DCT•– detected at 500 nm with 1 M chlorobenzene as an electron acceptor. Electron transfer occurs selectively from the higher excited state populated with 532 nm excitation (blue trace, reaction marked by a blue arrow), whereas the lowest excited state (orange trace) remains unreactive. (c) Simplified energy diagram. Excitation of DCT at 355 nm with a sacrificial electron donor (Dsac) present generates DCT•–. Using 1064 or 532 nm light, DCT•– is selectively excited into the first ([2*DCT•–]D1 ) or second ([2*DCT•–]D2 ) doublet excited state. In the presence of an electron acceptor with a reduction potential between those of the D1 and D2 states of DCT•–, electron transfer (ET) occurs selectively from [2*DCT•–]D2 . (d) Power dependency of the second laser pulse at 532 nm (blue trace) and 1064 nm (orange trace) using chlorobenzene. (Inset) Illustration of the two key observables: ΔODPP (PP = pump–probe, change in optical density before the second pump pulse) and ΔODPPP (PPP = pump–pump–probe, change in optical density after the second pump pulse).
To explore the anticipated anti-Kasha reactivity (Figure c), chlorobenzene was initially selected as the electron acceptor, with a reduction potential of −2.8 V versus SCE lying between the redox potentials of [2*DCT•–]D1 and [2*DCT•–]D2 . Under these conditions, excitation at 1064 nm, which populates the D1 state, should not induce ET with chlorobenzene. In contrast, 532 nm excitation populates the D2 state, providing sufficient reduction power expected to drive a fast ET reaction, resulting in the characteristic bleach of the transient absorption signal (Figure b).
Given that the absorption coefficient of DCT•– at 532 nm is ∼2.5 times higher than at 1064 nm, the 355 nm laser pulse energy was tuned to equalize the transient absorption signal intensities at both wavelengths. Since both transient absorption kinetics were monitored at 500 nm, a ∼2.5 times stronger signal was targeted for the 1064 nm excitation to compensate for the lower absorption at that wavelength. This ensures a valid comparison by maintaining equimolar photon absorbance at 1064 and 532 nm (see Supporting Information). Using a deaerated DMF solution containing DCT, DMA, and 1 M chlorobenzene, direct excitation of DCT with 355 nm pulses generated DCT•–. A subsequent 532 nm pulse caused an instantaneous bleach of the DCT•– absorbance, indicating ET from the [2*DCT•–]D2 state to chlorobenzene, as anticipated. In contrast, excitation at 1064 nm resulted in undetectable changes in the transient absorption signal (Figure b, orange trace), consistent with the insufficient reduction potential of the [2*DCT•–]D1 state for ET with chlorobenzene.
Photoreactivity after D1 excitation remained undetectable unless very high pulse energies of up to 410 mJ per pulse were applied, delivering ∼10 times more photons than 532 nm excitation (Figure S3). An excitation power-dependent study was conducted to distinguish between one- and two-photon excitation processes by monitoring the induced bleach of DCT•–. The bleach was quantified by comparing the changes in optical densities (ΔOD) at 500 nm, determined immediately before the second pump pulse (ΔODPP; PP = pump–probe) and shortly afterward (ΔODPPP; PPP = pump–pump–probe). , To avoid potential optical artifacts, the ΔODPPP values were extracted by averaging the signal in a time window of 75 ns starting approximately 50 ns after the second laser pulse. A plot of ΔODPP/ΔODPPP against the number of photons used reveals a linear relationship for 532 nm excitation, consistent with a monophotonic mechanism (Figure d, blue trace). In contrast, 1064 nm excitation showed a quadratic dependency, indicating a two-photonic mechanism (Figure d, orange trace). This suggests a two-photon absorption of DCT•– or further excitation of [2*DCT•–]D1 , both leading to the population of the [2*DCT•–]D2 state from which reaction with chlorobenzene is possible. These results indicate that anti-Kasha reactivity is accessible both via conventional monophotonic processes as well as via two-photon excitation events. While it is reasonable to assume that no decomposition product absorbs at 1064 nm, this assumption is less certain at 532 nm, where more organic chromophores typically absorb. , To address this, we performed a pseudoexcitation (or “photoaction”) spectrum by keeping the first pulse constant while varying the excitation wavelength of the second laser pulse between 420 and 510 nm. The observed bleach at different excitation wavelengths was compared to the UV–vis absorption spectrum of DCT•–, showing good alignment within the accessible wavelength range (Figure S5). This result confirms reactivity originating from the [2DCT•–]D2 excited state.
Exploring the Thermodynamic Limits
To explore the thermodynamic and kinetic limits of the anti-Kasha reactivity exhibited by DCT•–, we tested fluorobenzene as an electron acceptor, with a reaction free energy of −1.0 eV for photoinduced electron transfer from the D2 excited state (ΔG ET(D2) 0). However, no reaction was observable indicating that photoinduced electron transfer is kinetically outcompeted by internal conversion. This finding most likely reflects a too high activation barrier for ET resulting from an imbalance between driving-force and reorganization energy. To systematically explore the driving-force dependence of potential anti-Kasha reactivity and conventional Kasha reactivity, we conducted two-color pump–pump–probe experiments with electron acceptors having reduction potentials ranging from −3.0 to −1.2 V versus SCE (Figure e). While accurately determining the thermodynamic redox potentials of this family of quenchers is challenging due to their irreversible redox behavior, cyclic voltammetry has been shown to provide reasonable estimates.
3.
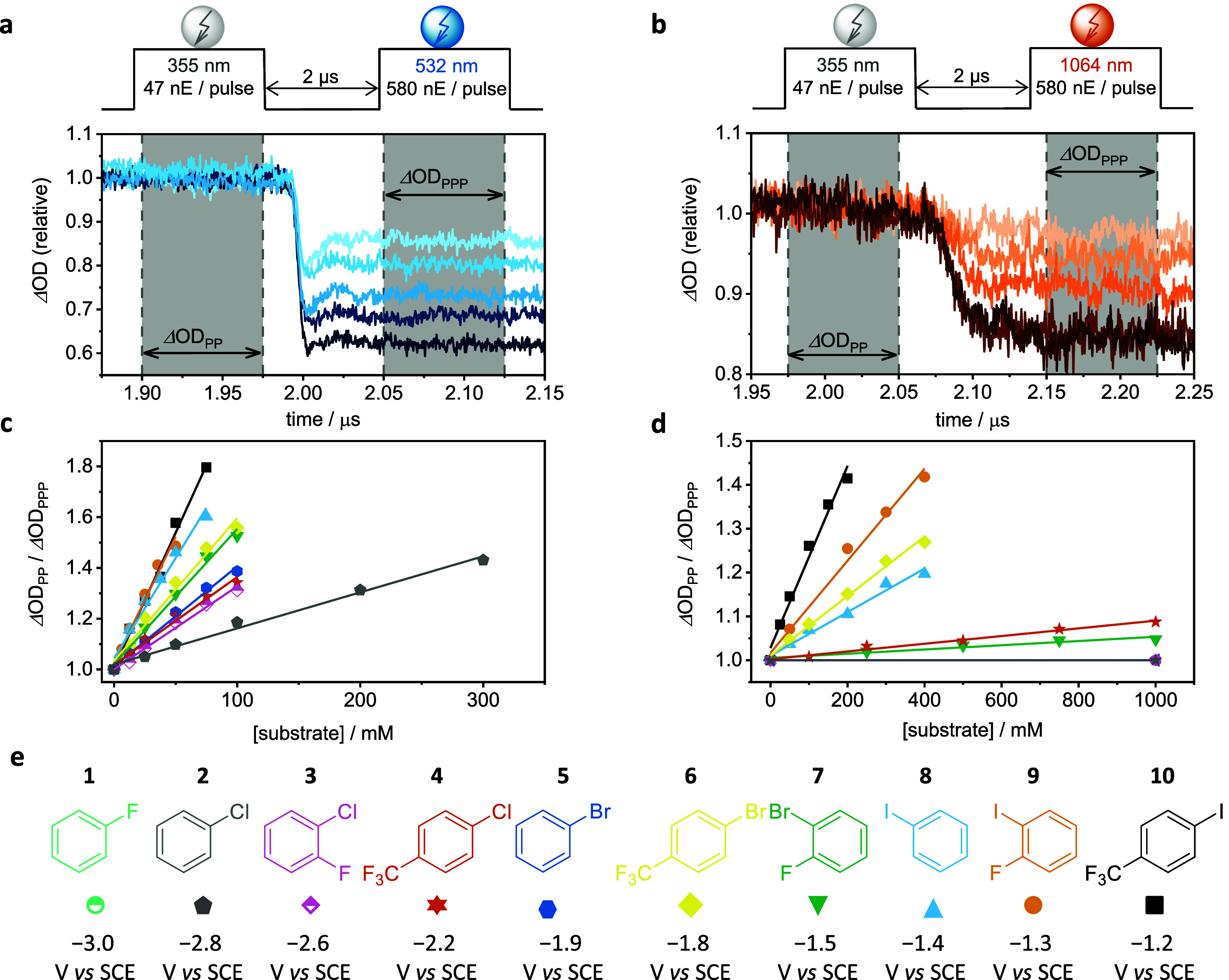
(a, b) Transient UV–vis absorption kinetic decay of DCT•– monitored at 500 nm in a two-color pump–pump–probe experiment, with the pulse scheme illustrated at the top. A 355 nm laser pulse (16 mJ) first generates DCT•–. After a delay of 2 μs, a second laser pulse of 532 nm (a) or 1064 nm (b) excites DCT•–. The experiment used an argon-saturated DMF solution containing 2 mM DCT, 200 mM DMA, and varying iodobenzene concentrations. The gray shaded areas indicate the range of the averaged signal intensity for ΔODPP (PP = pump–probe, changes in optical density immediately before the second pump pulse) and ΔODPPP (PPP = pump–pump–probe, change in optical density recorded after the second pump pulse). To avoid potential optical artifacts, the ΔODPPP values were extracted by averaging the signal over a 75 ns time window starting approximately 50 ns after the second laser pulse. (c, d), Stern–Volmer type plot based on the two-color pump–pump–probe experiments performed at varying electron acceptor concentrations. (e) Chemical structures of the electron acceptors used, with their reduction potential against saturated calomel electrode (SCE). Color- and symbol-codes correspond with panels (c, d).
Specifically, we compared the signal bleach induced by a pulse at 532 nm, exciting the D2 excited state, and 1064 nm, exciting the D1 excited state, at different electron acceptor concentrations. Exemplary experiments with iodobenzene as the electron acceptor are shown in Figure a,b, while results with other acceptors are presented in Figures S6–S20. Titration experiments with electron acceptors at 12.5 to 1000 mM concentrations revealed progressively larger bleaches. The effect of the 532 or 1064 nm pulse was quantified by comparing the changes in optical densities (ΔOD) at 500 nm, measured immediately before the pulse (ΔODPP, PP = pump–probe) and shortly afterward (ΔODPPP, PPP = pump–pump–probe). These values were determined by averaging signal intensities within the gray-shaded areas of Figure a,b. To avoid potential optical artifacts, the ΔODPPP values were extracted by averaging the signal over a 75 ns time window starting approximately 50 ns after the second laser pulse. A pseudo Stern–Volmer analysis was performed by plotting ΔODPP/ΔODPPP against the electron acceptor concentration (Figure c,d).
As the electron acceptor concentration increased, a linear Stern–Volmer relationship was observed for both excitation wavelengths, indicating a first-order reaction with respect to the electron acceptors (Table S1). For 532 nm excitation, the pseudo Stern–Volmer constant (K SV) showed a direct correlation with the increasing driving force (Figure a). For 1064 nm excitation (populating the [2*DCT•–]D1 state), overall significantly smaller K SV values were obtained (Figure b). For reaction free energies (ΔG ET(D1) 0) less negative than −0.9 eV, the K SV values were too small to be determined in most cases (Table S1). Evidently, far more effective photoreactivity is observed after excitation of DCT•– into its D2 excited state compared to D1 excitation, illustrating the power of anti-Kasha reactivity.
4.

(a) Stern–Volmer constants (K SV) obtained for D2 excitation using 532 nm are plotted against the estimated driving force for photoinduced electron transfer from the D2 excited state to the different acceptors (Figure e), with the pulse scheme illustrated above. (b) The K SV values obtained for direct D1 excitation using 1064 nm are plotted against the estimated driving force for photoinduced electron transfer from the D1 state to the different acceptors (Figure e) with the pulse scheme illustrated above. (c–f) The potential energy well diagrams illustrate four different kinetic regimes for photoinduced electron transfer (ET) between [2DCT•–]D2 (blue parabola) or [2DCT•–]D1 (orange parabola) and the electron acceptors from Figure e to form photoproducts P (= DCT and reduced acceptor, purple parabola). The key point is to achieve barrierless electron transfer, by increasing the amount of the reaction free energies (ΔG ET(D2) 0 or (ΔG ET(D1) 0) to the point that they equal the reorganization energy (λ). This point is reached in (d) for the D2 excited state and in (f) for the D1 excited state of DCT•–.
Decoding Anti-Kasha Behavior via Marcus Theory
The data in Figure a,b suggest a complex interplay between the driving force and the contributions of the D1 and D2 excited states to the overall reactivity. The energy-level diagram in Figure c cannot adequately explain this interplay. Instead, we use Marcus’ theory for electron transfer to qualitatively distinguish between four different regimes that account for the combined D1 and D2 photoreactivity data.
The first regime (Figure c) describes the situation for the weakest electron acceptors, where photoinduced electron transfer is exergonic from the D2 excited state but endergonic from the D1 state. In this regime, the driving force for electron transfer from the D2 excited state (ΔG ET(D2) 0) is assumed to be smaller than the reorganization energy (λ) leading to a significant activation barrier for electron transfer. This barrier likely accounts for the absence of observable D2 reactivity with fluorobenzene (1), the weakest acceptor investigated. This barrier persists despite a thermodynamic driving force of 1.0 eV from the D2 state of DCT•–, making photoinduced electron transfer kinetically uncompetitive with internal conversion (Figure c). The lifetime of the D2 excited state has not been determined but is likely in the picosecond range or shorter, corresponding to an internal conversion rate (k IC) on the order of 1012–1013 s–1. To observe ET from D2, the electron transfer rate (k ET) must compete with k IC (Figure c). This necessitates k ET to be near to its theoretical upper limit, which is expectable when the activation barrier is close to zero. This condition is met when the reaction free energy (ΔG ET(D2) 0) equals the total reorganization energy (λ) of the electron transfer reaction.
Overall (inner and outer sphere) reorganization energies are typically between 0.8 and 1.2 eV in molecular systems. , Hence, in such a simple picture activation free electron transfer is expected for driving forces in this range. This point is not reached with fluorobenzene (ΔG ET(D2) 0 ≈ −1.0 eV), where the experiment suggests k IC > k ET (Figures a and c). Additionally, reverse electron transfer from the primary photoproducts (purple parabola, Figure c) to a lower excited D1 state (orange parabola, Figure c), may further limit the observable reactivity. However, it is important to note that these two-color two-pulse experiments require an ET efficiency of 5–10% for detection. In contrast, under photoredox conditions with continuous light irradiation for hours, much smaller efficiencies can sustain reactivity.
The second regime (Figure d) presents a similar scenario, in which photoinduced electron transfer is exergonic from the D2 excited state but remains endergonic from the D1 state. In contrast to the first regime, the driving force of the D2 excited state (ΔG ET(D2) 0) is now comparable to the reorganization energy (λ), resulting in a nearly barrierless electron transfer. This regime is in line for chlorobenzene (2), where reactivity is solely observed from the D2 excited state with an estimate driving force of ΔG ET(D2) 0 ≈ −1.2 eV (Figure ), which evidently makes the electron transfer rate competitive with the internal conversion rate (Figure c). This aligns with previous studies, showing that k ET can reach values between 1012 and 1013 s–1 for driving forces above 1.0 eV. − Further, reverse charge shift to the D1 excited state in this regime (Figure d) is likely reduced compared to the first regime (Figure c) due to the higher activation barrier for this unwanted process.
The third regime (Figure e) represents a scenario where photoinduced electron transfer is thermodynamically accessible from both the D2 and the D1 excited state. Electron transfer from the D2 excited state remains near barrierless or is slightly inverted, while the ET rate from D1 remains limited due to a relatively small driving force (ΔG ET(D1) 0), introducing a significant activation barrier. This behavior is exemplified by the next five electron acceptors (3–7). Upon D2 excitation, fast ET is consistently observed (Figure a), compatible with the near-barrierless regime. In contrast, despite a driving force from D1 with ΔG ET(D1) 0 values ranging from −0.1 to −1.2 eV no significant reactivity is observed (Figure b). This indicates that for these acceptors k ET is significantly smaller compared to k IC due to the activation barrier imposed by insufficient driving force (Figure e). Given the D1 excited-state lifetime of DCT•– is ∼1.1 ps, the corresponding k IC is ∼ 1 × 1012 s–1, supporting this analysis similarly as discussed above (regime 1) for the D2 excited state where no reaction occurs with fluorobenzene (1). Photoreactivity from the D2 state with acceptors 3–7 occurs with pseudo Stern–Volmer constants (K SV) in a narrow range of 3 to 5 mM–1, with no evidence for a Marcus inverted driving-force effect, where k ET decreases when the reaction free energy significantly exceeds the reorganization energy. This inverted effect can weaken due to nuclear tunneling, and the commonly used harmonic potential-well picture becomes simplistic at high driving-forces. Hence, the relatively constant D2 photoreactivity with acceptors 3–7 is not necessarily surprising.
The fourth regime (Figure f) represents systems with the highest driving forces studied, where photoinduced electron transfer is feasible from both D2 and D1 excited states. As the driving force increases further, the D2 excited state falls more clearly into the Marcus inverted region, which in principle could result in a sizable activation barrier that could slow ET despite the strong thermodynamic driving force, but no deceleration is observed. The less reactive D1 excited state now enters the barrierless regime, where ET proceeds very rapidly. This scenario is reached with iodobenzene (8), 2-fluoro-1-iodobenzene (9), and 4-iodobenzotrifluoride (10), for which D1 photoreactivity is detectable in the ΔG ET(D1) 0 range from −1.3 to −1.5 eV (Figure b). Thus, the activation barrier becomes sufficiently low for D1 reactivity at a ΔG ET(D1) 0 value of −1.3 eV, similar to the threshold value of ΔG ET(D2) 0 of −1.2 eV necessary to observe D2 reactivity. For acceptors 8–10, the apparent D2 reactivity observed after excitation at 532 nm further increases compared to K SV values for acceptors 3–8 (Figure a). This is likely due to additional D1 reactivity after internal conversion from D2 to D1. Thus, reactivity in the fourth regime reflects contributions from both excited states, complicating the isolation of the higher excited states reactivity. For the most readily reducible electron acceptors (8–10), K SV values obtained after D2 excitation are about four times higher than those K SV values obtained after D1 excitation (Figure a,b). The electronic coupling between the reactant and product potential wells in Figure may differ depending on the involved D1 or D2 excited state, and even small differences in electronic coupling could influence the reaction kinetics here. Additionally, unlike classical (luminescence) quenching experiments, our two-color pump–pump–probe experiments monitor electron transfer including successful cage escape, since in-cage charge recombination regenerates the original species prior to the second laser pulse, resulting in no net signal change (Figure S22). As cage escape efficiency is known to be driving force dependent in some cases, − the D2 state can be expected to exhibit higher reactivity compared to D1, potentially explaining the enhanced reactivity observed for excited radicals under higher-energy light illumination. ,,
The Future of Anti-Kasha Reactivity
This study demonstrates that productive photoinduced electron transfer in solution can occur directly from a higher-energy excited state of an organic radical, challenging the long-standing principle that photochemical reactivity is limited to the lowest excited state of a given spin multiplicity. Due to the close relationship with Kasha’s rule (which states that emission typically originates from the lowest excited state), , the observed reactivity is nowadays often referred to as anti-Kasha reactivity. ,,, However, this terminology remains somewhat controversial, as Kasha’s rule was originally formulated to describe light emission, not chemical reactivity. Nevertheless, the kinetic parallels between emissive and reactive excited-state decay pathways justify the analogy, when reactivity arises from higher excited states within the same spin manifold. This distinction is important, as it differentiates the photoreactivity seen here from the more commonly observed reactivity involving S1 and T1 excited states. ,
While previous synthetic-oriented research has speculated on solution based anti-Kasha reactivity, ,, direct spectroscopic evidence has been lacking until now. Our findings highlight the critical role of preassociation in enabling static electron transfer on the picosecond time scale, countering the widespread belief that productive bimolecular photochemistry requires excited state lifetimes on the order of nanoseconds or longer. Such preassociation can be achieved through Coulombic interactions, − π stacking, ,, dipole–dipole interactions, , or spatial confinement within supramolecular structures, leading to ultrafast electron transfer. However, our study also suggests that for anti-Kasha reactivity, the driving force must closely approach the reorganization energy (λ) associated with the photoinduced electron transfer, ensuring that the reaction is competitive with nonradiative energy dissipation. This requirement highlights the delicate balance between thermodynamics and kinetics in picosecond photochemistry. In the cases studied here, driving forces of 1.2 eV or more were required for anti-Kasha reactivity. Given that reorganization energies for electron transfer in artificial molecular systems are often between 0.8 and 1.2 eV, this insight provides a guiding principle for designing anti-Kasha reactivity far beyond the presently known examples.
The reductive dehalogenations used here were not aimed at establishing another synthetic method or tackling more challenging substrates. Instead, their purpose was to define the principles for achieving a fundamentally different reaction mode, distinct from more than 99% of the established photochemistry, and this key conceptual goal has been achieved. To date, anti-Kasha reactivity has been confined mainly to intramolecular processes such as isomerization reactions, photodissociation processes or electron transfer in covalently bound donor–acceptor compounds. Our study lays the groundwork for a broader use of anti-Kasha reactivity by providing design principles for bimolecular reactions.
The implications of anti-Kasha reactivity extend far beyond academic interest. By harnessing reactivity from short-lived, higher-energy states, catalytic systems can achieve unprecedented reactivities, with potential applications in selective pollutant degradation and chiral resolution. These findings expand the scope of photoredox catalysis and lay the foundation for exploring unconventional excited-state reactivity in organic and inorganic systems. Ultimately, this work redefines the energetic landscape of photocatalysis, providing new perspectives for leveraging higher excited states in synthetic methodologies and solar energy conversion.
Supplementary Material
Acknowledgments
We thank D. Gejsnæs-Schaad from our group for preparing the abstract figure. O.S.W. acknowledges funding by the University of Basel.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.5c06115.
General procedures and equipment details, photophysical and photochemical data (PDF)
†.
Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
References
- Kasha M.. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950;9:14–19. doi: 10.1039/df9500900014. [DOI] [Google Scholar]
- Del Valle J. C., Catalan J.. Kasha’s rule: a reappraisal. Phys. Chem. Chem. Phys. 2019;21(19):10061–10069. doi: 10.1039/C9CP00739C. [DOI] [PubMed] [Google Scholar]
- Schultz D. M., Yoon T. P.. Solar synthesis: prospects in visible light photocatalysis. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. H., Twilton J., MacMillan D. W. C.. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund B., Wenger O. S.. Excited Organic Radicals in Photoredox Catalysis. JACS Au. 2025;5:426–447. doi: 10.1021/jacsau.4c00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotic A., Cerfontaine S., Slep L. D., Elias B., Troian-Gautier L., Cadranel A.. Anti-Dissipative Strategies toward More Efficient Solar Energy Conversion. J. Am. Chem. Soc. 2023;145:5163–5173. doi: 10.1021/jacs.2c11593. [DOI] [PubMed] [Google Scholar]
- East N. R., Naumann R., Förster C., Ramanan C., Diezemann G., Heinze K.. Oxidative two-state photoreactivity of a manganese(IV) complex using near-infrared light. Nat. Chem. 2024;16:827–834. doi: 10.1038/s41557-024-01446-8. [DOI] [PubMed] [Google Scholar]
- Goti G., Manal K., Sivaguru J., Dell’Amico L.. The impact of UV light on synthetic photochemistry and photocatalysis. Nat. Chem. 2024;16:684–692. doi: 10.1038/s41557-024-01472-6. [DOI] [PubMed] [Google Scholar]
- Turro N. J., Ramamurthy V., Cherry W., Farneth W.. The Effect of Wavelength on Organic Photoreactions in Solution. Reactions from Upper Excited States. Chem. Rev. 1978;78:125–145. doi: 10.1021/cr60312a003. [DOI] [Google Scholar]
- Chantry N., Cotic A., De Kreijger S., Di Forti R., Elias B., Troian-Gautier L., Cadranel A.. Nature of Anti-Dissipative High-Energy Excited States in Quaterpyridine-Bridged Ruthenium Complexes. Angew. Chem., Int. Ed. 2025:e202507738. doi: 10.1002/anie.202507738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbott E. D., Burnett N. L., Swierk J. R.. Mechanistic and kinetic studies of visible light photoredox reactions. Chem. Phys. Rev. 2023;4:031312. doi: 10.1063/5.0156850. [DOI] [Google Scholar]
- Sakizadeh J. D., Weiss R., Scholes G. D., Kudisch B.. Ultrafast Spectroscopy and Dynamics of Photoredox Catalysis. Annu. Rev. Phys. Chem. 2025;76:203–229. doi: 10.1146/annurev-physchem-082423-013952. [DOI] [PubMed] [Google Scholar]
- Rieth A. J., Gonzalez M. I., Kudisch B., Nava M., Nocera D. G.. How Radical Are ″Radical″ Photocatalysts? A Closed-Shell Meisenheimer Complex Is Identified as a Super-Reducing Photoreagent. J. Am. Chem. Soc. 2021;143:14352–14359. doi: 10.1021/jacs.1c06844. [DOI] [PubMed] [Google Scholar]
- Dunlop D., Ludvikova L., Banerjee A., Ottosson H., Slanina T.. Excited-State (Anti)Aromaticity Explains Why Azulene Disobeys Kasha’s Rule. J. Am. Chem. Soc. 2023;145:21569–21575. doi: 10.1021/jacs.3c07625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veys K., Escudero D.. Anti-Kasha Fluorescence in Molecular Entities: Central Role of Electron-Vibrational Coupling. Acc. Chem. Res. 2022;55:2698–2707. doi: 10.1021/acs.accounts.2c00453. [DOI] [PubMed] [Google Scholar]
- Awwad N., Bui A. T., Danilov E. O., Castellano F. N.. Visible-Light-Initiated Free-Radical Polymerization by Homomolecular Triplet-Triplet Annihilation. Chem. 2020;6:3071–3085. doi: 10.1016/j.chempr.2020.08.019. [DOI] [Google Scholar]
- Tetreault N., Muthyala R. S., Liu R. S. H., Steer R. P.. Control of the Photophysical Properties of Polyatomic Molecules by Substitution and Solvation: The Second Excited Singlet State of Azulene. J. Phys. Chem. A. 1999;103:2524–2531. doi: 10.1021/jp984407q. [DOI] [Google Scholar]
- Chosrowjan H., Tanigichi S., Okada T., Takagi S., Arai T., Tokumaru K.. Electron transfer quenching of S2 state fluorescence of Zn-tetraphenylporphyrin. Chem. Phys. Lett. 1995;242:644–649. doi: 10.1016/0009-2614(95)00790-B. [DOI] [Google Scholar]
- Petersson J., Eklund M., Davidsson J., Hammarström L.. Ultrafast Electron Transfer Dynamics of a Zn(II)porphyrin-Viologen Complex Revisited: S2 vs S1 Reactions and Survival of Excess Excitation Energy. J. Phys. Chem. B. 2010;114:14329–14338. doi: 10.1021/jp911686z. [DOI] [PubMed] [Google Scholar]
- Sugunan S. K., Robotham B., Sloan R. P., Szmytkowski J., Ghiggino K. P., Paige M. F., Steer R. P.. Photophysics of untethered ZnTPP-fullerene complexes in solution. J. Phys. Chem. A. 2011;115:12217–12227. doi: 10.1021/jp2082853. [DOI] [PubMed] [Google Scholar]
- Mataga N., Taniguchi S., Chosrowjan H., Osuka A., Yoshida N.. Ultrafast charge transfer and radiationless relaxations from higher excited state (S2) of directly linked Zn-porphyrin (ZP)-acceptor dyads: investigations into fundamental problems of exciplex chemistry. Chem. Phys. 2003;295:215–228. doi: 10.1016/j.chemphys.2003.09.005. [DOI] [Google Scholar]
- Nieto-Pescador J., Abraham B., Pistner A. J., Rosenthal J., Gundlach L.. Electronic state dependence of heterogeneous electron transfer: injection from the S1 and S2 state of phlorin into TiO2 . Phys. Chem. Chem. Phys. 2015;17:7914–7923. doi: 10.1039/C5CP00296F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozina M. V., Ionkin V. N., Ivanov A. I.. What factors control product yield in charge separation reaction from second excited state in zinc-porphyrin derivatives? J. Phys. Chem. A. 2012;116:1159–1167. doi: 10.1021/jp210988k. [DOI] [PubMed] [Google Scholar]
- Hayes R. T., Walsh C. J., Wasielewski M. R.. Competitive Electron Transfer from the S2 and S1 Excited States of Zinc meso-Tetraphenylporphyrin to a Covalently Bound Pyromellitimide: Dependence on Donor-Acceptor Structure and Solvent. J. Phys. Chem. A. 2004;108:2375–2381. doi: 10.1021/jp037176i. [DOI] [Google Scholar]
- Fujitsuka M., Cho D. W., Tojo S., Inoue A., Shiragami T., Yasuda M., Majima T.. Electron Transfer from Axial Ligand to S1 - and S2 -Excited Phosphorus Tetraphenylporphyrin. J. Phys. Chem. A. 2007;111:10574–10579. doi: 10.1021/jp076303y. [DOI] [PubMed] [Google Scholar]
- Pfund B., Gejsnæs-Schaad D., Lazarevski B., Wenger O. S.. Monitoring picosecond reactions of excited radical ion super reductants in photocatalysis. Nat. Commun. 2024;15:4738. doi: 10.1038/s41467-024-49006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimioulle R., Bach T.. Enantioselective Lewis Acid Catalysis of Intramolecular Enone [2 + 2] Photocycloaddition Reactions. Science. 2013;342:840–843. doi: 10.1126/science.1244809. [DOI] [PubMed] [Google Scholar]
- Crisenza G. E. M., Mazzarella D., Melchiorre P.. Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc. 2020;142:5461–5476. doi: 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti L., Crisenza G. E. M., Melchiorre P.. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019;58:3730–3747. doi: 10.1002/anie.201809984. [DOI] [PubMed] [Google Scholar]
- Ferrere S., Gregg A.. B. A. Photosensitization of TiO2 by [FeII(2,2′-bipyridine-4,4′-dicarboxylic acid)2(CN)2]:Band Selective Electron Injection fromUltra-Short-Lived Excited States. J. Am. Chem. Soc. 1998;120:843–844. doi: 10.1021/ja973504e. [DOI] [Google Scholar]
- Giokas P. G., Miller S. A., Hanson K., Norris M. R., Glasson C. R. K., Concepcion J. J., Bettis S. E., Meyer T. J., Moran A. M.. Spectroscopy and Dynamics of Phosphonate-Derivatized Ruthenium Complexes on TiO2 . J. Phys. Chem. C. 2013;117(2):812–824. doi: 10.1021/jp310155q. [DOI] [Google Scholar]
- Horsewill S. J., Hierlmeier G., Farasat Z., Barham J. P., Scott D. J.. Shining Fresh Light on Complex Photoredox Mechanisms through Isolation of Intermediate Radical Anions. ACS Catal. 2023;13:9392–9403. doi: 10.1021/acscatal.3c02515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Žurauskas J., Domański M., Hitzfeld P. S., Butera V., Scott D. J., Rehbein J., Kumar A., Thyrhaug E., Hauer J., Barham J. P.. Hole-mediated photoredox catalysis: tris(p-substituted)biarylaminium radical cations as tunable, precomplexing and potent photooxidants. Org. Chem. Front. 2021;8:1132–1142. doi: 10.1039/D0QO01609H. [DOI] [Google Scholar]
- Paulus B. C., Adelman S. L., Jamula L. L., McCusker J. K.. Leveraging excited-state coherence for synthetic control of ultrafast dynamics. Nature. 2020;582:214–218. doi: 10.1038/s41586-020-2353-2. [DOI] [PubMed] [Google Scholar]
- Rehm D., Weller A.. Kinetik und Mechanismus der Elektronübertragung bei der Fluoreszenzlöschung in Acetonitril. Ber. Bunsen-Ges. Phys. Chem. 1969;73:834–839. doi: 10.1002/bbpc.19690730818. [DOI] [Google Scholar]
- Kim S., Zeman C. J. T., Gobeze H. B., Duvva N., Schanze K. S.. Photoinduced electron transfer from the naphthalene diimide anion radical doublet excited state. Chem. Commun. 2025;61(29):5511–5514. doi: 10.1039/D5CC00190K. [DOI] [PubMed] [Google Scholar]
- Zeman C. J., Kim S., Zhang F., Schanze K. S.. Direct Observation of the Reduction of Aryl Halides by a Photoexcited Perylene Diimide Radical Anion. J. Am. Chem. Soc. 2020;142:2204–2207. doi: 10.1021/jacs.9b13027. [DOI] [PubMed] [Google Scholar]
- Zedler L., Muller C., Wintergerst P., Mengele A. K., Rau S., Dietzek-Ivanšić B.. Influence of the Linker Chemistry on the Photoinduced Charge-Transfer Dynamics of Hetero-dinuclear Photocatalysts. Chem. - Eur. J. 2022;28:e20220049. doi: 10.1002/chem.202200490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz Neto D. H., Pino T., Ha-Thi M.-H.. Mechanistic Investigations of Photocatalytic Systems by Pump-Pump-Probe Spectroscopy. ChemPhotoChem. 2025:2500113. doi: 10.1002/cptc.202500113. [DOI] [Google Scholar]
- Beckwith J. S., Aster A., Vauthey E.. The excited-state dynamics of the radical anions of cyanoanthracenes. Phys. Chem. Chem. Phys. 2021;24:568–577. doi: 10.1039/D1CP04014F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Deetz A. M., Hu J., Meyer G. J., Hu K.. Chloride Oxidation by One- or Two-Photon Excitation of N-Phenylphenothiazine. J. Am. Chem. Soc. 2022;144:17604–17610. doi: 10.1021/jacs.2c07107. [DOI] [PubMed] [Google Scholar]
- Radziszewski J. G.. Electronic absorption spectrum of phenyl radical. Chem. Phys. Lett. 1999;301:565–570. doi: 10.1016/S0009-2614(99)00050-0. [DOI] [Google Scholar]
- Bassan E., Gualandi A., Cozzi P. G., Ceroni P.. Design of BODIPY dyes as triplet photosensitizers: electronic properties tailored for solar energy conversion, photoredox catalysis and photodynamic therapy. Chem. Sci. 2021;12(19):6607–6628. doi: 10.1039/D1SC00732G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J. C., Lim C. H., Yang H., Ryan M. D., Musgrave C. B., Miyake G. M.. Organocatalyzed atom transfer radical polymerization driven by visible light. Science. 2016;352:1082–1086. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- Espinoza E. M., Clark J. A., Soliman J., Derr J. B., Morales M., Vullev V. I.. Practical Aspects of Cyclic Voltammetry: How to Estimate Reduction Potentials When Irreversibility Prevails. J. Electrochem. Soc. 2019;166:H3175–H3187. doi: 10.1149/2.0241905jes. [DOI] [Google Scholar]
- Sakamoto M., Cai X., Hara M., Tojo S., Fujitsuka M., Majima T.. Anomalous Fluorescence from the Azaxanthone Ketyl Radical in the Excited State. J. Am. Chem. Soc. 2005;127:3702–3703. doi: 10.1021/ja043212v. [DOI] [PubMed] [Google Scholar]
- Gray H. B., Winkler J. R.. Long-range electron transfer. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Closs G. L., Miller J. R.. Intramolecular Long-Distance Electron Transfer in Organic Molecules. Science. 1988;240:440–447. doi: 10.1126/science.240.4851.440. [DOI] [PubMed] [Google Scholar]
- Muller P.-A., Vauthey E.. Charge Recombination Dynamics of Geminate Ion Pairs Formed by Electron Transfer Quenching of Molecules in an Upper Excited State. J. Phys. Chem. A. 2001;105:5994–6000. doi: 10.1021/jp010015z. [DOI] [Google Scholar]
- Marzo L., Pagire S. K., Reiser O., König B.. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem., Int. Ed. 2018;57(32):10034–10072. doi: 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Rosspeintner A., Angulo G., Vauthey E.. Bimolecular photoinduced electron transfer beyond the diffusion limit: the Rehm-Weller experiment revisited with femtosecond time resolution. J. Am. Chem. Soc. 2014;136:2026–2032. doi: 10.1021/ja4118279. [DOI] [PubMed] [Google Scholar]
- Lin C., Kim T., Schultz J. D., Young R. M., Wasielewski M. R.. Accelerating symmetry-breaking charge separation in a perylenediimide trimer through a vibronically coherent dimer intermediate. Nat. Chem. 2022;14(7):786–793. doi: 10.1038/s41557-022-00927-y. [DOI] [PubMed] [Google Scholar]
- Delor M., Scattergood P. A., Sazanovich I. V., Parker A. W., Greetham G. M., Meijer A. J. H. M., Towrie M., Weinstein J. A.. Toward control of electron transfer in donor-acceptor molecules by bond-specific infrared excitation. Science. 2014;346:1492–1495. doi: 10.1126/science.1259995. [DOI] [PubMed] [Google Scholar]
- Wang C., Li H., Bürgin T. H., Wenger O. S.. Cage escape governs photoredox reaction rates and quantum yields. Nat. Chem. 2024;16:1151–1159. doi: 10.1038/s41557-024-01482-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin M. J., Dickenson J. C., Ripak A., Deetz A. M., McCarthy J. S., Meyer G. J., Troian-Gautier L.. Factors that Impact Photochemical Cage Escape Yields. Chem. Rev. 2024;124:7379–7464. doi: 10.1021/acs.chemrev.3c00930. [DOI] [PubMed] [Google Scholar]
- Draper F., DiLuzio S., Sayre H. J., Pham L. N., Coote M. L., Doeven H., Francis P. S., Connell T. U.. Maximizing Photon-to-Electron Conversion for Atom Efficient Photoredox Catalysis. J. Am. Chem. Soc. 2024;146:26587–27214. doi: 10.1021/jacs.4c07396. [DOI] [PubMed] [Google Scholar]
- Tian X., Karl T. A., Reiter S., Yakubov S., de Vivie-Riedle R., König B., Barham J. P.. Electro-mediated PhotoRedox Catalysis for Selective C(sp3)-O Cleavages of Phosphinated Alcohols to Carbanions. Angew. Chem., Int. Ed. 2021;60:20817–20825. doi: 10.1002/anie.202105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchenko A. P., Tomin V. I., Chou P.-T.. Breaking the Kasha Rule for More Efficient Photochemistry. Chem. Rev. 2017;117:13353–13381. doi: 10.1021/acs.chemrev.7b00110. [DOI] [PubMed] [Google Scholar]
- Lin Y., He Y., Wang Q., Feng J., Hou Y., Wang C.. Anti-Kasha’s rule for semiconductor photocatalytic reactions: the wavelength dependence of quantum efficiency. Phys. Chem. Chem. Phys. 2025;27(10):5006–5011. doi: 10.1039/D4CP03976A. [DOI] [PubMed] [Google Scholar]
- Pfund B., Hutskalova V., Sparr C., Wenger O. S.. Isoacridone dyes with parallel reactivity from both singlet and triplet excited states for biphotonic catalysis and upconversion. Chem. Sci. 2023;14:11180–11191. doi: 10.1039/D3SC02768F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund B., Schreier M. R., Steffen D. M., Wenger O. S.. Photocatalytic Regeneration of a Nicotinamide Adenine Nucleotide Mimic with Water-Soluble Iridium(III) Complexes. Inorg. Chem. 2023;62:7636–7643. doi: 10.1021/acs.inorgchem.2c03100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M., Bertrams M.-S., Sell A. C., Glaser F., Kerzig C.. Efficient Energy and Electron Transfer Photocatalysis with a Coulombic Dyad. J. Am. Chem. Soc. 2024;146:25799–25812. doi: 10.1021/jacs.4c08551. [DOI] [PubMed] [Google Scholar]
- Glaser F., Schmitz M., Kerzig C.. Coulomb interactions for mediator-enhanced sensitized triplet-triplet annihilation upconversion in solution. Nanoscale. 2023;16(1):123–137. doi: 10.1039/D3NR05265F. [DOI] [PubMed] [Google Scholar]
- Li P., Bourgois C., Glaser F., De Kreijger S., Cadranel A., Troian-Gautier L., Hu K.. Outcompeting Thermodynamics: Ion-Pairing and Coulombic Interactions to Trigger Perfluoroacetate Intra-Ionic Photooxidation for Perfluoroalkylation Reactions. J. Am. Chem. Soc. 2025;147:12082–12091. doi: 10.1021/jacs.5c00129. [DOI] [PubMed] [Google Scholar]
- Troian-Gautier L., Beauvilliers E. E., Swords W. B., Meyer G. J.. Redox Active Ion-Paired Excited States Undergo Dynamic Electron Transfer. J. Am. Chem. Soc. 2016;138:16815–16826. doi: 10.1021/jacs.6b11337. [DOI] [PubMed] [Google Scholar]
- De Kreijger S., Ripak A., Elias B., Troian-Gautier L.. Investigation of the Excited-State Electron Transfer and Cage Escape Yields Between Halides and a Fe(III) Photosensitizer. J. Am. Chem. Soc. 2024;146:10286–10292. doi: 10.1021/jacs.4c02808. [DOI] [PubMed] [Google Scholar]
- Jin P., Xu X., Yan Y., Hammecke H., Wang C.. Luminescent Fe(III) Complex Sensitizes Aerobic Photon Upconversion and Initiates Photocatalytic Radical Polymerization. J. Am. Chem. Soc. 2024;146:35390–35401. doi: 10.1021/jacs.4c14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzink M. J., Kidd J. B., Swords W. B., Yoon T. P.. Chiral Photocatalyst Structures in Asymmetric Photochemical Synthesis. Chem. Rev. 2022;122:1654–1716. doi: 10.1021/acs.chemrev.1c00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton S. T., Lee G., Moore C. E., Sevov C. S., Turro C.. Cyclometallated Co(III) Complexes with Lowest-Energy Charge Transfer Excited States Accessible with Visible Light. J. Am. Chem. Soc. 2025;147(16):13315–13327. doi: 10.1021/jacs.4c18299. [DOI] [PubMed] [Google Scholar]
- Jhun B. H., Jang J., Lee S., Cho E. J., You Y.. Efficient photoredox catalysis in C-C cross-coupling reactions by two-coordinated Au(I) complex. Nat. Commun. 2024;15(1):6586. doi: 10.1038/s41467-024-50979-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan D. A., Bim D., Silva B., Kazmierczak N. P., McNicholas B. J., Hadt R. G.. Elucidating the Mechanism of Excited-State Bond Homolysis in Nickel-Bipyridine Photoredox Catalysts. J. Am. Chem. Soc. 2022;144(14):6516–6531. doi: 10.1021/jacs.2c01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.