

Abstract
Harvesting energy from the human body is an area of growing interest. While several techniques have been explored, the focus in the field is converging on using Glucose Fuel Cells (GFCs) that use glucose oxidation reactions at an anode and oxygen reduction reactions (ORRs) at a cathode to create a voltage gradient that can be stored as power. To facilitate these reactions, catalysts are immobilized at an anode and cathode that result in electrochemistry that typically produces two electrons, a water molecule, and gluconic acid. There are two competing classes of these catalysts: enzymes, which use organic proteins, and abiotic options, which use reactive metals. Enzymatic catalysts show better specificity towards glucose, whereas abiotic options show superior operational stability. The most advanced enzymatic test showed a maximum power density of 119 μW/cm2 and an efficiency loss of 4% over 15 hours of operation. The best abiotic experiment resulted in 43 μW/cm2 and exhibited no signs of performance loss after 140 hours. Given the range of existing implantable devices’ power budget from 10μW to 100mW and expected operational duration of 10 years or more, GFCs hold promise, but considerable advances need to be made to translate this technology to practical applications.
Index Terms —: Abiotic Catalyst, Blood Sugar, Energy Harvesting, Enzymatic Fuel Cell, Glucose, Glucose Fuel Cell, Raney-Platinum
I. Introduction
THE US Food and Drug Administration recognizes Active Implantable Medical Devices (AIMDs) as surgically implanted medical devices that require an energy source to power[1]. In most cases, these energy sources are batteries that are either wirelessly recharged, or surgically replaced after a long lifespan up to ~10 years. AIMDs are a growing area of interest as artificial organs and advanced prostheses become more accepted as medical therapy[2]. The less sophisticated, but long-duration AIMDs such as pacemakers and neurostimulators for pain or sleep apnea typically consume 10 μW to 100 μW [3], [4], while daily recharged AIMDs such as cochlear implants have higher power requirements at 10 to 100 mW[5]. For life sustaining artificial organs where daily recharging is not a reasonable option or for high power applications such as neuroprostheses, the existing strategies of never recharging, or recharging frequently, may be insufficient. Instead, continuous recharging of the battery using power derived from the body may be the path forward.
A number of technologies show promise in being able to generate electrical energy from the human body, but most suffer from critical drawbacks[6]. Placing tiny hydroelectric turbines in the bloodstream[7], [8], has concerning implications for thrombosis and blood pressure both up and downstream from the turbines. Electricity generated by the thermal gradient between the body and the outside temperature[9], requires access to skin surface in order to function. Reclamation of excess kinetic energy in the limbs or torso[10], [11] generates power if the patient is in motion and thus is not helpful for sedentary or immobile users. Piezo electronics can generate charge based on the movement of organs such as the heart or lungs[12] but suffer from low energy yields (~0.25 μW/cm2) and poor scalability. The Hydrovoltaic Effect that generates electrical potential using water evaporating from carbon nanomaterials, could be used in conjunction with human breath to generate electricity. Benchtop testing has produced 150 nW, but implementation would require both the wearing of a special mask as well as appropriate atmospheric conditions to function optimally[13]. ATP fuel cells use Adenosine Triphosphate (ATP) as the energy-containing substrate[14], but because ATP is naturally formed inside of cells it has low concentrations in the interstitial tissue[15], reducing access by an implantable device. All of these technologies ultimately use energy that can be traced back to the same source: glucose.
Glucose Fuel Cell (GFC) technology is a family of energy conversion devices that chemically react with naturally occurring glucose (C6H12O6) in the human body to break their molecular bonds down and capture some of their stored energy[16] (Figure 1). Compared to other bioenergy harvesting methods (Figure 2), this option is the most directly analogous to the natural means of generating energy by the body. As such, it has a substantial amount of biological infrastructure supporting it, such as hormonal systems that regulate glucose levels, means of clearing broken down glucose, and methods of storing glucose for later use. This preexisting support system, in conjunction with the eliminating as many lossy energy conversions as possible, positions GFCs as the most elegant and efficient energy harvesting option available.
Figure 1:
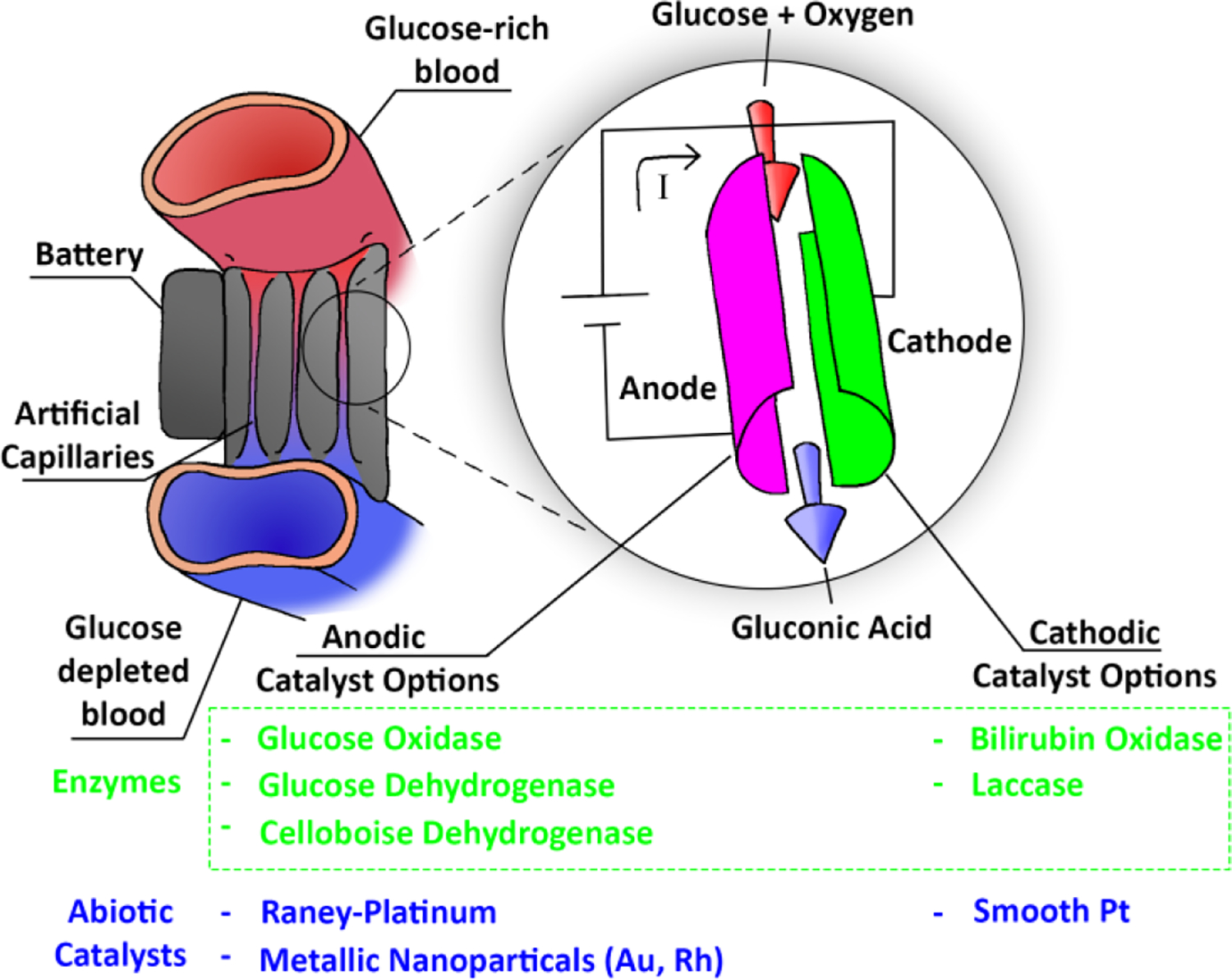
A conceptual overview of Glucose Fuel Cells. The artificial capillary configuration is one of many possible design strategies.
Figure 2:

Each arrow in the image above represents a conversion of energy. In each of these conversions, energy is lost. By selecting the pathway with the fewest lossy energy conversions, a more efficient system can be realized.
GFCs have a history reaching back to the 1960s[17]. They function by selectively catalyzing reactions at the anode and cathode electrodes within a glucose and oxygen rich system. The oxidation of glucose releases an electron, while the reduction of oxygen receives an electron, thereby creating a voltage gradient that drives current. In the past two decades alone, in vivo experiments have been performed with snails[18], lobsters[19], cockroaches[20], clams[21], rats[22]–[25], mice[26], and an orange[27].
Implanted GFCs have two primary challenges: longevity, and power density. An implanted device must maintain effectiveness over the implant’s entire lifespan of operational duration, typically years. Further, in order for this technology to become a viable alternative to batteries it will need to be able to generate the tens to hundreds of mW of power needed to keep artificial organs running continually. The additional challenge of biocompatibility precludes using many materials or compounds in a device, limiting design options.
Several review papers in the past decade have considered the field and draw attention to these shortcomings, often highlighting what they consider to be the most promising route towards overcoming the hurdles. In 2009, Willner et al. suggested that improved power outputs could be achieved by higher enzyme loading on the electrodes, alongside improving conductivity between the catalysts and the electrodes[28]. Rasmussen et al. published a review paper in 2016 that cited enzyme engineering and enzyme cascading as potential paths forward[17]. As recently as 2022, Tong Liu published a review in which they theorized that in biology, glucose decomposition is largely done by enzymes within a membrane structure, and that the lack of this structure could be the missing key[29].
Here we pose that while the recent approaches might reasonably improve power densities to serviceable levels, the expectation that any amount of engineering or structural design might allow an enzyme to survive immobilized on an electrode for an entire decade is unreasonable. The fact that Glucose Oxidase (GOx), one of the most common enzyme selections for glucose oxidation in a GFC, has a half-life of less than two hours in the liver and spleen and rat models[30] conveys the enormity of the task at hand. Conversely, several recent papers have shown abiotic catalyst options such as platinum to have promise as glucose oxidation agents. Platinum catalysts boast a number of advantages over enzymatic ones, most notably the drastically increased stability. This paper posits that shifting the focus from enzymes to abiotic catalysts is the mostly likely path toward overcoming the major challenges in the field.
II. Theory
The principle concept behind GFCs is that glucose, an energy-rich organic molecule that is the backbone of human metabolism, can be broken down via a chemical reaction known as a redox reaction, releasing some of its energy in the process. That released energy can then be captured to power AIMDs. The molecular breakdown, occurring at the anode of a GFC device, results in electrons moving away from the site of the reaction. At the cathode of the device, the secondary reaction occurs, requiring an electron and thereby creating a voltage gradient. This gradient between the anode and the cathode generates a current which is delivered to an energy storage device, such as a battery or capacitor[31] and used by the AIMD as needed for its operation. The low voltages generally produced by GFCs can be boosted to higher voltages and regulated to drive more complex systems[32]. For this process to work, both the anode and the cathode require some catalyst that will facilitate their respective halves of the redox reaction, as well as any necessary mediators, which are supporting materials that will immobilize or conductively connect this catalyst to the electrodes that form the base of the system (Figure 3). The catalysts, electrode materials, and immobilization mediators and techniques vary, but all of them perform the same underpinning reaction at the anode: the oxidation of glucose.
Figure 3:
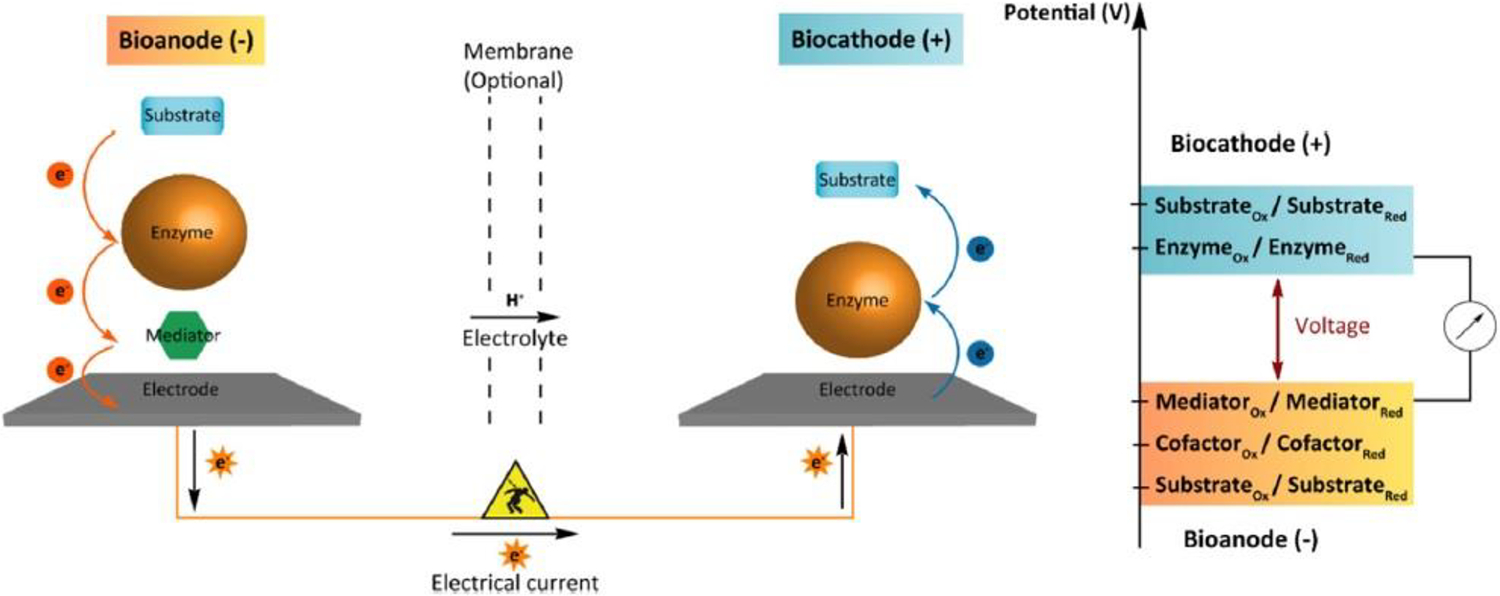
A fuel cell overview. The anodic substrate in a GFC is glucose, while the cathodic substrates are oxygen and positively charged hydrogen ions. The mediator is an optional component, depending on enzyme selection. Used with permission from Rasmussen et al., 2016
Oxidation of glucose, occurring at the anode of a GFC, is half of a redox reaction that releases electrons to the electrode[33]. There are multiple pathways for this reaction depending on the catalyst used, but in all cases the resulting products are Gluconic Acid (C6H12O7), protons, and electrons. The four main catalysts used on the anode side of a GFC are Glucose Oxidase (GOx)[20], [22], [25], [26], [34]–[37], Glucose Dehydrogenase (GDH)[18], [19], [21], [24], [27], [38], [39], Cellobiose Dehydrogenase (CDH)[23], [40], [41], and Raney-type Platinum (Pt)[42]–[44]. Note that GOx, GDH, and CDH are all enzymes, whereas platinum is an abiotic catalyst.
At the cathode, there needs to be a reduction reaction which receives electrons from the electrode[33]. Like the anode, there are multiple options for catalysts that have been investigated, but each of them consumes O2 and 4H+ to make 2 H2O. H+ is generated at the anode and blood has high concentrations of O2. Similarly, H2O is ubiquitous in the human body, so the generation of additional H2O at the cathode will not interrupt normal biological functions. GFC cathodes have been attempted using Laccase[18], [19], [21], [24], [25], [27], [35], [36], Bilirubin Oxidase (BOx)[20], [23], [26], [34], [37], [38], [40], [41], and Pt[43].
Whether at the anode or the cathode, the strategies used to optimize enzymatic GFCs and those used for abiotic catalyst designs differ in their focus. Enzymatic systems often strive to protect the organic catalysts and improve their electrical conductivity with the electrode by using mediators, external frameworks, and enzyme engineering efforts. In contrast, abiotc systems that use reactive metals such as platinum, gold[45], or rhodium[46], can use a number of strategies that are not applicable to enzymes, such as adjusting alloying ratios of the various metals, varying the fabrication methods to improve porosity or other selected material properties, or even attempting to use synthetic, enzyme-like structure with abiotic material at the reactive sites.
III. Ongoing Challenges
GFCs have not made the translation from research to commercial application due to their continued struggles with longevity and power density, especially in the context of in-vivo applications. These challenges can be seen illustrated in the Voltage Retention and Power Density columns in Table I. Most GFC experiments have very poor long-term efficacy, with the half-life of the fuel cell being measured in hours instead of the needed years of operation. Further, the power output per square centimeter of fluid-interfacing device is typically less than 100 μW. Power density on this scale is likely insufficient to power a meaningfully sophisticated implant. Finally, while not a challenge that explicitly needs to be overcome during initial experimentation, the question of biocompatibility severely constrains the possible practical solutions. The set of permissible materials that can be used as electrodes, mediators, catalysts, and structural componentry safely inside of a biological organism is very narrow, closing a lot of doors to otherwise promising choices.
Table I:
Comparison of selected published GFC experiments. Note that not all experiments were conducted in biologically representative environments.
| Test Environment | Anode | Cathode | Open Circuit Voltage (V) | Power Density (μW/cm2) | Voltage Retention (time) | Source |
|---|---|---|---|---|---|---|
| In vitro (Glucose in phosphate-buffered saline) | Pt | Pt | 0.09 | 43 | 100% (140 h) | Simons, 2022[43] |
| In vitro (glucose solution) | Pt | - | 0.21 | 0.42 | 100% (70 h) | Do, 2019[44] |
| In vitro (human blood) | GOx Peroxidase Hydrophilic MAF-7 | BOx | 0.34 | 119 | 96% (15 h) | Yimamumaimaiti, 2020[34] |
| In vivo (cockroach) | GOX Trehalase (Tre) | BOx | 0.2 | 55 | 95% (2.5 h) | Rasmussen, 2012[20] |
| In vitro (soda) | Rhodium Nanozyme | BOx | 135 | 88% (2.78 h) | Zhao, 2023[46] | |
| In vitro (glucose solution) | Pt-Au | - | 0.34 | 95700 | 72.6% (3 h) | Torigoe, 2018[42] |
| In vivo (rat) | GOx Catalase Graphite Ubiquinone | Graphite Quinone Hydroquinone Polyphenol Oxidase (PPO) | 0.275 | - | 70% (2.5 h) | Cinquin, 2010[22] |
| In vivo (mouse) | GOx | BOx | 0.34 | 300 | 55% (5 days) | Lee. 2020[26] |
| In vitro (human serum) | NAD-GDH | BOx | 0.73 | 53.9 | 54% (7 days) | Gao, 2007[47] |
| In vivo (clam) | PQQ-GDH | Laccase | 0.4 | 40 | 50% (48 h) | Szczupak, 2012[21] |
| In vitro (human blood) | PAC-GDH (gold electrode with polyaniline, AuNP, cysteamine, and PQQ-GDH) | - | 0.1 | 65 | 50% (4.5 h) | Gineitytė, 2019[39] |
| In vitro (human serum) | CDH | BOx | 0.58 | 4 | 50% (2 h) | Coman, 2010[40] |
| In vitro (human blood) | CDH | Box | 0.65 | 3 | 50% (2 h) | Wang, 2012[41] |
| In vivo (rat) | CDH | Box | 0.4 | 2 | 50% (2 h) | Andoralov, 2013[23] |
| In vivo (snail) | PQQ-GDH | Laccase | 0.53 | 30 | 50% (0.2 h) | Halámková, 2012[18] |
| In vitro | GOx | Laccase | 0.075 | 0.196 | 48.6% (40 days) | Rewatkar, 2020[36] |
| In vitro (human blood) | PQQ-GDH | BOx | 0.65 | 68.1 | 25% (3 h) | Cadet, 2016[38] |
| In vitro (human serum) | FAD-GDH | BOx | 0.56 | 40.3 | 24% (13 h) | Milton, 2015[48] |
| In vivo (lobster) | PQQ-GDH | Laccase | 0.6 | 640 | - | MacVittie, 2012[19] |
| In vivo (rat) | PQQ-GDH | Laccase | 0.14 | 0.175 | - | Castorena-Gonzalez, 2013[24] |
| In vivo (rat) | GOx | Laccase | 0.57 | 193.5 | - | Zebda, 2013[25] |
| In vivo (orange) | PQQ-GDH FAD-dependent Fructose Dehydrogenase (FAD-FDH) | Laccase | 0.6 | 90 | - | MacVittie, 2015[27] |
| In vitro (Phosphate buffer w/ 0.05mol/l of glucose) | GOx Catalase | Laccase | 0.95 | 1,300 | - | Zebda, 2011[35] |
A. Efficacy Retention
Current AIMDs such as pacemakers typically have an operational lifespan of greater than ten years[4]. This one-decade threshold is a reasonable target for the functional duration of GFCs. Table I, arranged by operational duration, shows two solutions at the top that suffered no loss of functionality over time for 140 hours in the longest running experiment. Both used abiotic catalysts, suggesting that these options might conceivably be able to achieve this target. Currently the enzymatic GFC solutions are unable to sustain power delivery without drop-off for longer than 15 hours. There are two main reasons why a GFC might lose efficacy over time: loss of structural stability, and loss of functional stability. Structural stability issues include enzyme delamination and enzyme rearrangement[49], and generally only impact enzymatic catalyst options. Functional stability issues can be caused by reaction site inhibition or by enzyme degradation. Again, enzyme degradation is not a risk for non-enzymatic based systems. Synthetic enzymes should be considered to suffer from most, if not all, of the same shortcomings of natural enzymes.
1). Structural Stability
Enzyme delamination, a term for when enzymes detach from the surface to which they were immobilized, is a cumulative problem because no further immobilization can occur post-manufacturing. At reactant saturation, each lost enzyme directly translates to loss of reactions per second and thus loss of power. Causes for enzyme delamination depend on the environment in which the device is placed; however, pH, temperature, and mechanical erosion from blood flow or other factors can all play a part[49].
Enzyme rearrangement is a similar phenomenon in that it involves the enzymes that were immobilized on the device surface shifting or reorienting in deleterious ways and cannot be easily corrected without rebuilding the device. Some enzymes are only effective if oriented in a specific way with respect to the electrode and the reactant supply[50]. Enzymes cannot be oriented such that access of the substrates to the reaction sites is blocked, as one example. Enzymes must also be oriented so that proximity of the electrode to the electrooxidation/electroreduction sites is sufficiently close to allow the electron to cross the gap[51], [52]. In some cases, multiple enzymes must exist near each other in specific ratios to facilitate enzyme cascading reactions. These are typically called coenzymes. Enzyme rearrangement of necessary coenzymes away from each other, even if they remain bonded to the electrode, can limit the intended synergies between the two enzymes. GOx is frequently coenzymed with catalase[35] or peroxidase[34] so that the produced hydrogen peroxide (H2O2) is disposed of before it can cause GOx inhibition. Loss or rearrangement of these enzymes with relation to each other could result in increased GOx inhibition.
These structural integrity problems have been addressed through the addition of cross-linking or protective membranes[49]. 1-pyrenebutanoic acid succinimidyl ester (PBSE)[18], [21], poly-(3,4-ethylenedioxythiophene) (PEDOT) coating[26], and Poly (ethylene glycol) diglycidyl ether (PEGDGE)[26] have all been used as agents to improve the structural stability of an enzymatic GFC. Dong et al. trialed a model in which the enzymes were trapped between two layers of carbon nanotubes[26] to improve retention. Yimamumaimaiti et al. employed a metal-oxide framework, MAF-7, interlaced with single walled carbon nanotubes to both improve enzyme retention and improve the conductivity of the system[34]. Besanger et al. published on how entrapping enzymes on diglyceryl silane derived sol-gel glasses improves their long-term stability and catalytic efficient in all tested cases[53], while a few years prior Mateo et al. discussed how a multifunctional support containing epoxy groups has been shown to drastically improve covalent bonding between the protein and the support[54]. However, while adding these cross-linkers seems to increase the longevity of the GFC from hours to days, they still largely fall short of expectations.
Single-material abiotic catalysts such as Raney-Platinum do not suffer from delamination or rearrangement failure modes by virtue of being a single continuous plane of reactive material.
2). Functional Stability
An alternative contribution to loss of operation over time is associated with the loss of function of catalysts themselves. Enzyme functionality is highly sensitive to pH, temperature, and local reactants. Enzymes can be further damaged by macromolecular proteins that are present in the bloodstream, such as high ionic strength, low-molecular weight proteolytic enzymes[34].
Catalyst inhibition can be caused on a micro-scale by a specific reactant permanently disabling the reaction site, or on a macro-scale by larger biological entities such as entire blood cells adhering to the surface of the electrode, mechanically blocking access to the reactive region[41]. Many of the most commonly selected enzymes for GFCs have inhibiting agents that are present in the blood, though in some cases the inhibition is transient rather than permanent. Hydrogen peroxide inhibits GOx[34], [35], [55], [56], CDH[49], BOx[57], and laccase[50], irreversibly so in BOx’s case. GOx and CDH also produce hydrogen peroxide, exacerbating the problem. Evidence suggests that calcium inhibits Pyrroloquinoline Quinone (PQQ)-GDH[38], zinc sulfate inhibits CDH[41], and chloride ions inhibit both BOx[48] and Raney-Pt[58]. Indeed, Harris et al. state that any molecule below 10 kiloDaltons can potentially inhibit GOx[59]; Wang et al. similarly claim that chloride ions, zinc ions, and uric and ascorbic acids can inhibit not only GOx but most GFC enzymes[41]. Even non-enzymatic catalysts are subject to inhibition. Reaction intermediaries like carbon monoxide may adsorb to the electrode under certain conditions[44]. Reaction products like gluconic acid have been suggested to inhibit Raney-Platinum anodes[58], though other sources imply that this is due to the Raney-Pt actually oxidizing the gluconic acid further, releasing additional electrons before freeing up the reaction site again [60]. Rhodium based synthetic enzymes have been theorized to be inhibited by carbonic acid[46].
Finding solutions to the problem of catalyst inhibition is therefore critical for reliability. One option is to engineer enzymes that will be resistant to inhibition from the offending substance. This has been tried with PQQ-GDH, with an attempt to narrow the set of substrates that the enzyme is sensitive to[61]. Another option is to try to find new enzymes that may counteract the inhibition effects. A common approach is to build the electrode with coenzymes that will destroy the offending substrates before they can do damage; this works well when the inhibiting agent is a common metabolic byproduct such as hydrogen peroxide[34], [35], but is less effective when the inhibitor is a chloride ion. Furthermore, coenzymes occupy space on the electrode that would otherwise have been used for increased catalyst loading, directly costing the system reactions per second per unit area.
Mechanical or chemical barriers are another potential solution; dialysis membranes[22], [25], [38] have been used to moderate effect, provided that they are appropriately sized to limit the passage of the inhibitor but not the desired substrates. If the molecular weight of both sets are similar, this option becomes less potent. The previously mentioned MAF-7 metal-oxide framework that Yimamumaimaiti et al. used was deliberately chosen not just to improve structural stability of the system, but also to protect the enzymes from much of the blood’s hostile environment. MAF-7 is hydrophilic which allows for glucose to follow hydrophilic channels to the GOx while the framework keeps at bay many known GOx inhibitors[34]. Other attempts at encapsulating the enzymes in porous structures for protection include the use of multiwalled carbon nanotubes[26], [35], [48], compression into buckypaper[24], [27], and a 3D gold/gold-nanoparticle structure[41]. Nafion coating of a Raney-Platinum anode at least partially mitigates this risk of inhibition by gluconic acid, as the negatively charged Nafion repels the similarly negatively charged gluconic acid[58].
Another option is to introduce a transient catalyst that would clear out any offending inhibitors and, where possible, return an inhibited enzyme to its functional state. For example, introducing catalase into a system in which there is hydrogen peroxide-inhibited laccase has been shown to liberate the laccase from the offending substrates[50]. The downside of this method is that it is temporary and must be periodically repeated with a period that can be as short as an hour.
Finally, there is the risk that the enzyme will simply denature. Enzymes are constantly being created and destroyed in biological systems, and many do not have a particularly long shelf life. GOx, for example, lasts for about 2 hours in the blood of rat models[30]. Cells contain complex biological machinery to constantly replenish their degrading enzymes. No proposal to replicate this machinery in a GFC has been yet proposed, and this natural degradation of enzymes appears to be a potent flaw in the enzymatic GFC model. In contrast, Raney-Platinum catalysts are not proteins and are therefore not at risk of degradation.
Raney-Platinum anodes were shown to exhibit the formation of precipitates after 140 hours of operation, which scanning electron microscopy and energy dispersive x-ray spectroscopy indicated were salts that were forming as a result of the glucose reactions with the phosphate buffered saline solution that was the solvent of the test solution[43]. With enough time these salts could create a film over the electrode, preventing further glucose interaction with the Pt. It is unlikely that this specific problem would translate to the in-vivo applications as it appears to be a side effect of the experimental bench preparation.
B. Power Density
The most sophisticated AIMDs currently on the market require daily or weekly battery recharges due to their average power consumption rate of around 100 mW[5]. In order for a GFC to achieve a power output rate equivalent to this demand, it will be necessary to optimize for all of the variables in the power output equation.
Power output can be viewed as a function of reactions per second, which in turn is the product of the total number of catalytic sites and the number of reactions per second per site. The number of catalytic sites is a mechanical matter, governed by total surface area and loading density. The reactions per second per site is a chemical limit, influenced by reaction velocity. Reactions per second is not the only variable, however: the produced electrical current is influenced by the resistivity between the enzyme and electrode. Finally, there must be sufficient substrate concentration and replenishment rates in the vicinity of the GFC in order to continually feed the reactions. Each of these areas must be considered when selecting a catalyst, choosing a fabrication method, and designing a device.
1). Surface Area and Catalyst Density
For enzymatic GFCs, catalytic sites per unit area is the product of enzyme loading density, i.e. the number of enzymes immobilized per unit area[62], and the total area, whereas for abiotic catalysts it can be a fixed value if the entire electrode is made of reactive catalytic material. The native benefit of having effectively maximum reactive site density is another strong benefit that abiotic catalysts can have over enzymatic ones. Both total area and number of enzymes per unit area, if applicable, can be influenced by the structural design of the device. Material selection can increase surface area due to nano-scale ridges, grooves, or other textures. Carbon nanohorns, for example, are a subclass of carbon nanotubes that have horn-shaped protrusions on the ends of the tubes, adding to the functional surface area without requiring additional electrode real estate[63], [64]. Other forms of porous electrode mediators and structural elements (metal-organic frameworks, Au/AuNP cages) that also serve to increase surface area have been discussed above.
Beyond material selection, there are design choices that can be made to increase the available real estate for electrode surface area within a limited three-dimensional envelope. One biologically inspired notion is to replicate the capillary system that nature has evolved. The adult human has around 6,000 m2 of capillary surface area, or sixty million square centimeters[65]. With adult humans having a volume of roughly sixty thousand cubic centimeters[66], that is an average ratio of 1000:1 cm2 of capillary surface area per cm3. Utilizing a similar structure could allow for huge electrode surface area in a relatively small volume. While miniaturization at this scale has not been thoroughly explored, there have been examples of GFCs being built into artificial blood vessels[67].
Additionally, the catalyst and mediator selection, as well as the device manufacturing methodology, can impact how many enzymes are immobilized per unit area. Electrostatic binding techniques have been demonstrated to be more effective at tightly packing PQQ-GDH enzymes onto gold electrodes and increased surface coverage from 16% to 49%[68]. This electrostatic technique, often referred to as a ‘layer-by-layer’ technique due to the multiple layers of electrode substrate that get deposited to increase static charge, has also been employed with GOx[62] and with Pt nanoparticle[69]. It has the potential drawback of increasing the thickness of the immobilizing film coating the electrode itself, which can increase electrical resistance, reducing total current.
2). Reaction Velocity
The third parameter that impacts the total reactions per second of a complete GFC is the number of reactions per second of each individual catalyst. To maximize power output, selecting a catalyst that operates very quickly makes logical sense. kCAT represents the rate constant of a catalyst, which is the reaction rate in mM/s when the catalyst exists in an environment with substrate saturation. Raney-platinum anodes and other abiotic options are difficult to characterize, as there is no discrete enzyme that can be assessed for molar throughput. As listed in the BRaunschweig ENzyme DAtabase (BRENDA)[70], the kCAT values for the most commonly used anodic enzymes are:
Note that Tables II and III only include anodic enzyme options. Enzymes used at the cathode, such as BOx and Laccase, are more difficult to find appropriate kCAT values for on the grounds that GFCs do not use these enzymes in their traditional fashion. BOx, for example, does not react with bilirubin in a GFC, and so published literature on the enzyme’s rate constant are not applicable. In lieu of converting bilirubin to biliverdin[51], the electrode simply provides BOx with the electron it needs, and the ORR, typically concomitant with the bilirubin oxidation[52], occurs alone.
Table II:
Anode kCAT values, per BRENDA
| Enzyme | kCAT (mM/s) |
|---|---|
| Gox | 44 |
| PQQ-GDH | 74 |
| FAD-GDH | 10 |
| NAD-GDH | 3 |
| CDH* | 17.4 |
CDH values from BRENDA relate to its reactions with cellodextrins, etc. and not with glucose, a non-primary substrate. This value instead comes from Geiss, 2021.
Table III:
Anode KM values, per BRENDA
| Enzyme | KM (mM) |
|---|---|
| Gox | 213 |
| PQQ-GDH | 321 |
| FAD-GDH | 39 |
| NAD-GDH | 16 |
| CDH* | 138 |
CDH values from BRENDA relate to its reactions with cellodextrins, etc. and not with glucose, a non-primary substrate. This value instead comes from Geiss, 2021.
3). Catalyst Resistivity
In addition to considering the number of reactions per second of the entire system, power output is also impacted by the conductivity along the entire circuit from anode to cathode. For enzymatic GFCs, any resistance against electron movement from the enzyme onto the electrode can decrease the system’s overall efficiency. Many structural stability mediators (doped PBSE[18], [21], PEDOT[26], PEGDGE[26]) also serve as mediated electron transfer conveyors, setting up conductive pathways for the electron to follow with minimal resistance. These and other redox-active hydrogels, cofactor monolayers, or crosslinking polymers can boost electrical communication significantly[28]. Electrode material choice itself is also a factor; carbon nanotubes are a popular choice due to their high conductivity and surface area. Other mediators serve as orienting agents in cases where enzyme orientation has a meaningful impact on the resistance between the reaction site and the electrode, such as the use of anthracene with BOx and laccase[50]. By carefully balancing reaction potential, electrical resistance, and reactions per second, it is be possible to improve power density though material selection alone. Once again, abiotic catalysts that comprise the entirety of the electrode benefit from being conductive metal from start to finish, removing much of the concern surrounding resistivity.
4). Substrate Concentration and Mass Turnover
kCAT is an important catalytic property to consider when selecting an enzyme, but it only relates to reaction velocity at substrate saturation. It is not necessarily the case that the substrates will be at saturation in an in vivo environment, and so the specific substrate concentration requirements of each catalyst must also be considered. In GFCs KM, the Michaelis constant, represents the amount of glucose needed for the catalyst to operate at 50% of maximum speed. This differs by catalyst, and is one of the factors that is often optimized for in enzyme engineering research. One such engineering example saw improvements in the performance of the anodic enzyme CDH, reducing its KM by two-thirds[71]. Table III shows the baseline KM values for the main GFC catalysts discussed here, as reported by BRENDA[70].
While with kCAT, higher values indicate a more desirable outcome (reactions per second), with KM a lower number is preferable, as it indicates that the catalyst is functional even in lower densities of the reactants. There is a correlation between the kCAT and KM values, as a higher max reaction velocity naturally necessitates an interaction with higher substrate concentration. For this reason, the ambient glucose concentration levels need to be considered with respect to the selected catalyst’s KM value.
Glucose concentration levels depleted by GFCs would need to be replenished through some means, the rate of which may bottleneck the maximum power output of the device. Glucose concentration in blood is different from its concentration in the Interstitial Fluid (IF). The tradeoff is that lacing a GFC in the IF would be far simpler surgically but the placement suffers from lack of blood circulation, instead relying on diffusion for glucose replenishment. Evidence of the reduced IF placement effectiveness can be seen in in vivo GFC experiments using the IF of lobsters[19] and snails[18], in which the power output of the devices would drop by 50% after only 15 minutes, but after a 30 minute pause would return to full strength when restarted. The likely explanation for this observation is that the depletion of glucose was faster than its replenishment.
In an average adult, the fasting blood glucose concentration is between 3.9 mM and 5.6 mM[72], which when compared to the values in Table III are clearly not high enough to push any of the standard enzymatic options to saturation. IF glucose levels under steady state conditions will usually match blood glucose levels, but are the first to drop when the cells need a sudden uptake of energy, and only slowly recover as glucose diffuses through the blood vessel walls[73]. Furthermore, without the heart to actively circulate the fluid in the IF space, glucose movement is entirely reliant on simple diffusion. With a diffusion coefficient (D) of around 8.3e-10 m2s−1 [74], [75], the average time t for a glucose molecule to cross any distance × can be calculated with[76]:
| Eq. 1 |
The average cell in the human body is about 100 μm away from the nearest capillary wall[77], [78], and using exclusively simple diffusion and Eq. 1, glucose can cross that gap in about 6 seconds. However, GFCs installed in the IF space would likely have a much greater average distance from the capillary walls; Zebda et al. use a 5 mm diameter, 6 mm thick electrode in the IF space[25]. The time needed to get the average glucose molecule even half way across this disc would be over an hour. This GFC configuration would also deprive any local cells of needed glucose.
Conversely, installing the GFC in the bloodstream eliminates these diffusion concerns immediately, as the heart actively circulates blood through the GFC. In addition to the forced flow, red blood cells, equipped with membrane-bridging GLUT1 proteins[79], have a tremendously high diffusion rate, reaching a glucose concentration equilibrium across the cell wall in 2 seconds[79], as opposed to about 5 minutes across the blood vessel wall[80]. While all reviewed in vivo experiments have utilized the IF as the glucose supply, MacVittie et al. did raise the prospect of bloodstream GFCs. They ran three different glucose concentrations through a system mimicking a human capillary at two different speeds as part of an exploration on the impacts of various physiological conditions on GFC function[19].
In addition to the requirements that there be sufficient glucose in the vicinity of the catalyst to support desired reaction velocities, there is a need to ensure that any supporting chemicals required to see the reaction occur remain in the area in sufficient concentrations. For example, Nicotine Adenine Dinucleotide (NAD)-GDH does not have the NAD redox cofactor attached to the enzyme[17], [55] and therefore NAD must exist in sufficient concentrations near the NAD-GDH electrode in order for it to function. As another example, Cinquin et al. 2010 experimented with using a cathode made of compressed graphite discs containing polyphenol oxidase (PPO) and quinhydrone, described by the team as an equimolar mixture of quinone and hydroquinone[22]. In this study, the cathode was placed inside a dialysis bag with a cut-off weight of 100 g/mol, described as a technique necessary to prevent the diffusion of quinhydrone away from the cathode. The anode and cathode together were then placed inside of a larger dialysis bag with a cut-off of 7000 g/mol, to ensure that the GOx and catalase of the anode did not escape the area. Whether or not this solution is effective, the approach is arguably less practical than others that do not require the same level of control over the reactants’ isolation.
5). Reaction Efficiency
In addition to the above listed considerations when designing a GFC device, it is worth taking a wider view in considering the efficiency of energy harvesting. All anodic options in Table I use the oxidation or dehydrogenation of β-D-glucose at its first hydroxyl group. This frees up two electrons and produces gluconolactone (C6H10O6). This is not how nature typically extracts energy from glucose, and indeed collecting only two electrons per sugar molecule leaves a tremendous amount of energy untapped.
Glycolysis, the first sequence in the long process by which humans naturally break down glucose for energy, is an elaborate process involving ten different enzymes and as many steps[81]. Glycolysis yields only two electrons as well as two pyruvate molecules. The pyruvate converts to acetyl-CoA via decarboxylation, and then proceeds into the Krebs Cycle, the second sequence in our metabolic sequence. The Krebs Cycle is similarly complex, utilizing another nine enzymes. Once completed, this process liberates all 24 available electrons in the original glucose molecule, reducing it down to just six each of CO2 and H2O molecules. Artificial replication of the entire nineteen enzyme aerobic respiration pathway is likely too complex a system for cost-effective GFC devices, and as such other means of harvesting energy from glucose must be considered.
The extraction of two of the 24 electrons via oxidation or dehydrogenation is a serviceable start, but it does waste 11/12ths of the energy[82], and thus it would be preferable to use all parts of the glucose. There has been minimal published research into the potential of extracting further energy from the gluconolactone or gluconic acid byproducts of current GFCs, despite the large amount of energy remaining within. Gluconolactone Oxidase (GLO) is an enzyme that converts gluconolactone into erythorbic acid (C6H8O6)[83], a stereoisomer of Vitamin C. Not naturally occurring in humans due to a genetic divergence[84], GLO oxidizes the byproduct of most of the common enzymatic choices for GFC anodes. Gluconolactone Dehydrogenase is another gluconolactone catalyst, fairly comparable alternative to GLO[85], [86]. By pulling another two electrons out of each glucose, co-immobilizing GLO could reduce the necessary glucose concentration needed for reactant saturation.
Indeed, this approach was followed to its logical extreme by Xu and Minteer, who proposed a six-enzyme cascade that could theoretically reduce a glucose molecule down to just H2O and CO2, plus all 24 electrons[82] (Figure 4). Their recorded values demonstrated vastly improved power density (though other limitations of their test setup resulted in a fairly weak power density when compared to most line items in Table I), and further verified that there was meaningful CO2 product, suggesting that the enzyme cascade was propagating to the optimal energy efficiency. Unfortunately, there are inherent complications that come with attempting to coenzyme a large number of different enzymes: reaction velocity differences can require careful ratios of enzyme to enzyme, and mass transportation limitations can slow the collective reaction speed down entirely as the local area gets gridlocked with reactants[87]. Additionally, the need for so many different enzymes limits the maximum number of any given one enzyme, reducing total glucose throughput.
Figure 4:

A six-enzyme cascade that could reduce glucose all the way down to CO2, extracting the maximum amount of energy per glucose molecule. Adapted with permission from Xu, 2012
Platinum may also be able to oxidize glucose beyond just the first two electrons. Experimentation has indicated that certain Pt alloys can oxidize gluconic acid in much the same way that it oxidized glucose[60]. One paper suggests that platinum can actually fully oxidize glucose, presenting testing results in which they calculate that each glucose molecule released an average of 17.5 of the 24 electrons[88], which would suggest tremendous potential efficiency. Subsequent research has cautioned that while Raney-Pt can indeed oxidize gluconic acid, its adsorption rate is far lower than that of glucose under the same conditions[44], possibly as much as ten times less[89]. Such starkly different turnover rates may prevent a single Raney-Pt anode from being able to oxidize glucose further than gluconic acid. However, platinum’s demonstrated ability to oxidize gluconic acid may be able to be optimized and exploited with novel Pt configurations or coatings.
IV. Platinum as a Catalyst
Abiotic catalysts do not use enzymes, and as such do not benefit from their narrow specificity. However, the benefit of not relying on organic proteins is that abiotic catalysts can have substantially higher durability, allowing for more aggressive fabrication and sterilization methods, as well as increased lifespan within aggressive environments such as the human bloodstream. Abiotic catalyst stability far exceeds enzymatic catalyst stability[43], [44]. Abiotic catalysts can also, in some cases, be built directly onto printed circuit boards, facilitating the miniaturization of device design[43].
Abiotic glucose sensors have been built using iridium and gold[90], as well as platinum[42]–[44] and several platinum group metals, including palladium[91], [92] and rhodium[93]. In some cases the abiotic catalyst may be structured as nanoparticles that are bound to an electrode[93] whereas in other cases the electrode itself is made of the catalytic metal[43], simplifying the design and boasting tremendous reaction site density. Here we focus on the mechanisms of action using platinum as it is by far the most explored of the abiotic catalysts for GFCs, and can act as both the cathode and the anode.
A. Platinum as an Anode
Platinum is a reactive, non-toxic metal that can oxidize glucose without the need for mediators, enzymes, or other additions. It has the immediate drawback of also being a cathodic catalyst, meaning that as soon as it oxidizes the glucose, it also reduces the products down to water, completing the redox reaction in the same location and thus preventing any voltage gradient from being established. This can be overcome through the use of coatings or through changes to the Pt structure, ultimately by ensuring the local oxidation outperforms the reduction [44].
Smooth Pt has been assessed as a GFC anode candidate as far back as 1969 when Giner and Malachesky published a paper characterizing its performance under a range of temperatures, pHs, and other parameters[60]. The reaction pathway is theorized to start with the glucose adsorbing onto the Pt at C1, taking the place of the hydrogen there[94]. This is followed by Pt-C1 bond being attacked by an H2O molecule, which loses one of its hydrogens in the process[95]. The newly formed gluconic acid is more weakly bonded to the Pt, and thus detaches. The process of detaching the hydrogens results in them being split from their electrons and released from the catalyst as well.
Seven years after Giner and Malachesky, Gebhardt et al. published on a novel method of fabricating Pt electrodes that improved their glucose reaction activity by almost 20-fold[88]. This method produced what they called Raney-Platinum, which is created when an anodized platinum alloy foil has the non-Pt element (e.g. Nickel[88] and Zinc[58]) dissolved out of it, making Raney-Platinum porous. In the years since Gebhardt, other methods of fabricating porous Pt have been developed and explored: e-beam evaporation, sputtering, thermal or electrodeposition, wet chemical methods, or through the use of nanoparticles[44], [69]. This porosity is one of the key methods of overcoming the cathodic ORR occurring at the Pt surface by creating cavities that become oxygen deprived at their depths[96].
Abiotic GFC architecture typically involves a porous Pt anode and a denser, smoother Pt that acts as the cathode. The connection between these two poles can by standard copper circuitry that conducts electrons, or in the case of some ceramic GFC designs, a proton-conducting ceria[43]. In either case, the charged ion flow between the two platinum surfaces is what generates the current. The stoichiometry is thus[42], [43]:
| Eq. 2 |
| Eq. 3 |
| Eq. 4 |
The fuel, glucose and oxygen, are converted into power and gluconic acid.
Experimentally, one of the most exciting results seen using Pt GFCs is that the long-term stability is orders of magnitude better than its enzymatic competitors. Whereas the best enzyme-based systems see a decrease in voltage output of about 50% in the span of a week[26], [34], [47], Simons et al. ran a bank of Pt ceramic GFCs for 140 hours with no stated loss of performance[43]. Do et al. similarly ran for 70 hours with no performance decline[44]. Under non-biologically viable conditions (400 mM glucose concentration vs human maximum healthy value of 5.6 mM, flow rate of 50 cm3/min vs human capillary nominal flow rate of 5 cm3/min), a gold-platinum alloy was shown to achieve almost 100 mW/cm2, [42]. While glucose concentrations this high are not found in in vivo settings, they can be achieved in external GFC devices for potential non-medical applications, and several of the other optimized parameters from the study can serve to inform future Pt GFC designs. For example, the power output scaled with flow rate up to 50 cm3/min before leveling off, suggesting that after that the fundamental limit is one of reaction and not replenishment. Since human capillaries typically have a flow rate of one tenth of that, it can be inferred that a hard limit to output results from a failure to replenish glucose at the rate of consumption. This insight can guide future design decisions. Furthermore, this study underscores the ability of abiotic GFCs to have power densities that rival those of enzymatic GFCs.
In addition to the previously mentioned drawback of being a complete redox catalyst, there is also evidence that Pt can be inhibited by its byproduct, gluconic acid[58]. The inclusion of a Nafion coating on the anode surface at least partially mitigates this risk, as the negatively charged Nafion repels the similarly negatively charged gluconic acid. Ongoing work attempts to address this problem through alternative material preparation and fabrication techniques, coatings, or alternate electrolyte pairing. One such study examines the effect of the annealing temperature used to create the Pt-Ni alloy that serves as the base of the Raney-Pt structure[44]. Despite a renewed interest in Pt-based abiotic GFCs, in vivo experiments with Pt anodes have not yet been conducted.
B. Platinum as a Cathode
At the cathode, platinum acts as an ORR agent. This may be the most well-studied cathodic option due to its use in the catalytic converters of internal combustion engines[97]. In the automotive field, as with the biomedical one, there is a need for highly efficient catalysts that use a minimum amount of platinum, given the cost and rarity of the substance[98]. To that end, despite having been an established ORR agent for many decades, there is continued work into means of improving Pt electrocatalytic efficiency.
The ORR pathway is well understood. Incident O2 binds to the Pt surface and is split into its two component atoms. These then bind to the free hydrogen protons and conducted electrons generated at the anode to produce water through the following reactions[99]:
| Eq. 5 |
The * indicates an oxygen atom that is bound to the surface of the catalyst. Efforts to reduce the amount of Pt needed without impacting reaction efficiency have been a subject of interest given the scarcity of the catalyst. Alloying with transition metals such as cobalt, gallium, copper, and others dilutes the volume of Pt used with minimal impact to operational efficiency[100]. A more advanced means of combining these metals uses an alloyed or even pure transition metal core encased in a pure platinum shell, creating a nanoparticle that can be immobilized on an electrode surface. This shell/core configuration protects the non-precious metal center from potentially hazardous environments and increases the Pt surface area with the reacting solution. Further, it does not suffer from strain effects that result from alloys with mismatched lattice structures.
Other mechanical configurations of Pt have been explored for improved surface area per unit volume. Nanowires, porous nanotubes, hollow nanoparticles, and mesoporous double gyroid networks have each been evaluated for Pt use efficiency[101]. Similarly, a range of Pt nanoparticle shapes and sizes have been screened to find the most cost-effective option available. Li et al. assessed a single shaped (cubo-octahedral) nanoparticle at multiple sizes (2 nm through 7 nm), as well as multiple shapes (cube, octahedral, and cubo-octahedral) of nanoparticles at a single size (~7 nm), in a methodical attempt to find an ideal Pt nanoparticle shape[102]. Interestingly, their findings suggest that while there is an impact due to size, it is difficult to validate a shape effect independent of the nanoparticle’s original morphology. The shape property tends to dilute over time from the formation of surface oxides. It is worth noting that Li et al.’s testing was done in a highly acidic environment, as their focus was not for in vivo applications. Similar testing on polyhedron, cubic, and truncated cubic Pt nanoparticles was undertaken by a different team[103] which found a pronounced shape effect, with 7nm nanocubic Pt strongly outperforming some of the other parameter combinations.
V. Conclusion
While GCF technology holds promise for implantable medical devices, in its current state it does not yet indicate near-term returns and as such, research associated with this work remains in the academic space. The glucose fuel cell research field has both broadened and deepened its scope in the past decade as interest in novel energy sources and implantable medical devices grow. This work has examined a number of these efforts with an eye toward viability as a stable, long-term implant. The ongoing struggles to harvest meaningful quantities energy from these devices, as well as their relatively rapid decease in functionality, underscores the shortcomings that need to be overcome before GCFs can be used reliably in human patients.
Almost all recent advances in this field have been focused on enzymatic GFCs, which boast highly specific reactivity with glucose, as well as benefit from being cheap and biologically harvestable. However, a growing interest in abiotic catalytic options, most notably Raney-Platinum, present an attractive alternative path. These Pt based GFCs can be smaller, have denser reaction sites per unit area, can be sterilized using heat or caustic chemicals, are immune to enzyme delamination and rearrangement, do not inherently need mediators, can be built directly onto copper circuitry, and may even be able to harvest more than just two electrons per glucose molecule. Crucially, they are also substantially more stable than enzymatic GFCs. While they have some drawbacks, such as being expensive, potentially inhibited by their own byproduct, and sensitive to oxygen and glucose simultaneously, these are challenges that are solvable, whereas the challenge of getting enzymes to survive and function for a continuous decade may be a far larger undertaking.
Contributor Information
Robert G. Gloeb-McDonald, an engineer living in Manchester, NH, USA.
Gene Fridman, Johns Hopkins University, Baltimore, MD, USA..
References
- [1].“Recognized Consensus Standards.” Accessed: May 29, 2023. [Online]. Available: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfstandards/detail.cfm?standard__identification_no=37708
- [2].Menciassi A and Iacovacci V, “Implantable biorobotic organs,” APL Bioeng, vol. 4, no. 4, p. 040402, Nov. 2020, doi: 10.1063/5.0032508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].“Reactiv8 Manual,” FDA.gov. Accessed: May 29, 2023. [Online]. Available: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190021C.pdf
- [4].“MEDTRONIC AZURE XT SR MRI SURESCAN W2SR01 DEVICE MANUAL Pdf Download,” ManualsLib. Accessed: Jul. 10, 2023. [Online]. Available: https://www.manualslib.com/manual/2262716/Medtronic-Azure-Xt-Sr-Mri-Surescan-W2sr01.html
- [5].“Nucleus 7 processor,” Cochlear. Accessed: May 29, 2023. [Online]. Available: https://www.cochlear.com/us/en/professionals/products-and-candidacy/nucleus/nucleus-sound-processors/nucleus-7-processor
- [6].Amar A, Kouki A, and Cao H, “Power Approaches for Implantable Medical Devices,” Sensors (Basel, Switzerland), vol. 15, pp. 28889–28914, Nov. 2015, doi: 10.3390/s151128889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pfenniger A, Vogel R, Koch VM, and Jonsson M, “Performance Analysis of a Miniature Turbine Generator for Intracorporeal Energy Harvesting,” Artificial Organs, vol. 38, no. 5, pp. E68–E81, 2014, doi: 10.1111/aor.12279. [DOI] [PubMed] [Google Scholar]
- [8].Haeberlin A et al. , “Intracardiac Turbines Suitable for Catheter-Based Implantation—An Approach to Power Battery and Leadless Cardiac Pacemakers?,” IEEE Transactions on Biomedical Engineering, vol. 67, no. 4, pp. 1159–1166, Apr. 2020, doi: 10.1109/TBME.2019.2932028. [DOI] [PubMed] [Google Scholar]
- [9].Ren W et al. , “High-performance wearable thermoelectric generator with self-healing, recycling, and Lego-like reconfiguring capabilities,” Science Advances, vol. 7, no. 7, p. eabe0586, Feb. 2021, doi: 10.1126/sciadv.abe0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cai M, Liao W-H, and Cao J, “A smart harvester for capturing energy from human ankle dorsiflexion with reduced user effort,” Smart Mater. Struct, vol. 28, no. 1, p. 015026, Dec. 2018, doi: 10.1088/1361-665X/aaed66. [DOI] [Google Scholar]
- [11].Chan HH-T, Gao F, Chung BL-H, Liao W-H, and Cao J, “Knee energy harvester with variable transmission to reduce the effect on the walking gait,” Smart Mater. Struct, vol. 30, no. 8, p. 085024, Jul. 2021, doi: 10.1088/1361-665X/ac0bfe. [DOI] [Google Scholar]
- [12].Dagdeviren C et al. , “Conformal piezoelectric energy harvesting and storage from motions of the heart, lung, and diaphragm,” Proceedings of the National Academy of Sciences, vol. 111, no. 5, pp. 1927–1932, Feb. 2014, doi: 10.1073/pnas.1317233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xue G et al. , “Water-evaporation-induced electricity with nanostructured carbon materials,” Nature Nanotech, vol. 12, no. 4, Art. no. 4, Apr. 2017, doi: 10.1038/nnano.2016.300. [DOI] [PubMed] [Google Scholar]
- [14].Sundaresan VB, Sarles SA, and Leo DJ, “Stack of BioCells Converting ATP to Electrical Power and Possible Applications,” MRS Online Proceedings Library (OPL), vol. 950, p. 0950, ed 2006, doi: 10.1557/PROC-0950-D01-03. [DOI] [Google Scholar]
- [15].Gorman MW, Feigl EO, and Buffington CW, “Human plasma ATP concentration,” Clin Chem, vol. 53, no. 2, pp. 318–325, Feb. 2007, doi: 10.1373/clinchem.2006.076364. [DOI] [PubMed] [Google Scholar]
- [16].Leech D, Kavanagh P, and Schuhmann W, “Enzymatic fuel cells: Recent progress,” Electrochimica Acta, vol. 84, pp. 223–234, Dec. 2012, doi: 10.1016/j.electacta.2012.02.087. [DOI] [Google Scholar]
- [17].Rasmussen M, Abdellaoui S, and Minteer SD, “Enzymatic biofuel cells: 30 years of critical advancements,” Biosensors and Bioelectronics, vol. 76, pp. 91–102, Feb. 2016, doi: 10.1016/j.bios.2015.06.029. [DOI] [PubMed] [Google Scholar]
- [18].Halámková L, Halámek J, Bocharova V, Szczupak A, Alfonta L, and Katz E, “Implanted Biofuel Cell Operating in a Living Snail,” Journal of the American Chemical Society, vol. 134, pp. 5040–3, Mar. 2012, doi: 10.1021/ja211714w. [DOI] [PubMed] [Google Scholar]
- [19].MacVittie K et al. , “From ‘cyborg’ lobsters to a pacemaker powered by implantable biofuel cells,” Energy Environ. Sci, vol. 6, pp. 81–86, Dec. 2012, doi: 10.1039/C2EE23209J. [DOI] [Google Scholar]
- [20].Rasmussen M, Ritzmann RE, Lee I, Pollack AJ, and Scherson D, “An Implantable Biofuel Cell for a Live Insect,” J. Am. Chem. Soc, vol. 134, no. 3, pp. 1458–1460, Jan. 2012, doi: 10.1021/ja210794c. [DOI] [PubMed] [Google Scholar]
- [21].Szczupak A, Halámek J, Halámková L, Bocharova V, Alfonta L, and Katz E, “Living Battery–Biofuel Cells Operating in vivo in Clams,” Energy & Environmental Science, vol. 5, p. 8891, Oct. 2012, doi: 10.1039/C2EE21626D. [DOI] [Google Scholar]
- [22].Cinquin P et al. , “A Glucose BioFuel Cell Implanted in Rats,” PLoS One, vol. 5, no. 5, p. e10476, May 2010, doi: 10.1371/journal.pone.0010476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Andoralov V et al. , “Biofuel Cell Based on Microscale Nanostructured Electrodes with Inductive Coupling to Rat Brain Neurons,” Scientific reports, vol. 3, p. 3270, Nov. 2013, doi: 10.1038/srep03270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Castorena-Gonzalez J et al. , “Biofuel Cell Operating in Vivo in Rat,” Electroanalysis, vol. 25, Jul. 2013, doi: 10.1002/elan.201300136. [DOI] [Google Scholar]
- [25].Zebda A et al. , “Single Glucose Biofuel Cells Implanted in Rats Power Electronic Devices,” Sci Rep, vol. 3, no. 1, Art. no. 1, Mar. 2013, doi: 10.1038/srep01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee DY et al. , “Two-Ply Carbon Nanotube Fiber-Typed Enzymatic Biofuel Cell Implanted in Mice,” IEEE Trans Nanobioscience, vol. 19, no. 3, pp. 333–338, Jul. 2020, doi: 10.1109/TNB.2020.2995143. [DOI] [PubMed] [Google Scholar]
- [27].MacVittie K, Conlon T, and Katz E, “A wireless transmission system powered by an enzyme biofuel cell implanted in an orange,” Bioelectrochemistry, vol. 106, pp. 28–33, Dec. 2015, doi: 10.1016/j.bioelechem.2014.10.005. [DOI] [PubMed] [Google Scholar]
- [28].Willner I, Yan Y-M, Willner B, and Tel-Vered R, “Integrated Enzyme-Based Biofuel Cells–A Review,” Fuel Cells, vol. 9, no. 1, pp. 7–24, 2009, doi: 10.1002/fuce.200800115. [DOI] [Google Scholar]
- [29].Liu T, “Glucose Fuel Cells and Membranes: A Brief Overview and Literature Analysis,” Sustainability, vol. 14, no. 14, Art. no. 14, Jan. 2022, doi: 10.3390/su14148376. [DOI] [Google Scholar]
- [30].Samoszuk M, Ehrlich D, and Ramzi E, “Preclinical safety studies of glucose oxidase.,” J Pharmacol Exp Ther, vol. 266, no. 3, pp. 1643–1648, Sep. 1993. [PubMed] [Google Scholar]
- [31].Sode K, Yamazaki T, Lee I, Hanashi T, and Tsugawa W, “BioCapacitor: A novel principle for biosensors,” Biosensors and Bioelectronics, vol. 76, pp. 20–28, Feb. 2016, doi: 10.1016/j.bios.2015.07.065. [DOI] [PubMed] [Google Scholar]
- [32].Katz E, “Implantable Biofuel Cells Operating In Vivo—Potential Power Sources for Bioelectronic Devices,” Bioelectronic Medicine, vol. 2, pp. 1–12, Jun. 2015, doi: 10.15424/bioelectronmed.2014.00011. [DOI] [Google Scholar]
- [33].“Definition of redox - NCI Dictionary of Cancer Terms - NCI.” Accessed: May 29, 2023. [Online]. Available: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/redox
- [34].Yimamumaimaiti T, Lu X, Zhang J-R, Wang L, and Zhu J-J, “Efficient Blood-toleration Enzymatic Biofuel Cell via In Situ Protection of an Enzyme Catalyst,” ACS Applied Materials & Interfaces, Aug. 2020, doi: 10.1021/acsami.0c11186. [DOI] [PubMed] [Google Scholar]
- [35].Zebda A, Gondran C, Le Goff A, Holzinger M, Cinquin P, and Cosnier S, “Mediatorless high-power glucose biofuel cells based on compressed carbon nanotube-enzyme electrodes,” Nat Commun, vol. 2, no. 1, Art. no. 1, Jun. 2011, doi: 10.1038/ncomms1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rewatkar P and Goel S, “3D Printed Bioelectrodes for Enzymatic Biofuel Cell: Simple, Rapid, Optimized and Enhanced Approach,” IEEE Transactions on NanoBioscience, vol. 19, no. 1, pp. 4–10, Jan. 2020, doi: 10.1109/TNB.2019.2941196. [DOI] [PubMed] [Google Scholar]
- [37].Heller A, “Potentially implantable miniature batteries,” Anal Bioanal Chem, vol. 385, no. 3, pp. 469–473, Jun. 2006, doi: 10.1007/s00216-006-0326-4. [DOI] [PubMed] [Google Scholar]
- [38].Cadet M et al. , “An enzymatic glucose/O2 biofuel cell operating in human blood,” Biosens Bioelectron, vol. 83, pp. 60–67, Sep. 2016, doi: 10.1016/j.bios.2016.04.016. [DOI] [PubMed] [Google Scholar]
- [39].Gineitytė J, Meškys R, Dagys M, and Ratautas D, “Highly efficient direct electron transfer bioanode containing glucose dehydrogenase operating in human blood,” Journal of Power Sources, vol. 441, p. 227163, Nov. 2019, doi: 10.1016/j.jpowsour.2019.227163. [DOI] [Google Scholar]
- [40].Coman V et al. , “A Direct Electron Transfer-Based Glucose/Oxygen Biofuel Cell Operating in Human Serum,” Fuel Cells, vol. 10, no. 1, pp. 9–16, 2010, doi: 10.1002/fuce.200900121. [DOI] [Google Scholar]
- [41].Wang X et al. , “Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes,” Biosensors and Bioelectronics, vol. 31, no. 1, pp. 219–225, Jan. 2012, doi: 10.1016/j.bios.2011.10.020. [DOI] [PubMed] [Google Scholar]
- [42].Torigoe K et al. , “High-Power Abiotic Direct Glucose Fuel Cell Using a Gold–Platinum Bimetallic Anode Catalyst,” ACS Omega, vol. 3, no. 12, pp. 18323–18333, Dec. 2018, doi: 10.1021/acsomega.8b02739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Simons P, Schenk SA, Gysel MA, Olbrich LF, and Rupp JLM, “A Ceramic-Electrolyte Glucose Fuel Cell for Implantable Electronics,” Advanced Materials, vol. 34, no. 24, p. 2109075, 2022, doi: 10.1002/adma.202109075. [DOI] [PubMed] [Google Scholar]
- [44].Do UP, Seland F, Wang K, and Johannessen EA, “Raney-platinum thin film electrodes for the catalysis of glucose in abiotically catalyzed micro-glucose fuel cells,” J Mater Sci, vol. 54, no. 22, pp. 14143–14156, Nov. 2019, doi: 10.1007/s10853-019-03907-9. [DOI] [Google Scholar]
- [45].Chen J et al. , “Glucose-oxidase like catalytic mechanism of noble metal nanozymes,” Nat Commun, vol. 12, no. 1, Art. no. 1, Jun. 2021, doi: 10.1038/s41467-021-23737-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhao P et al. , “Glucose Oxidase-like Rhodium Single-Atom Nanozymes: A Mimic Platform for Biometabolism and Electrometabolism of Glucose Oxidation at Neutral pH,” ACS Energy Lett., vol. 8, no. 4, pp. 1697–1704, Apr. 2023, doi: 10.1021/acsenergylett.3c00444. [DOI] [Google Scholar]
- [47].Gao F, Yan Y, Su L, Wang L, and Mao L, “An enzymatic glucose/O2 biofuel cell: Preparation, characterization and performance in serum,” Electrochemistry Communications, vol. 9, pp. 989–996, May 2007, doi: 10.1016/j.elecom.2006.12.008. [DOI] [Google Scholar]
- [48].Milton RD, Lim K, Hickey DP, and Minteer SD, “Employing FAD-dependent glucose dehydrogenase within a glucose/oxygen enzymatic fuel cell operating in human serum,” Bioelectrochemistry, vol. 106, pp. 56–63, Dec. 2015, doi: 10.1016/j.bioelechem.2015.04.005. [DOI] [PubMed] [Google Scholar]
- [49].Geiss AF et al. , “Engineering the Turnover Stability of Cellobiose Dehydrogenase toward Long-Term Bioelectronic Applications,” ACS Sustainable Chem. Eng, vol. 9, no. 20, pp. 7086–7100, May 2021, doi: 10.1021/acssuschemeng.1c01165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Milton RD, Giroud F, Thumser AE, Minteer SD, and Slade RCT, “Bilirubin oxidase bioelectrocatalytic cathodes: the impact of hydrogen peroxide,” Chem. Commun, vol. 50, no. 1, pp. 94–96, Nov. 2013, doi: 10.1039/C3CC47689H. [DOI] [PubMed] [Google Scholar]
- [51].Shleev S et al. , “Direct electron transfer between copper-containing proteins and electrodes,” Biosens Bioelectron, vol. 20, no. 12, pp. 2517–2554, Jun. 2005, doi: 10.1016/j.bios.2004.10.003. [DOI] [PubMed] [Google Scholar]
- [52].Ramírez P et al. , “Direct electron transfer from graphite and functionalized gold electrodes to T1 and T2/T3 copper centers of bilirubin oxidase,” Biochim Biophys Acta, vol. 1777, no. 10, pp. 1364–1369, Oct. 2008, doi: 10.1016/j.bbabio.2008.06.010. [DOI] [PubMed] [Google Scholar]
- [53].Besanger TR et al. , “Screening of Inhibitors Using Enzymes Entrapped in Sol−Gel-Derived Materials,” Anal. Chem, vol. 75, no. 10, pp. 2382–2391, May 2003, doi: 10.1021/ac026370i. [DOI] [PubMed] [Google Scholar]
- [54].Mateo C, Fernández-Lorente G, Abian O, Fernández-Lafuente R, and Guisán JM, “Multifunctional Epoxy Supports: A New Tool To Improve the Covalent Immobilization of Proteins. The Promotion of Physical Adsorptions of Proteins on the Supports before Their Covalent Linkage,” Biomacromolecules, vol. 1, no. 4, pp. 739–745, Dec. 2000, doi: 10.1021/bm000071q. [DOI] [PubMed] [Google Scholar]
- [55].Ferri S, Kojima K, and Sode K, “Review of Glucose Oxidases and Glucose Dehydrogenases: A Bird’s Eye View of Glucose Sensing Enzymes,” J Diabetes Sci Technol, vol. 5, no. 5, pp. 1068–1076, Sep. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Venugopal R and Saviue BA, “The effect of oxygen upon the kinetics of glucose oxidase inactivation,” The Canadian Journal of Chemical Engineering, vol. 71, no. 6, pp. 917–924, 1993, doi: 10.1002/cjce.5450710613. [DOI] [Google Scholar]
- [57].Milton RD, Giroud F, Thumser AE, Minteer SD, and Slade RCT, “Glucose oxidase progressively lowers bilirubin oxidase bioelectrocatalytic cathode performance in single-compartment glucose/oxygen biological fuel cells,” Electrochimica Acta, vol. 140, pp. 59–64, Sep. 2014, doi: 10.1016/j.electacta.2014.02.058. [DOI] [Google Scholar]
- [58].Kerzenmacher S, Schroeder M, Brämer R, Zengerle R, and Von Stetten F, “Raney-platinum film electrodes for potentially implantable glucose fuel cells. Part 1: Nickel-free glucose oxidation anodes,” Journal of Power Sources, vol. 195, no. 19, pp. 6516–6523, Oct. 2010, doi: 10.1016/j.jpowsour.2010.04.039. [DOI] [Google Scholar]
- [59].Harris JM, Reyes C, and Lopez GP, “Common Causes of Glucose Oxidase Instability in In Vivo Biosensing: A Brief Review,” J Diabetes Sci Technol, vol. 7, no. 4, pp. 1030–1038, Jul. 2013, doi: 10.1177/193229681300700428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Giner J and Malachesky P, Anodic Oxidation of Glucose, Artificial Heart Program Conference ; proceedings, Washington, D.C., June 9–13, 1969, p 839–846. Bethesda, Md.: National Institutes of Health; For sale by the Supt. of Docs., U.S. Govt. Print. Off., Washington, 1969. Accessed: May 29, 2023. [Online]. Available: https://catalog.hathitrust.org/Record/100718875 [Google Scholar]
- [61].Hamamatsu N et al. , “Modified substrate specificity of pyrroloquinoline quinone glucose dehydrogenase by biased mutation assembling with optimized amino acid substitution,” Appl Microbiol Biotechnol, vol. 73, no. 3, pp. 607–617, Dec. 2006, doi: 10.1007/s00253-006-0521-4. [DOI] [PubMed] [Google Scholar]
- [62].Gao Q, Guo Y, Zhang W, Qi H, and Zhang C, “An amperometric glucose biosensor based on layer-by-layer GOx-SWCNT conjugate/redox polymer multilayer on a screen-printed carbon electrode,” Sensors and Actuators B: Chemical, vol. 153, no. 1, pp. 219–225, Mar. 2011, doi: 10.1016/j.snb.2010.10.034. [DOI] [Google Scholar]
- [63].Zhang Z, Han S, Wang C, Li J, and Xu G, “Single-Walled Carbon Nanohorns for Energy Applications,” Nanomaterials, vol. 5, no. 4, pp. 1732–1755, Oct. 2015, doi: 10.3390/nano5041732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kagkoura A and Tagmatarchis N, “Carbon Nanohorn-Based Electrocatalysts for Energy Conversion,” Nanomaterials (Basel), vol. 10, no. 7, p. 1407, Jul. 2020, doi: 10.3390/nano10071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li SZ and Jain A, Eds., “Capillary Blood Vessel,” in Encyclopedia of Biometrics, Boston, MA: Springer US, 2009, pp. 178–178. doi: 10.1007/978-0-387-73003-5_133. [DOI] [Google Scholar]
- [66].Bianconi E et al. , “An estimation of the number of cells in the human body,” Annals of Human Biology, vol. 40, no. 6, pp. 463–471, Nov. 2013, doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- [67].Guan H-S et al. , “Artificial blood vessel biofuel cell for self-powered blood glucose monitoring,” Nanotechnology, vol. 33, no. 2, p. 025404, Oct. 2021, doi: 10.1088/1361-6528/ac2d47. [DOI] [PubMed] [Google Scholar]
- [68].Kim Y-P, Park S, Lee D, and Kim H-S, “Electrochemical glucose biosensor by electrostatic binding of PQQ-glucose dehydrogenase onto self-assembled monolayers on gold,” Journal of Applied Electrochemistry, vol. 42, Jun. 2012, doi: 10.1007/s10800-012-0409-1. [DOI] [Google Scholar]
- [69].Kloke A, Köhler C, Samba R, Zengerle R, and Kerzenmacher S, “Cyclic Electrodeposition of PtCu Alloy: Facile Fabrication of Highly Porous Platinum Electrodes,” Advanced materials (Deerfield Beach, Fla.), vol. 24, pp. 2916–21, Jun. 2012, doi: 10.1002/adma.201200806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].“BRENDA Enzyme Database.” Accessed: May 29, 2023. [Online]. Available: https://www.brenda-enzymes.org/index.php
- [71].Jayakumar K, Reichhart TMB, Schulz C, Ludwig R, Felice AKG, and Leech D, “An Oxygen Insensitive Amperometric Glucose Biosensor Based on An Engineered Cellobiose Dehydrogenase: Direct versus Mediated Electron Transfer Responses,” ChemElectroChem, vol. 9, no. 13, p. e202200418, 2022, doi: 10.1002/celc.202200418. [DOI] [Google Scholar]
- [72].“Indicator Metadata Registry Details.” Accessed: May 29, 2023. [Online]. Available: https://www.who.int/data/gho/indicator-metadata-registry/imr-details/2380
- [73].Kulcu E, Tamada JA, Reach G, Potts RO, and Lesho MJ, “Physiological Differences Between Interstitial Glucose and Blood Glucose Measured in Human Subjects,” Diabetes Care, vol. 26, no. 8, pp. 2405–2409, Aug. 2003, doi: 10.2337/diacare.26.8.2405. [DOI] [PubMed] [Google Scholar]
- [74].Kreft M, Lukšič M, Zorec TM, Prebil M, and Zorec R, “Diffusion of d-glucose measured in the cytosol of a single astrocyte,” Cell. Mol. Life Sci, vol. 70, no. 8, pp. 1483–1492, Apr. 2013, doi: 10.1007/s00018-012-1219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Koirala RP, Dawanse S, and Pantha N, “Diffusion of glucose in water: A molecular dynamics study,” Journal of Molecular Liquids, vol. 345, p. 117826, Jan. 2022, doi: 10.1016/j.molliq.2021.117826. [DOI] [Google Scholar]
- [76].Cushman-Roisin B, “Environmental Transport and Fate.” Accessed: Jul. 02, 2023. [Online]. Available: https://cushman.host.dartmouth.edu/courses/engs43/Chapter2.pdf
- [77].Sarveswaran K, Kurz V, Dong Z, Tanaka T, Penny S, and Timp G, “Synthetic Capillaries to Control Microscopic Blood Flow,” Sci Rep, vol. 6, no. 1, Art. no. 1, Feb. 2016, doi: 10.1038/srep21885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Alberts B, Johnson A, Lewis J, Raff M, Roberts K, and Walter P, “Blood Vessels and Endothelial Cells,” in Molecular Biology of the Cell. 4th edition, Garland Science, 2002. Accessed: Jul. 02, 2023. [Online]. Available: https://www.ncbi.nlm.nih.gov/books/NBK26848/ [Google Scholar]
- [79].Guizouarn H and Allegrini B, “Erythroid glucose transport in health and disease,” Pflugers Arch - Eur J Physiol, vol. 472, no. 9, pp. 1371–1383, Sep. 2020, doi: 10.1007/s00424-020-02406-0. [DOI] [PubMed] [Google Scholar]
- [80].Cengiz E and Tamborlane WV, “A Tale of Two Compartments: Interstitial Versus Blood Glucose Monitoring,” Diabetes Technol Ther, vol. 11, no. Suppl 1, p. S-11–S-16, Jun. 2009, doi: 10.1089/dia.2009.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Fuller GG and Kim JK, “Compartmentalization and metabolic regulation of glycolysis,” Journal of Cell Science, vol. 134, no. 20, p. jcs258469, Oct. 2021, doi: 10.1242/jcs.258469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Xu S and Minteer SD, “Enzymatic Biofuel Cell for Oxidation of Glucose to CO2,” ACS Catal., vol. 2, no. 1, pp. 91–94, Jan. 2012, doi: 10.1021/cs200523s. [DOI] [Google Scholar]
- [83].Salusjärvi T, Kalkkinen N, and Miasnikov AN, “Cloning and Characterization of Gluconolactone Oxidase of Penicillium cyaneo-fulvum ATCC 10431 and Evaluation of Its Use for Production of d-Erythorbic Acid in Recombinant Pichia pastoris,” Appl Environ Microbiol, vol. 70, no. 9, pp. 5503–5510, Sep. 2004, doi: 10.1128/AEM.70.9.5503-5510.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nishikimi M, Koshizaka T, Ozawa T, and Yagi K, “Occurrence in humans and guinea pigs of the gene related to their missing enzyme l-gulono-γ-lactone oxidase,” Archives of Biochemistry and Biophysics, vol. 267, no. 2, pp. 842–846, Dec. 1988, doi: 10.1016/0003-9861(88)90093-8. [DOI] [PubMed] [Google Scholar]
- [85].Takahashi T, Yamashita H, Kato E, Mitsumoto M, and Murakawa S, “Purification and Some Properties of d-Glucono-γ-lactone Dehydrogenase,” Agricultural and Biological Chemistry, vol. 40, no. 1, pp. 121–129, Jan. 1976, doi: 10.1080/00021369.1976.10861999. [DOI] [Google Scholar]
- [86].Harada Y, Shimizu M, Murakawa S, and Takahashi T, “Identification of FAD of d -Gluconolactone Dehydrogenase: d -Erythorbic Acid Producing Enzyme of Penicillium cyaneo-fulvum,” Agricultural and Biological Chemistry, vol. 43, no. 12, pp. 2635–2636, Dec. 1979, doi: 10.1080/00021369.1979.10863876. [DOI] [Google Scholar]
- [87].Lapinsonnière L, Picot M, and Barrière F, “Enzymatic versus Microbial Bio-Catalyzed Electrodes in Bio-Electrochemical Systems,” ChemSusChem, vol. 5, no. 6, pp. 995–1005, 2012, doi: 10.1002/cssc.201100835. [DOI] [PubMed] [Google Scholar]
- [88].Gebhardt U, Rao JR, and Richter GJ, “A special type of raney-alloy catalyst used in compact biofuel cells,” J Appl Electrochem, vol. 6, no. 2, pp. 127–134, Mar. 1976, doi: 10.1007/BF00615377. [DOI] [Google Scholar]
- [89].Vassilyev Yu. B., Khazova OA, and Nikolaeva NN, “Kinetics and mechanism of glucose electrooxidation on different electrode-catalysts: Part I. Adsorption and oxidation on platinum,” Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, vol. 196, no. 1, pp. 105–125, Dec. 1985, doi: 10.1016/0022-0728(85)85084-1. [DOI] [Google Scholar]
- [90].Kerzenmacher S, Ducrée J, Zengerle R, and von Stetten F, “Energy harvesting by implantable abiotically catalyzed glucose fuel cells,” Journal of Power Sources, vol. 182, no. 1, p. 1, 2008. [Google Scholar]
- [91].Fernandes TA, Kurhe DK, Chavan AA, and Jayaram RV, “Recovery and reuse of palladium from spent glucometer electrochemical test strips,” Hydrometallurgy, vol. 165, pp. 199–205, Oct. 2016, doi: 10.1016/j.hydromet.2015.09.002. [DOI] [Google Scholar]
- [92].Koskun Y, Şavk A, Şen B, and Şen F, “Highly sensitive glucose sensor based on monodisperse palladium nickel/activated carbon nanocomposites,” Analytica Chimica Acta, vol. 1010, pp. 37–43, Jun. 2018, doi: 10.1016/j.aca.2018.01.035. [DOI] [PubMed] [Google Scholar]
- [93].Elouarzaki K, Le Goff A, Holzinger M, Thery J, and Cosnier S, “Electrocatalytic Oxidation of Glucose by Rhodium Porphyrin-Functionalized MWCNT Electrodes: Application to a Fully Molecular Catalyst-Based Glucose/O2 Fuel Cell,” J. Am. Chem. Soc, vol. 134, no. 34, pp. 14078–14085, Aug. 2012, doi: 10.1021/ja304589m. [DOI] [PubMed] [Google Scholar]
- [94].Beden B, Largeaud F, Kokoh KB, and Lamy C, “Fourier transform infrared reflectance spectroscopic investigation of the electrocatalytic oxidation of d-glucose: Identification of reactive intermediates and reaction products,” Electrochimica Acta, vol. 41, no. 5, pp. 701–709, Apr. 1996, doi: 10.1016/0013-4686(95)00359-2. [DOI] [Google Scholar]
- [95].Neha N, Rafaïdeen T, Faverge T, Maillard F, Chatenet M, and Coutanceau C, “Revisited Mechanisms for Glucose Electrooxidation at Platinum and Gold Nanoparticles,” Electrocatalysis, vol. 14, no. 1, pp. 121–130, Jan. 2023, doi: 10.1007/s12678-022-00774-y. [DOI] [Google Scholar]
- [96].Kerzenmacher S, Kräling U, Schroeder M, Brämer R, Zengerle R, and Von Stetten F, “Raney-platinum film electrodes for potentially implantable glucose fuel cells. Part 2: Glucose-tolerant oxygen reduction cathodes,” Journal of Power Sources, vol. 195, no. 19, pp. 6524–6531, Oct. 2010, doi: 10.1016/j.jpowsour.2010.04.049. [DOI] [Google Scholar]
- [97].Haldar SK, “Chapter 1 - Introduction,” in Platinum-Nickel-Chromium Deposits, Haldar SK, Ed., Elsevier, 2017, pp. 1–35. doi: 10.1016/B978-0-12-802041-8.00001-8. [DOI] [Google Scholar]
- [98].Liu J et al. , “High performance platinum single atom electrocatalyst for oxygen reduction reaction,” Nat Commun, vol. 8, no. 1, Art. no. 1, Jul. 2017, doi: 10.1038/ncomms15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ma Z et al. , “Enhancing Oxygen Reduction Activity of Pt-based Electrocatalysts: From Theoretical Mechanisms to Practical Methods,” Angewandte Chemie International Edition, vol. 59, no. 42, pp. 18334–18348, 2020, doi: 10.1002/anie.202003654. [DOI] [PubMed] [Google Scholar]
- [100].Wang L, Holewinski A, and Wang C, “Prospects of Platinum-Based Nanostructures for the Electrocatalytic Reduction of Oxygen,” ACS Catal., vol. 8, no. 10, pp. 9388–9398, Oct. 2018, doi: 10.1021/acscatal.8b02906. [DOI] [Google Scholar]
- [101].Raciti D, Liu Z, Chi M, and Wang C, “Recent Development of Platinum-Based Nanocatalysts for Oxygen Reduction Electrocatalysis,” in Nanomaterials for Fuel Cell Catalysis, Ozoemena KI and Chen S, Eds., in Nanostructure Science and Technology., Cham: Springer International Publishing, 2016, pp. 253–280. doi: 10.1007/978-3-319-29930-3_6. [DOI] [Google Scholar]
- [102].Li D et al. , “Functional links between Pt single crystal morphology and nanoparticles with different size and shape: the oxygen reduction reaction case,” Energy & Environmental Science, vol. 7, no. 12, pp. 4061–4069, 2014. [Google Scholar]
- [103].Wang C, Daimon H, Onodera T, Koda T, and Sun S, “A General Approach to the Size- and Shape-Controlled Synthesis of Platinum Nanoparticles and Their Catalytic Reduction of Oxygen,” Angewandte Chemie International Edition, vol. 47, no. 19, pp. 3588–3591, 2008, doi: 10.1002/anie.200800073. [DOI] [PubMed] [Google Scholar]