

Abstract
This article describes a DNA-based vaccination strategy evaluated ex vivo with human cells. The vaccine was prepared by transferring tumor-derived genomic DNA to PCI-13 cells, a highly immunogenic tumor cell line (“recipient cell”), which had been genetically modified to secrete IL-2 (PCI-13/IL-2). PCI-13 cells expressed class I MHC determinants (HLA-A2) shared with the tumor from which the DNA was obtained as well as allogeneic determinants. DNA from a gp100+ melanoma cell line was transduced into gp100− PCI-13/IL-2 cells (PCI-13/IL-2/DNA). A T cell line specific for the gp100 epitope responded to PCI-13/IL-2/DNA cells by IFN-γ-secretion measured in enzyme-linked immunospot assays. The T cell line also recognized the gp100 epitope presented by dendritic cells that ingested PCI-13/IL-2/DNA cells, which had been induced by UVB irradiation to undergo apoptosis. After up-take and processing of apoptotic PCI-13/IL-2/DNA cells, the dendritic cells primed normal peripheral blood lymphocytes to generate effector T cells specific for the tumor donating the DNA. The results indicate that tumor epitopes encoded in such DNA are expressed in recipient cells and can induce tumor-specific T cells. The findings support translation of this vaccination strategy to a phase I trial in patients with cancer.
Keywords: dendritic cells‖cancer vaccines‖cancer immunity
The strategy of genetic modification of tumor cells to render them more immunogenic has been widely used in the development of antitumor vaccines (1–5). However, insertion of genes into tumor cells is technically challenging, requiring the use of vectors and isolates from primary neoplasms. We tested a different vaccination strategy, based on the transfer of genomic DNA from the patient's own tumor to a “recipient” cell line, which had been preselected for attributes desirable in a vaccine. The recipient cell line transduced with the tumor-derived DNA constitutes the vaccine.
This vaccination strategy is based on the hypothesis that genes encoding weakly immunogenic tumor-associated antigens (TAA) will be expressed in a highly immunogenic form by the recipient cell. It is now accepted that cancer cells express an array of TAA. TAA are products of mutated or dysregulated genes (6). Transfer of genomic DNA from a tumor cell into a recipient cell results in integration of the DNA and stable expression of the transferred genes. A variety of genes are expressed, providing a variety of immunogenic TAA, which need not be a priori purified or identified. The selected recipient cell is semiallogeneic to the DNA donor (shares at least one MHC class I allele), which allows for MHC-restricted antigen presentation and allostimulation (7). Modification of the recipient cell to secrete IL-2 facilitates in vivo expansion of vaccine-specific T lymphocytes (8, 9).
Our preclinical in vivo studies performed in four different syngeneic murine models of tumor growth (melanoma, breast carcinoma, lymphoma, and squamous cell cancer) support the validity of this approach (10–13). In tumor-prevention models, significant inhibition of tumor growth was observed (10–13). In tumor-therapy models, mice with palpable, established tumors survived significantly longer than unimmunized tumor-bearing mice. In some instances, they rejected the neoplastic cells and survived indefinitely (10–13). The immunity was tumor-type specific, as a vaccine prepared by transfer of DNA from breast cancer, for example, was not effective against melanoma (9–12).
Based on our results obtained in animal models, pilot clinical trial of a DNA-based vaccine for patients with oral carcinoma was designed. The recipient cell is PCI-13/IL-2, a squamous cell carcinoma of the head and neck (SCCHN) cell line established in our laboratory and genetically modified to secrete IL-2 (14). Before implementing this phase I clinical protocol, a series of ex vivo experiments was performed to demonstrate that PCI-13/IL-2 can express genes encoded by the transferred tumor-derived DNA. The results indicated that a vaccine prepared by transfer of DNA from SCCHN cells into the recipient cells resulted in ex vivo activation and generation of tumor-reactive T cells.
Materials and Methods
Cells.
HLA-A2+ SCCHN cell lines, PCI-13 and PCI-1, were established from tumor biopsies as described (15). OSC-19 is an SCCHN cell line donated by E. Yamamoto, Kanazawa Univ., Kanazawa, Japan (16). PCI-13 cells were transfected with the hIL-2 gene, using the retroviral DFG-IL-2-Neo vector (PCI-13/IL-2), as described (14). The cells maintained in culture for more than 4 years secrete IL-2 (5–15 ng/106 cells/24 h).
Human HLA-A2+ melanoma cell lines, Mel 526 (gp100+) and SML-2 (gp100−⋅), were donated by W. J. Storkus (Univ. of Pittsburgh). HLA-A2+ gastric carcinoma cell line was established in our laboratory (17). The embryonal renal cell line, 293, was obtained from the American Type Culture Collection.
The cells were cultured in flasks (Costar) in DMEM or RPMI media 1640 supplemented with 10% (vol/vol) of heat-inactivated FBS, 2 mM l-glutamine, 100 μg/ml of streptomycin, and 100 units/ml of penicillin (Life Technologies, Grand Island, NY) at 37°C in a humidified 5% CO2 in air. For passage, the cells were detached from plastic with 0.05% trypsin/0.02% EDTA solution (Life Technologies).
The gp100209-217-specific, HLA-A2-restricted T cell line (no. 1520) was obtained from S. Rosenberg (National Cancer Institute) and maintained in AIM-V medium supplemented with 5% (vol/vol) of human AB serum and 100 units/ml of rIL-2 (Chiron–Cetus). The line was periodically sensitized with irradiated gp100+ Mel 526 cells.
Peripheral blood mononuclear cells (PBMC) were obtained from HLA-A2+ platelet donors by Ficoll–Hypaque centrifugation of leukapheresis products. These PBMC were used for generation of dendritic cells (DC) or T cell lines.
Cytokines and Abs.
IL-4 and granulocyte macrophage colony-stimulating factor were obtained from Schering-Plough; tumor necrosis factor-α was obtained from Knoll (Whippany, NJ); IL-6 was obtained from Sandoz Pharmaceutical; IL-2 was obtained from Chiron–Cetus; IL1-β and IL-7 were obtained from R & D Systems. MHC class I-specific mAbs (W6.32), HLA-A2-specific mAbs (HB95; BB7.3), and MHC class II-specific mAbs (L243) were obtained from A. DeLeo (Univ. of Pittsburgh).
DC.
Human monocyte-derived DC were generated as described by Sallusto and Lanzavecchia (18) with modification. Briefly, PBMC suspended in AIM-V medium (107/ml) were incubated for 1 h at 37°C in T75 flasks (Falcon/Becton Dickinson). Plastic-adherent cells were cultured in AIM-V medium supplemented with 1,000 units/ml of IL-4 and 1,000 units/ml of granulocyte macrophage colony-stimulating factor for 6 days at 37°C/5% CO2 in air. The DC were harvested on day 6 in cold Hanks' solution (Life Technologies), washed, and resuspended at a concentration of 2 × 106 cells per ml in AIM-V medium for coincubation with DNA-transduced recipient cells in the presence of IL-1-β, IL-6, and tumor necrosis factor-γ, as described (19).
Induction of Apoptosis.
PCI-13-IL-2 cells transduced with tumor-DNA (PCI-13-IL-2/DNA) and cultured in AIM-V medium received 1,500 μW/cm2 of UVB (UVB bulb BLE-GT 302; Spectronic, Westbury, NY) for 15 min (19). The irradiated cells were incubated for 18 h, and then apoptotic tumor cells (ATC) were cocultured with DC at the ATC:DC cell ratio of 1:5 for 18 h.
T Cell Generation.
In vitro sensitization with irradiated recipient cells transduced with tumor DNA or with DC that ingested ATC was used to generate tumor-reactive T cells. PBMC (as a source of autologous DC and T cells) were coincubated with stimulators (tumor cells or DC/ATC) for 7 or 14 days and then tested in enzyme-linked immunospot (ELISPOT) assays for responses to the tumor cells that donated the DNA. T cell cultures were performed in AIM-V medium supplemented with IL-2 (20 units/ml) and IL-7 (10 ng/ml).
ELISPOT Assay.
ELISPOT assays were performed as described (20). Responder (R) T cells (1 × 103 to 5 × 103) were plated in 96-well plates with nitrocellulose membrane inserts (Millipore) coated with 50 μl of the capture Ab (10 μg/ml in l× PBS, clone MABl-DlK; MABTECH, Stockholm). Stimulator (S) cells (PCI-13/IL-2/DNA or DC/ATC) were then added at the R:S ratio of 20:1. After a 24-h incubation, cells were removed by washing the plates 6 times with 0.05% (wt/vol) Tween-20 in PBS (Fisher Scientific). The detection Ab (2 μg/ml, clone Mab7-B6-l; MABTECH) was added to each well. The plates were incubated for 2 h and the washing steps were repeated. After a 1-h incubation with avidin-peroxidase (Vectastain Elite Standard ABC kit; Vector Laboratories), the plates were washed. Aliquots (100 μl) of aminoethylcarbazole staining solution (Sigma) were added to each well to develop the spots. The reaction was stopped after 4–6 min by washing with water. The spots were counted with computer-assisted image analysis (ELISPOT 4.14.3, Zeiss). When experimental values were significantly different from the mean number of spots against nonpulsed DC (background values), as determined by a two-tailed Wilcoxon rank sum test, the background values were subtracted from the experimental values. The coefficient of variation for the assay was determined to be 15% (n = 50).
For Ab blocking, stimulator cells were incubated for 30 min with W6.32 Ab, HLA-A2 (BB7.2) Ab, or HLA-DR Ab, or with mouse IgG1 (clone S1-68.1, PharMingen) used as control.
DNA Isolation.
Genomic DNA was isolated with an endotoxin-free purification kit (Qiagen, Valencia, CA).
Transduction of PCI-13/IL-2 Cells with Tumor-Derived DNA.
DNA was obtained from Mel 526, OSC-19, or PCI-1 cells. Mel 526 or 293 cells were genetically modified to express enhanced green fluorescence protein (EGFP). The tumor-derived DNA was transferred into PCI-13/IL-2 recipient cells by one of several methods.
(i) Lipofection.
DNA (10–20 μg) was mixed with 1 μg of pBabe-puro (from M. L. Collins, Univ. College, London) or pTK-hygromycin plasmid (CLONTECH). The mixture was then combined with lipofectin (Life Technologies) and added to PCI-13/IL-2 cells in the log phase of growth. Eighteen hours later, the selection medium containing either 3 μg/ml of puromycin (Sigma) or 100 μg/ml of hygromycin (CLONTECH) was added. Surviving colonies of transduced PCI-13/IL-2/DNA cells were expended and used as stimulators in ELISPOT assays.
(ii) Calcium phosphate.
Transfection medium (DMEM plus 10% FCS containing 25 μM chloroquine; Sigma) was added to semiconfluent tumor cell monolayers. DNA aliquots (10–100 μg) were diluted with water, mixed with 2M CaCl2 and 2× Hanks' balanced salt solution by bubbling, and added to the recipient cells. The cells were then incubated at 37°C in a humidified atmosphere of 5% CO2 in air for 10 h before selection.
(iii) Ballistomagnetic gene delivery.
A modified ballistic system (PDS-1000/He; Bio-Rad) was used for ex vivo DNA transfer (21). Gold particles (0.8–1.6 μm; ABCR, Karlsruhe, Germany) were coated with DNA from 293 cells modified to specify EGFP and superparamagnetic beads (65 nm; Miltenyi Biotec, Auburn, CA) at the ratio of 1:3 and propelled at 1,550 psi into tumor cells plated in 30-mm2 dishes. The ballistomagnetic transfer was performed as described (21). Immediately after gene transfer, fresh medium was added and the cells were incubated for 24 or 48 h before microscopic examination.
When DNA was derived from tumor cells modified with the EGFP gene, the recipient cells were examined for EGFP expression in the fluorescent microscope 24–48 h after transfection or after selection.
Retroviral Transfection of Tumor Cells.
For the EGFP gene transfer, confluent tumor monolayers plated in 100-mm2 Petri dishes were incubated at 37°C with 4 ml of the viral supernatant containing polybrene (8 μg/ml). The DFG-EGFP-zeocin retroviral vector was constructed as described (4). The culture medium was replaced after 3 h, and antibiotic selection medium was added after 48 h. Stable selection was completed after 10–14 days, and expression of the EGFP was monitored by fluorescent microscopy.
Recombinant Adenovirus.
E1- and E3-deleted adenovirus was constructed at the Univ. of Pittsburgh Cancer Institute Vector Core Laboratory by using Cre-lox recombinant (22) with reagents generously provided by S. Hardy (Somatics, Alameda, CA). A SnaB1/HpaI fragment containing part of the cytomegalovirus (CMV) promoter, the EGFP N1 cDNA, and part of the SV40 poly(A) derived from EGFP N1 (CLONTECH) was inserted into the shuttle vector pAdlox. The SalI-EGFP-NotI fragment of pAdlox-EGFP was replaced by a SalI-hGp100-NotI fragment from pCI-hGp100 (provided by S. Wagner, Univ. of Essen, Essen, Germany) to generate pAdlox-hGp100. Recombinant adenoviruses were generated by cotransfection of SfiI-digested pAdlox EGFP or pAdlox-hGp100 and ψ5 helper virus DNA into the packaging cell line CRE8, as described (23). Recombinant adenoviruses were propagated on CRE8 or 293 cells, purified by cesium chloride density gradient centrifugation and subsequent dialysis, and stored at −70°C. For infection, purified adenovirus was added directly to tumor monolayers for 45 min.
Statistical Analysis.
Wilcoxon's rank sum and Student's t tests were used. The difference of P < 0.05 was considered significant.
Results
This study consists of a series of ex vivo experiments performed with various tumor cells serving as DNA donors and PCI-13/IL-2 cells, an HLA-A2+ SCCHN cell line previously transfected with the IL-2 gene, as DNA recipients (14). The choice of PCI-13/IL-2 cells was dictated by their intended use as the “recipient cell” in a clinical phase I study. The purpose of the experiments is to show that tumor-derived DNA is expressed by PCI-13/IL-2 cells and that products of TAA genes are presented to T lymphocytes directly by these recipients or by DC after their processing.
Transfer of Tumor DNA Leads to Expression of Encoded Genes by Human Embryonal Kidney Cells.
For these experiments, 293 cells were first stably transfected with a retroviral construct containing a gene for EGFP. DNA isolated from the modified 293 cells was then transferred to parental 293 cells, and after 48 h in culture, the transduced cells were tested for expression of EGFP by fluorescence microscopy. As shown in Fig. 1, clusters of cells expressing EGFP were present among transduced cells, and 2–5% of the cells stained strongly. Flow cytometry confirmed that 5% of the recipient cells expressed EGFP (data not shown). Thus, the phenotype of the recipient cell was altered by transfer of genomic DNA from the modified 293 cells, and the transferred EGFP gene was expressed as a protein by the DNA recipient.
Figure 1.
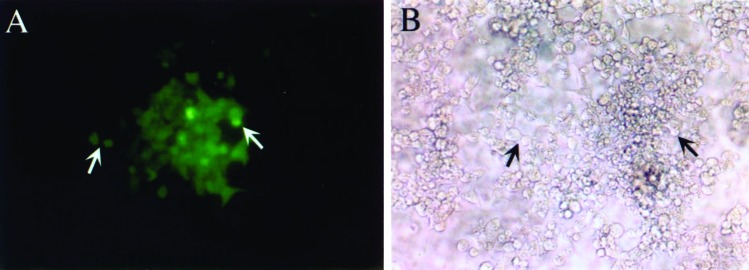
Expression of EGFP in a DNA-recipient cell. In A, a cluster of parental 293 cells, which were transduced by calcium phosphate coprecipitation with DNA containing the EGFP gene and are expressing EGFP, as seen in a fluorescent microscope (arrows). In B, the same cluster of cells seen in a light microscope. (Magnification: ×100.)
The Products of Transferred Genes Are Recognized by Epitope-Specific T Lymphocytes.
To determine whether the product of a gene encoded by the transferred DNA is expressed in the recipient cell in a form appropriate for recognition by specific T cells, PCI-13/IL-2 (gp100−) cells were transduced with DNA derived from HLA-A2+, gp100+ Mel 526 cells. The HLA-A2-restricted and gp100209-217 epitope-specific T cell line no. 1520 was used as responder in ELISPOT assays. PCI-13/IL-2 cells transduced with Mel 526-derived DNA induced IFN-γ production in T cells, which was significantly inhibited by the HLA-A2 Ab but not the HLA-DR Ab (Fig. 2A). This inhibition by HLA-A2 Ab of IFN-γ production provided the confirmation of gp100-specific response. The T cells did not respond to unmodified PCI-13/IL-2 cells nor to the gp100(−/low) melanoma cell line, SML-2 (Fig. 2B). The number of spots observed with gp100+Mel 526 + T cells (positive control in Fig. 2B) was ≈30 times higher than that seen with PCI-13/IL-2/DNA + T cells, consistent with the likelihood that not every PCI-13/IL-2/DNA cell expressed gp100 in sufficient amounts or long enough to trigger a response. The results indicated that gp100− PCI-13/IL-2 cells transduced with DNA derived from gp100+ melanoma expressed and presented this epitope to gp100-specific T cells.
Figure 2.

In ELISPOT assays, the gp100209–217 epitope-specific T cells recognize PCI-13/IL-2/MEL DNA (recipient cells transduced with DNA derived from gp100+ Mel 526) but not the nontransduced PCI-13/IL-2 (A). The negative controls (no T cells or T cells alone) had a very low background (A). In B, positive and additional negative controls from the same experiment are shown. Note that gp100-negative melanoma (SLM-2) is not recognized, whereas gp100+ Mel 526, the target for these T cells, induced a large number of spots. HLA-A2 or HLA-DR Abs were used to block the MHC-restricted responses in ELISPOT assays, as described in Materials and Methods. The results were obtained in the same ELISPOT assay for IFN-γ production. A representative experiment of four performed is shown. The results in this and all other ELISPOT assays shown are means ± SD from quadruplicate wells.
To demonstrate further that PCI-13/IL-2 cells process and present proteins coded for by the transferred genes, they were infected with recombinant adenovirus incorporating EGFP or gp100 genes. After infection with the Adlox EGFP/adenovirus, the majority of PCI13/IL-2 cells expressed EGFP (data not shown). These cells were then used as stimulators in ELISPOT assays and tested for the ability to induce responses in the gp100-specific T cell line. As shown in Fig. 3, these T cells reacted strongly to PCI-13/IL-2 cells infected with adenovirus carrying the gp100 gene, whereas all controls were negative. These results confirm that PCI-13/IL-2 cells synthesize and present gp100209–217 epitope encoded by the transferred DNA.
Figure 3.

PCI-13/IL-2 cells infected with recombinant adenovirus carrying a gene for gp100 express the gp100 epitope and are able to present it to gp100-specific T cells. The results are from a representative ELISPOT assay of three performed. PCI-13/IL-2 cells pulsed with the gp100 peptide or PCI-13/IL-2 infected with adenovirus modified with the EGFP gene did not stimulate the T cells. The asterisk indicates a significant increase in the numbers of spots over controls (P < 0.0001).
DC Fed with Apoptotic PCI-13/IL-2 Cells Transduced with DNA from Mel 526 Cells Present the gp100 Epitope to gp100-Specific T Cells.
It is expected that in vivo, delivery of irradiated PCI-13/IL-2/Mel DNA as vaccine would be accompanied by the up-take of tumor cells by DC. To model this in vivo setting, we investigated whether the gp100209-217 epitope expressed in the DNA-transduced recipient cells (see above) could be cross-presented by DC. UVB-treated PCI-13/IL-2/Mel DNA cells were coincubated with HLA-A2+ DC. We reported that exposure to UVB induced apoptosis in PCI-13 cells (19). As shown in Fig. 4, gp100-specific T cells stimulated in ELISPOT assays with DC, which had internalized PCI-13/IL-2/Mel DNA, produced IFN-γ (20–25 spots per 1,000 cells). This response was nearly completely inhibited by HLA-A2 or MHC class I Abs. As a positive control, DC that have internalized Mel 526 cells also stimulated the T cells to produce IFN-γ (30–35 spots per 1,000 cells), and this response was significantly inhibited by the HLA class I Abs (Fig. 4). In contrast, the response of the T cells to DC, which were fed with PCI-13/IL-2 (not transduced with Mel 526 DNA), was non-MHC restricted and was not blocked by the MHC class I Abs. These results indicated that the T cells could recognize the gp100 epitope presented by DC, which had ingested PCI-13/IL-2 transduced with DNA derived from Mel 526 cells.
Figure 4.

ELISPOT assays demonstrate that gp100-specific T cells respond by IFN-γ secretion to HLA-A2+ DC, which had internalized and processed apoptotic PCI-13/IL-2/Mel DNA cells. The recognition is MHC class I- and HLA-A2-restricted, as evidenced by its inhibition by anti-MHC Abs. DC ingesting Mel 526 also stimulate IFN-γ production by these T cells. A representative ELISPOT experiment of four performed is shown. The asterisks indicate significant differences (P < 0.001).
In subsequent experiments, Me l 562 cells were stably infected with the retroviral DFG/EGFP/zeo vector. All tumor cells expressed EGFP after selection. DNA obtained from these cells was transferred with the ballistomagnetic gene delivery method to the recipient PCI-13/IL-2 cells (21). Twenty-four or 48 h later, 1–2% of the recipient cells were found to express EGFP (Fig. 5A). The recipient cells were then enriched in cells containing DNA/EGFP gp100 beads by magnetic selection (21). Next, these selected cells were treated with UVB and fed to HLA-A2+ DC. In ELISPOT assays, we again found that DC fed with DNA-containing recipient cells were able to stimulate gp100-specific T cells, and that this response was inhibited by anti HLA-A2 Abs as well as by w6.32 Abs (Fig. 5B). Thus, magnetic enrichment in recipient cells containing transferred Mel DNA induced a stronger gp100-specific T cell response (over 120 spots per 1,000 cells) than that observed in the experiment shown in Fig. 4, which did not involve enrichment. This result indicated that the ability of the recipient cells to stimulate gp100-specific T cells depended on the proportion of DNA-transfected PCI-13/IL-2 cells in the population.
Figure 5.

In A, PCI-13/IL-2 cells transduced by the ballistomagnetic method with DNA obtained from the EGFP+/Mel 562 cells. Twenty-four to 48 h after transduction, 2% of PCI-13/IL-2 cells were fluorescent (Inset shows one such cell, ×400). After enrichment by magnetic selection, apoptosis was induced by UVB irradiation in recipient cells containing Mel DNA/EGFP, and ATC were internalized and processed by HLA-A2+ DC. In B, these DC were used as stimulators in ELISPOT assays. The responders were gp100 epitope-specific T cells. DC-processed PCI-13/IL-2/Mel DNA, but not DC-processed PCI-13/IL-2, induced MHC class I-restricted responses in these T cells. The gp100-specific response was significantly inhibited (*, P = 0.01) by HLA-A2 and w6.32 Abs. A representative experiment of three performed is shown.
PCI-13/IL-2 Cells Transduced with DNA Derived from HLA-A2+ SCCHN Prime Tumor-Specific Responses ex Vivo.
The following ex vivo model was used to demonstrate that DNA-transduced recipient cells induce generation of tumor-specific T cell lines from PBMC of normal donors. HLA-A2+ SCCHN cell lines, PCI-1 or OSC-19 served as donors of tumor DNA, and the HLA-A2+ PCI-13/IL-2 cells were the DNA recipients. PBMC obtained from an HLA-A2+ platelet donor were used as a source of DC and of responding T cells. As indicated in Fig. 6 A–C, T cell lines capable of recognizing the DNA donor were generated in these cultures from presumably naive T cells in PBMC of a normal donor. The tumor-specific responses were partially blocked by anti-HLA-A2 and w6.32 Abs. In Fig. 6 A and C, responses induced by the recipient cells were also blocked by w6.32 Abs, an indication for an allogeneic but not HLA-A2-specific antitumor response. These experiments, performed with PBMC of two normal donors, demonstrate specificity at the sensitization as well as recognition levels. T cells sensitized to the recipient cells expressing PCI-1 DNA recognized PCI-1 and PCI-13/IL-2 but not OSC-19 or HLA-A2+ gastric carcinoma cell line (HR) targets (Fig. 6A). Those sensitized to PCI-13/IL-2/OSC-19 DNA recognized OSC-19 and PCI-13/IL-2, not PCI-1 or HR (Fig. 6C). Those sensitized to PCI-13/IL-2 recognized only PCI-13/IL-2 targets (Fig. 6C).
Figure 6.

PCI-13/IL-2 cells were transduced with DNA obtained from HLA-A2+ SCCHN, PCI-1or OSC-19, or used as nontransduced recipient cells. They underwent UVB-induced apoptosis and were fed to HLA-A2+ DC which, in turn, were incubated with autologous PBMC. Three T cell lines were generated in 14-day in vitro sensitization cultures (T1, T2, and T3). Each T cell line was stimulated with a range of tumor cell targets in ELISPOT assays. In A, T1 cells responded to PCI-1, the DNA donor, and to PCI-13/IL-2. These responses were inhibited (P < 0.01; see asterisks) by anti-MHC Abs. In B, T2 cells responded only to PCI-13/IL-2, and the response was blocked by anti-MHC Abs. In C, T3 cells recognized OSC-19, the DNA donor, and PCI-13/IL-2 cells, and these responses were also partially inhibited by anti-class I MHC Abs. A representative experiment of four performed is shown. HR is an HLA-A2+ gastric carcinoma used as a negative control.
In general, the T cell lines generated from PBMC of two normal donors, with the DNA transfer strategy and DC for TAA presentation, had the following characteristics. (i) Only T cell lines generated in the presence of autologous DC processing the recipient cells transduced with tumor DNA (PCI-13/IL-2/DNA) specifically recognized the tumor donating the DNA, as confirmed by Ab-blocking experiments. (ii) T cell lines generated in the presence of DC processing nontransduced recipient cells in ELISPOT assays recognized only the recipient cells but not other HLA-A2+ targets.
Discussion
Cancer cells are genetically unstable and are characterized by an expression pattern of genes that differs from that expressed in nonmalignant cells of the same tissue origin in the same individual. Certain of these altered genes encode epitopes that, under appropriate circumstances, can be recognized by immune cells. These epitopes can be transferred to recipient cells with total cellular DNA and they induce generation of immune responses specific for the tumor donating the DNA.
In vivo experiments performed by us in several murine models of tumor growth validated the principle that tumor-derived DNA transduced into recipient cells enhances their antigenicity and may lead to tumor rejection (9–12). Although these in vivo experiments provided a compelling rationale for the application of this strategy to patients with cancer, concerns remained about the ability of the DNA-transduced recipient cell of human origin to induce generation of tumor-specific T cell lines. In a series of ex vivo experiments, we found that the antigenic phenotype of the recipient cell was altered by DNA transfer and that DNA-transduced recipient cells fed to HLA-A2-matched DC not only successfully activated tumor-specific T cells but also generated tumor-specific T cells from PBMC of HLA-A2+ normal donors.
A major advantage of this vaccination strategy is that the recipient of tumor DNA can be selected for special properties such as expression of defined class I MHC determinants. The recipient cell can be modified to secrete immune-augmenting cytokines or to express costimulatory molecules and shared TAA, all of which facilitate generation of robust immune responses (9–12). Efforts to increase immunogenicity of TAA-based vaccines have ranged from using adjuvants or cytokines to delivery of multiple epitopes or genetically modified tumor cells (24). Allogeneic MHC determinants are known to be strong immune adjuvants (6). PCI-13/IL-2 cells selected as DNA recipients expressed class I MHC determinants that were both syngeneic and allogeneic. The former provided a restriction element (HLA-A2) for direct presentation of TAA to the HLA-A2-restricted T lymphocytes. The recipient cells also expressed and secreted IL-2 and were able to process and present HLA-A2-restricted epitopes (gp100) encoded by the transduced DNA to the gp100-specific T cells. Another advantage of a vaccine prepared by DNA transfer is that TAA do not have to be purified or defined in a complex process of antigen discovery. Also, because the recipient cells replicate the transferred DNA as they divide, their number can be expanded for multiple immunizations. The DNA-based vaccines combine attributes that are likely to make them more immunogenic than conventional TAA-based vaccines.
When DC pulsed with recipient cells transduced with tumor-derived DNA were coincubated with PBMC, T cell lines specific for the DNA donor were generated. To facilitate up-take of DNA-transduced recipient cells by DC, UVB irradiation was used to induce apoptosis (19). The ATC were readily ingested by immature DC, and after maturation, these DC primed T cell precursors in PBMC of normal HLA-A2+ donors to develop into tumor-specific effector cells. These experiments illustrated the usefulness of the DC/ATC system for modeling of cellular interactions that might occur in vivo after vaccination. It is expected that in vivo, DNA-based vaccines will primarily engage DC residing in the skin or in lymph nodes draining the vaccination site for processing and presentation to T cells of epitopes encoded by the transferred tumor DNA.
In our experiments, ELISPOT assays were used to detect and quantitate TAA-specific T cells. These assays have been developed to measure the frequency of antigen-responsive T cells in populations (20, 25). In addition they allow for distinction of allo- from tumor-specific responses by blocking with MHC class I or class II Abs. Thus, T cells generated in the presence of DNA-transduced recipient cells or DC fed with DNA-transduced recipient cells recognized the tumor donating the DNA, and this recognition was prevented by HLA-A2 Abs, w6.32 Abs, and, in some cases, also HLA-DR Abs. As expected, the T cells also recognized alloantigens expressed by the recipient cell, and this alloresponse was not inhibited by HLA-A2 Abs but by anti-MHC class I Abs alone.
Overall, our results added to our previous in vivo results in tumor-bearing mice (10–13), strongly support the therapeutic potential of vaccines prepared by transfer of tumor DNA into a selected, highly immunogenic cell line. They make a convincing argument for application of this vaccination strategy to Phase I clinical trials in patients with cancer.
Acknowledgments
This work was supported in part by National Institutes of Health Grants RO-1 CA63513 and PO-1 DE12321 (to T.L.W.).
Abbreviations
- TAA
tumor-associated antigens
- SCCHN
squamous cell carcinoma of the head and neck
- PBMC
peripheral blood mononuclear cells
- DC
dendritic cells
- ATC
apoptotic tumor cells
- ELISPOT
enzyme-linked immunospot
- EGFP
enhanced green fluorescence protein
References
- 1.Nabel G J, Gordon D, Bishop D K, Nickoloff B J, Yang Z-Y, Acuga A, Cameron M J, Nabel E G, Chang A E. Proc Natl Acad Sci USA. 1996;93:15388–15393. doi: 10.1073/pnas.93.26.15388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun Y, Jurgousky K, Moller P, Alijagic S, Dorbic T, Georgieva J, Wittig B, Schadendorf D. Gene Ther. 1998;5:481–490. doi: 10.1038/sj.gt.3300619. [DOI] [PubMed] [Google Scholar]
- 3.Greten T, Jaffee E J. Clin Oncol. 1999;17:1047–1060. doi: 10.1200/JCO.1999.17.3.1047. [DOI] [PubMed] [Google Scholar]
- 4.Tuting T, Gambotto A, Barr J, Davis I D, Storkus W J, Zavodny P J, Narula S, Tahara H, Robbins P D, Lotze M T. Gene Ther. 1997;4:1053–1060. doi: 10.1038/sj.gt.3300509. [DOI] [PubMed] [Google Scholar]
- 5.Townsend S E, Allison J P. Science. 1993;259:368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 6.Boon T, Coulie P G, Van der Bruegger P. Curr Opin Immunol. 1997;9:684–693. [Google Scholar]
- 7.Hui K M, Sim T F, Foo T T, Oei A-A. J Immunol. 1989;143:3835–3843. [PubMed] [Google Scholar]
- 8.Fearon E R, Pardoll D M, Itaya T, Golumbek P T, Levitsky H I. Cell. 1990;60:397–403. doi: 10.1016/0092-8674(90)90591-2. [DOI] [PubMed] [Google Scholar]
- 9.Cavallo F, Pierro F D, Giovarelli M, Gulino A, Vacca A, Stoppacciaro A, Forni M, Modesti A, Forni G. Cancer Res. 1993;53:5067–5070. [PubMed] [Google Scholar]
- 10.dez Oeten B F, Carr-Brendel V, Cohen E P. J Immunol. 1998;160:2915–2922. [PubMed] [Google Scholar]
- 11.dez Oeten B F, Carr-Brendel V, Markovic D, Taylor-Papadimitriou J, Cohen E P. J Immunol. 1999;162:6934–6941. [PubMed] [Google Scholar]
- 12.Sun T, dez Oeten B F, Carr-Brendel V, Cohen E P. Cancer Gene Ther. 1998;5:110–118. [PubMed] [Google Scholar]
- 13.Cohen E P. Trends Mol Med. 2001;7:175–179. doi: 10.1016/s1471-4914(01)01961-x. [DOI] [PubMed] [Google Scholar]
- 14.Nagashima S, Reichert T, Kashii Y, Suminami Y, Chikamatsu K, Whiteside T L. Cancer Gene Ther. 1997;4:366–376. [PubMed] [Google Scholar]
- 15.Heo D S, Snyderman C H, Gollin S M, Pan S, Walker E, Deka R, Barnes E L, Johnson J T, Herberman R B, Whiteside T L. Cancer Res. 1989;49:5167–5175. [PubMed] [Google Scholar]
- 16.Kawashiri S, Kumagai S, Kojimak K, Harada H, Yamamoto E. J Cancer Oral Oncol (Europe) 1995;31B:216–221. doi: 10.1016/0964-1955(95)00027-f. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu Y, Weidmann E, Iwatsuki S, Herberman R B, Whiteside T L. Cancer Res. 1991;51:6153–6162. [PubMed] [Google Scholar]
- 18.Sallusto F, Lanzavecchia A. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann T K, Meidenbauer N, Dworacki G, Kanaya H, Whiteside T L. Cancer Res. 2000;60:3542–3549. [PubMed] [Google Scholar]
- 20.Asai T, Storkus W J, Whiteside T L. Clin Diagn Lab Immunol. 2000;7:145–154. doi: 10.1128/cdli.7.2.145-154.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wittig B, Marten A, Dorbic T, Weineck S, Min H, Miemitz S, Trojaneck B, Flieger D, Kruopis S, Albers A, et al. Hum Gene Ther. 2001;12:267–278. doi: 10.1089/10430340150218404. [DOI] [PubMed] [Google Scholar]
- 22.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps M L. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranieri E, Herr W, Gambotto A, Olson W, Rowe D, Robbins P D, Kierstead L S, Watkins S C, Gesualdo L, Storkus W J. J Virol. 1999;73:10416–10425. doi: 10.1128/jvi.73.12.10416-10425.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Offringa R, Van der Burg S H, Ossendorp F, Toes R E M, Melief C J M. Curr Opin Immunol. 2000;12:576–583. doi: 10.1016/s0952-7915(00)00145-x. [DOI] [PubMed] [Google Scholar]
- 25.Meidenbauer N, Harris D T, Spitler L E, Whiteside T L. Prostate. 2000;43:88–100. doi: 10.1002/(sici)1097-0045(20000501)43:2<88::aid-pros3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]