

Abstract
Benzophenones are among the most useful photocrosslinking agents in biology. We have evolved an orthogonal aminoacyl-tRNA synthetase/tRNA pair that makes possible the in vivo incorporation of p-benzoyl-l-phenylalanine into proteins in Escherichia coli in response to the amber codon, TAG. This unnatural amino acid was incorporated with high translational efficiency and fidelity into the dimeric protein glutathione S-transferase. Irradiation resulted in efficient crosslinking (>50%) of the protein subunits. This methodology may prove useful for discovering and defining protein interactions in vitro and in vivo.
All organisms use the same common 20 amino acids as building blocks for the biosynthesis of proteins. The ability to augment the genetically encoded amino acids with unnatural amino acids containing orthogonal chemical handles, photocrosslinking groups, fluorescent probes, redox active groups, or heavy atoms would provide powerful tools for manipulating and probing protein function in vitro, in cells and perhaps in whole organisms. Recently, we reported that by adding new components to the translational machinery of Escherichia coli, additional amino acids could be site-specifically incorporated with high translational fidelity into proteins in vivo (1). We now show that this methodology can be used to site specifically incorporate photocrosslinking amino acids into a protein in E. coli.
Benzophenones have been used extensively as photophysical probes to identify and map peptide–protein interactions (2). In contrast to aryl azides, diazoesters, and diazarenes, they are chemically stable and can be routinely manipulated under ambient light. Upon excitation at 350–360 nm, wavelengths that avoid protein damage, they preferentially react with otherwise unactivated carbon-hydrogen bonds (C—H) (3, 4). Furthermore, in contrast to other photocrosslinking groups, benzophenones do not photodissociate and their photoexcited triplet state readily relaxes in the absence of a suitable C—H bond with which to react. The reversible excitation of benzophenones allows repeated excitation by long-wavelength UV light and thus excellent crosslinking yields (5). Early uses of benzophenones included the functionalization of remote C—H bonds in steroids; mapping the conformations of flexible chains in solution, micelles, and membranes (6, 7); and mapping the nucleotide-binding sites in ATPases (8).
In 1986, DeGrado and coworkers (5) demonstrated that the photocrosslinking amino acid p-benzoyl-L-phenylalanine (pBpa) (Fig. 1A) could be site specifically incorporated into synthetic peptides by solid-phase peptide synthesis. Peptides incorporating this amino acid have been used extensively to probe peptide–protein interactions in numerous systems in vitro (2). These experiments have established that pBpa is a very efficient photocrosslinker, affording between 50% and 100% crosslinking between the peptides and their receptors. These experiments also demonstrated that the crosslinks are formed selectively between the benzophenone moiety and a limited number of amino acids on the target protein that are proximal to the benzophenone moiety in the peptide–protein complex (9).
Fig 1.

(A) The chemical structure of pBpa. (B) The active site of B. stearothermophilus (Bs) tyrosyl-tRNA synthetase with bound tyrosine. The homologous residues to those mutated in the library used are shown. (Bs:Y34, N123, D176, F177, and L180 correspond to Mj:Y32, E107, D158, I159, and L162). (C) The selection scheme used to isolate a p-benzoyl-L-phenylalanyl-tRNA synthetase.
Although benzophenones have been widely used to crosslink small molecules or peptides to proteins, no simple method exists to incorporate benzophenones site specifically into larger proteins not accessible by solid-phase peptide synthesis. We have previously demonstrated that benzophenone-containing amino acids can be incorporated into proteins by using an in vitro suppression methodology that relies on chemically misacylated tRNAs (10, 11), but the yields are low and the proteins are generated in vitro. Here we report the selection of an orthogonal aminoacyl-tRNA synthetase/tRNA pair for the in vivo incorporation of the photocrosslinker pBpa, into proteins in response to the amber codon, TAG. The amino acid is incorporated in good yield with high fidelity and has been used to crosslink efficiently monomers of a dimeric complex.
Methods
Selection of Mutant Synthetase.
A library of Methanococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS) mutants (on pBK plasmids, under the control of E. coli GlnRS promoter and terminator on pBR322-derived kanamycin-resistant plasmids) were transformed into DH10B E. coli containing pYC-JYCUA by electroporation, yielding approximately 1010 independent transformants. Transformants were recovered in SOC for 1 h before resuspension in 100 ml of glycerol minimal media with leucine (GMML) supplemented with kanamycin, tetracycline, and chloramphenicol and pBpa (Bachem) at 30, 20, 60 mg/liter and 1 mM, respectively. Cells were grown for 40 h to an OD >1 with shaking at 37°C, 250 rpm. Plasmid DNA was extracted and the pBK library was resolved from pREP(2)/YC-JYCUA by agarose gel electrophoresis. The faster-mobility pBK band was extracted and used to transform DH10B containing pJ17B3 (this plasmid contains the toxic barnase gene with amber mutations at Gln-2, Asp-44, Gly-65 under the control of an arabinose promoter and an rrnB terminator and mutRNA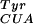 on a lpp promoter and rrnC terminator) by electroporation. Transformants were recovered in 1 ml of LB for 1 h before plating on LB-agar supplemented with kanamycin, chloramphenicol, and arabinose at 30, 50 mg/liter and 0.2% wt/vol, respectively. After 12 h at 37°C colonies were scraped into liquid LB with the same supplements and amplified for 1 h. Plasmid DNA was extracted, and the pBK library members were resolved from pJ17B3 by agarose gel electrophoresis. The selected pBK mutants were transformed into DH10B containing pREP(2)/YC-JYCUA [this reporter contains genes for chloramphenicol acetyltransferase (CAT), T7 RNA polymerase, green fluorescent protein (GFP), and mutRNA
on a lpp promoter and rrnC terminator) by electroporation. Transformants were recovered in 1 ml of LB for 1 h before plating on LB-agar supplemented with kanamycin, chloramphenicol, and arabinose at 30, 50 mg/liter and 0.2% wt/vol, respectively. After 12 h at 37°C colonies were scraped into liquid LB with the same supplements and amplified for 1 h. Plasmid DNA was extracted, and the pBK library members were resolved from pJ17B3 by agarose gel electrophoresis. The selected pBK mutants were transformed into DH10B containing pREP(2)/YC-JYCUA [this reporter contains genes for chloramphenicol acetyltransferase (CAT), T7 RNA polymerase, green fluorescent protein (GFP), and mutRNA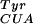 ] and plated on GMML-agar supplemented with kanamycin, tetracycline, and chloramphenicol and pBpa at 30, 20, 60 mg/liter and 1 mM, respectively. Plates were incubated at 37°C for 48 h, after which time the pBK plasmid was isolated and applied to further rounds of positive and negative selection on agar plates.
] and plated on GMML-agar supplemented with kanamycin, tetracycline, and chloramphenicol and pBpa at 30, 20, 60 mg/liter and 1 mM, respectively. Plates were incubated at 37°C for 48 h, after which time the pBK plasmid was isolated and applied to further rounds of positive and negative selection on agar plates.
Characterization of Mutant Synthetases.
Individual synthetase clones in DH10B/pREP(2)/YC-JYCUA were used to inoculate 0.5 ml of LB supplemented with kanamycin and tetracycline to 30 and 20 mg/liter. After 20 h growth (37°C, 300 rpm) cells were diluted 104-fold in dH20 and replica-spotted on two sets of GMML plates. The plates in one set were supplemented with kanamycin and tetracycline at 30 and 20 mg/liter, respectively, and chloramphenicol at concentrations ranging from 0 to 150 mg/liter. The plates in the second set were identical with the first, except that they were supplemented with 1 mM pBpa.
After 48 h the IC50 of chloramphenicol resistance in the presence and absence of pBpa was calculated from the concentration of chloramphenicol at which half the number of colonies on the plates with no chloramphenicol were visible. GFP expression in the presence and absence of pBpa was imaged by using a Storm PhosphorImager (Molecular Dynamics). Mutant synthetase genes exhibiting the strongest amino acid dependence in both GFP signal and chloramphenicol resistance were isolated and sequenced by standard methods.
Protein Expression.
Plasmid PYC/SjGSTmut, which contains the mutant Schistosoma japonica glutathione S-transferase (SjGST) gene on an arabinose promoter and rrnB terminator, mutRNA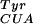 on a lpp promoter and rrnC terminator, and a tetracycline resistance marker was cotransformed with a pBK vector expressing MjBpaRS-1 into DH10B E. coli. Cells were amplified in 10 ml of 2× YT containing kanamycin at 30 mg/liter and tetracycline at 25 mg/liter before being washed in PBS and used to inoculate 1 liter of liquid GMML with the appropriate antibiotics and pBpa to 1 mM. Protein expression was induced at an OD600 of 0.6 by the addition of arabinose to 0.2% followed by 5 h growth. Cells were harvested by centrifugation, and protein was purified by virtue of a C-terminal hexahistidine tag by using nickel-nitrilotriacetic acid affinity chromatography.
on a lpp promoter and rrnC terminator, and a tetracycline resistance marker was cotransformed with a pBK vector expressing MjBpaRS-1 into DH10B E. coli. Cells were amplified in 10 ml of 2× YT containing kanamycin at 30 mg/liter and tetracycline at 25 mg/liter before being washed in PBS and used to inoculate 1 liter of liquid GMML with the appropriate antibiotics and pBpa to 1 mM. Protein expression was induced at an OD600 of 0.6 by the addition of arabinose to 0.2% followed by 5 h growth. Cells were harvested by centrifugation, and protein was purified by virtue of a C-terminal hexahistidine tag by using nickel-nitrilotriacetic acid affinity chromatography.
Sperm whale myoglobin was expressed and purified from cells containing pBAD/JYAMB-4TAG in an analogous manner to SjGST, except that induction was constitutive with 0.002% arabinose. Samples for mass spectrometry were desalted on a NAP-10 column (Amersham Pharmacia) and purified by HPLC. To verify the incorporation of pBpa, the protein mass was ascertained by electrospray-ionization ion trap mass spectrometry.
Mutant SjGST Cloning.
Mutant SjGST genes were assembled by overlapping PCR, with pGEX-3 (Amersham Pharmacia) as a template. All PCRs were performed by using the Expand PCR kit (Roche Diagnostics) according to the manufacturer's instructions. The resulting genes were digested with NcoI and KpnI restriction enzymes and cloned into predigested, dephosphorylated pBADJYC vector between the same restriction sites and in-frame with a C-terminal hexahistidine tag. All final constructs were confirmed by DNA sequencing.
Photoactivated Crosslinking.
Crosslinking reactions were performed in a 96-well microtiter plate (Nunclon, Nalge Nunc, Denmark) by using 100 μl of SjGST (in 50 mM NaH2PO4/300 mM NaCl/250 mM imidazole) at 4°C. Samples were irradiated at 365 nm by using a handheld UV lamp (115V, 60 Hz, 0.2 A; Spectronics, Westbury, NY), for 1 or 5 min. Samples were removed from the wells and diluted with SDS loading buffer before resolution of products by SDS/PAGE on a 10–20% gradient gel. SjGST was transferred to poly(vinylidene difluoride) (Bio-Rad) and probed by Western blot by using goat anti-GST (Amersham Pharmacia) and a secondary mouse anti-goat horseradish peroxidase conjugate (Sigma). The signal was developed by using SUPER SIGNAL WEST (Pierce) and visualized by exposure on hyperfilm (Amersham Pharmacia).
Results and Discussion
The MjTyrRS was used as a starting point for the generation of an orthogonal synthetase that incorporates pBpa, but not any of the 20 common amino acids. This synthetase does not aminoacylate any endogenous E. coli tRNAs with tyrosine (12), but does aminoacylate a mutant tyrosine amber suppressor (mutRNA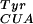 ) (13). We used a MjTyrRS library of mutants in which five residues (Tyr-34, Glu-107, Asp-158, Ile-159, Leu-162) were randomized (1). These residues were chosen on the basis of the crystal structure of Bacillus stearothermophilus TyrRS complexed with tyrosyl adenylate (14) in which homologous residues (Tyr-34, Asn-123, Asp-176, Phe-177, Leu-180) are within 6 Å of the para position of the aryl ring of bound tyrosine (Fig. 1B). The mutant TyrRS library was passed through a positive selection based on suppression of an amber stop codon at a permissive site (Asp-112) in the CAT gene. Cells transformed with the synthetase library, mutRNA
) (13). We used a MjTyrRS library of mutants in which five residues (Tyr-34, Glu-107, Asp-158, Ile-159, Leu-162) were randomized (1). These residues were chosen on the basis of the crystal structure of Bacillus stearothermophilus TyrRS complexed with tyrosyl adenylate (14) in which homologous residues (Tyr-34, Asn-123, Asp-176, Phe-177, Leu-180) are within 6 Å of the para position of the aryl ring of bound tyrosine (Fig. 1B). The mutant TyrRS library was passed through a positive selection based on suppression of an amber stop codon at a permissive site (Asp-112) in the CAT gene. Cells transformed with the synthetase library, mutRNA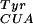 , and the CAT mutant were challenged to grow in the presence of 1 mM pBpa and chloramphenicol. Surviving cells contained synthetases capable of charging the orthogonal tRNA with either a natural or unnatural amino acid. These synthetase genes were transferred into cells containing mutRNA
, and the CAT mutant were challenged to grow in the presence of 1 mM pBpa and chloramphenicol. Surviving cells contained synthetases capable of charging the orthogonal tRNA with either a natural or unnatural amino acid. These synthetase genes were transferred into cells containing mutRNA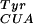 and a variant of the gene encoding the toxic barnase protein, which contains three amber mutations at permissive sites (Gln-2, Asp-44, Gly-65) (1). Growth of these cells in the absence of pBpa selected against synthetases capable of using natural amino acids.
and a variant of the gene encoding the toxic barnase protein, which contains three amber mutations at permissive sites (Gln-2, Asp-44, Gly-65) (1). Growth of these cells in the absence of pBpa selected against synthetases capable of using natural amino acids.
After five rounds of positive and negative selection (Fig. 1C) the surviving synthetase plasmids were transformed into a reporter strain in which the production of full-length CAT and T7 RNA polymerase depend on suppression of amber stop codons in the CAT and T7 RNA polymerase gene, respectively (S. W. Santoro and P.G.S., unpublished data). Because the T7 RNA polymerase drives expression of the GFP these cells can be fluorometrically screened. Ninety-six clones were screened for pBpa-dependent chloramphenicol resistance and GFP fluorescence (Fig. 2A). Six distinct synthetases conferred chloramphenicol resistance on E. coli with IC50 values of 120 and 5 mg/liter in the presence and absence of 1 mM pBpa, respectively; they also showed pBpa-dependent GFP fluorescence. The large difference between the chloramphenicol resistance in the presence and absence of pBpa suggests a substantial in vivo specificity of the selected synthetase/tRNA pairs for insertion of pBpa over all 20 natural amino acids found in the cell in response to an amber codon.
Fig 2.

Selection and characterization of a BpaRS. (A) After five rounds of selection, the Bpa-dependent GFP fluorescence of 96 independent clones (48 shown) was screened by replica spotting [DH10B cells cotransformed with individual selected synthetase genes and pREP(2)/YC-JYCUA] onto GMML agar plates in the presence (Upper Left) and absence (Upper Right) of 1 mM pBpa. Upon excitation with a blue laser, amino acid-dependent GFP expression was clearly visible (Lower Right vs. Lower Left). (B) Bpa-dependent expression of sperm whale myoglobin in response to an amber codon at the fourth position in the gene for this protein. The protein yield was similar to that observed when suppressing the amber codon with MjTyrRS and mutRNA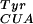
To measure the fidelity and efficiency of pBpa incorporation, the codon for Ser-4 in sperm whale myoglobin (containing a C-terminal His-6 tag) was converted to an amber codon. In the presence of both M. jannaschii BpaRS-1/mutRNA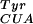 and pBpa, full-length myoglobin was produced with a purified yield of 2 mg/liter of culture. This yield is similar to that found when MjTyrRS/mutRNA
and pBpa, full-length myoglobin was produced with a purified yield of 2 mg/liter of culture. This yield is similar to that found when MjTyrRS/mutRNA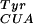 pair suppresses the same amber codon (Fig. 2B). No myoglobin protein was detectable by silver stain or Western blot against the C-terminal His-6 tag on myoglobin if any of the three components responsible for specific amber suppression with pBpa (amino acid, synthetase, or tRNA) were withheld. These data provide further evidence that the selected synthetase is very selective for pBpa. Electrospray-ionization ion trap mass spectrometry of the mutant myoglobin gave a mass of 18,519 ± 0.5, which is identical with the calculated mass of 18,519.0 for the pBpa-containing protein. This result confirms the incorporation of pBpa at a single site in the protein. No masses were observed in the mass spectra corresponding to natural amino acid incorporation providing additional evidence for the high-fidelity incorporation of pBpa.
pair suppresses the same amber codon (Fig. 2B). No myoglobin protein was detectable by silver stain or Western blot against the C-terminal His-6 tag on myoglobin if any of the three components responsible for specific amber suppression with pBpa (amino acid, synthetase, or tRNA) were withheld. These data provide further evidence that the selected synthetase is very selective for pBpa. Electrospray-ionization ion trap mass spectrometry of the mutant myoglobin gave a mass of 18,519 ± 0.5, which is identical with the calculated mass of 18,519.0 for the pBpa-containing protein. This result confirms the incorporation of pBpa at a single site in the protein. No masses were observed in the mass spectra corresponding to natural amino acid incorporation providing additional evidence for the high-fidelity incorporation of pBpa.
The selected synthetases show interesting sequence convergence (Table 1). Tyr-32 of MjTyrRS is converted to alanine or glycine in five of the six mutant synthetase clones. Asp-158 of the MjTyrRS is converted to threonine in five of the six selected mutants, whereas Ile-159 is converted to serine in four of the six mutants. Serine or proline substitutions dominate at position 107 of MjTyrRS; Leu-162 is conserved in four of the six mutants. A consensus set of mutations (32:Gly, Ala/107:Ser, Pro/158:Thr/159:Ser/162:Leu) emerges from this analysis.
Table 1.
Sequence convergence of selected synthetases
| MjTyrRS
|
Residue | ||||
|---|---|---|---|---|---|
| 32 Tyr | 107 Glu | 158 Asp | 159 Ile | 162 Leu | |
| MjBpaRS-1 | Gly | Ser | Thr | Ser | Leu |
| MjBpaRS-2 | Ala | Leu | Thr | Ala | Leu |
| MjBpaRS-3 | Gly | Pro | Thr | Ser | Leu |
| MjBpaRS-4 | Trp | Ser | Thr | Ser | Arg |
| MjBpaRS-5 | Ala | Ser | Thr | Ser | Ala |
| MjBpaRS-6 | Gly | Pro | Gly | Thr | Leu |
To begin to understand how these mutations alter the specificity of the synthetase, the x-ray crystal structure of the B. stearothermophilus tyrosyl-tRNA synthetase with bound tyrosyl adenylate (Fig. 1B) was used to model the complex between the pBpa adenylate and the selected consensus for M. jannaschii p-benzoyl-L-phenylalanyl-tRNA synthetase (MjBpaRS). In the wild-type tyrosyl synthetase the side chain of Tyr-32 accepts a hydrogen bond from the phenolic hydroxyl of bound tyrosine. In the mutant synthetases the selected alanines and glycines are unable to act as hydrogen bond donors, which likely disfavors the binding of tyrosine. In addition, the smaller size of the selected amino acids at this position in the active site may create a cavity that accommodates the second aryl ring of pBpa. In the wild-type tyrosyl-tRNA synthetase the carboxylate of Asp-158 also accepts a hydrogen bond from the phenolic hydroxyl of bound tyrosine. In this position the selected mutants contain threonine, which may act as a hydrogen bond donor to the side-chain carbonyl oxygen of pBpa. The selection of serine at position 159 may provide a second hydrogen bond donor for the ketone substrate.
To demonstrate the utility of this methodology for mapping protein–protein interactions, a model crosslinking experiment was performed with GST. This protein is a dimer of two identical subunits that have been crosslinked nonspecifically by using gluteraldehyde (15). The crystal structure of the dimeric SjGST (16) was used to identify two sites to substitute with pBpa: residue Phe-52, which is buried in the dimer interface of the crystal structure, and residue Tyr-198, which is solvent-exposed (Fig. 3A).
Fig 3.

Photocrosslinking reactions with pBpa. (A) The residues of SjGST mutated to pBpa. Monomers of the dimer are shown in blue and red. The side chain of Phe-52 is shown in orange for each monomer (Left). The side chain of residue Tyr-198 is shown in orange (Right). (B) Surface-specific protein–protein crosslinking. SjGST Phe-52-pBpa or SjGST Tyr-198-pBpa were irradiated for 0, 1, and 5 min with a 360-nm lamp, and monomer and covalent dimer were resolved by SDS/PAGE.
The codons corresponding to Phe-52 or Tyr-198 in the gene for the 27-kDa protein SjGST, were replaced with amber codons. The orthogonal synthetase/tRNA pair was then used to site specifically incorporate pBpa into SjGST in E. coli at these sites. The yield of purified protein from these mutants was 0.1 mg/liter of culture. (For comparison, the yield of protein from wild-type GST, expressed from a gene containing no amber codons, under the same conditions was ≈0.4 mg/liter.) Upon irradiation with long-wavelength UV radiation, purified SjGST (Phe-52-Bpa) was converted to a covalently linked homodimer as judged by denaturing SDS/PAGE (Fig. 3B). More than 50% of the SjGST present was crosslinked in 5 min. In contrast, control experiments with either wild-type SjGST or SjGST containing pBpa at residue 198, which lies outside the dimer interface, shows no detectable crosslinking in response to UV irradiation. These results demonstrate that site-specific pBpa substitution can be used to define amino acids involved in a protein–protein interaction.
In conclusion, we have developed a tool for site specifically incorporating pBpa into proteins expressed in E. coli with high translational fidelity and efficiency. The method reported herein can in principle be applied to any protein that is expressed in E. coli, irrespective of its size or sequence. Moreover, because the unnatural amino acid is incorporated genetically, it may be possible to map protein–protein interactions in vivo (J.W.C. and P.G.S., unpublished work) by using this methodology.
Acknowledgments
We thank Stephen W. Santoro for the vector pREP(2)/YC-JYCUA and Ryan A. Mehl for assistance with protein purification. J.W.C. is supported by a Damon Runyon Cancer Research Foundation Fellowship DRG 1707-02. This work was supported by the Department of Energy (DE-FG03 00FR45812), the National Institutes of Health (GM62159), and the Skaggs Institute. This is manuscript no. 14951-CH of the Scripps Research Institute.
Abbreviations
pBpa, p-benzoyl-l-phenylalanine
GMML, glycerol minimal media with leucine
GFP, green fluorescent protein
CAT, chloramphenicol acetyltransferase
GST, glutathione S-transferase
MjTyrRS, Methanococcus jannaschii tyrosyl-tRNA synthetase
SjGST, Schistosoma japonica GST
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wang L., Brock, A., Herberich, B. & Schultz, P. G. (2001) Science 292, 498-500. [DOI] [PubMed] [Google Scholar]
- 2.Dorman G. & Prestwich, G. D. (1994) Biochemistry 33, 5661-5673. [DOI] [PubMed] [Google Scholar]
- 3.Galardy R. E., Craig, L. C. & Printz, M. P. (1973) Nat. New Biol. 242, 127-128. [DOI] [PubMed] [Google Scholar]
- 4.Turro N. J., (1978) Modern Molecular Photochemistry (Benjamin/Cummings Publishing, Menlo Park, CA).
- 5.Kauer J. C., Erickson-Viitanen, S., Wolfe, H. R. & DeGrado, W. F. (1986) J. Biol. Chem. 261, 10695-10700. [PubMed] [Google Scholar]
- 6.Breslow R. (1986) Adv. Enzymol. Relat. Areas Mol. Biol. 58, 1-60. [DOI] [PubMed] [Google Scholar]
- 7.Breslow R. (1980) Acc. Chem. Res. 13, 170-177. [Google Scholar]
- 8.Williams N., Ackerman, S. H. & Coleman, P. S. (1986) Methods Enzymol. 126, 667-682. [DOI] [PubMed] [Google Scholar]
- 9.Keutmann H. T. & Rubin, D. A. (1993) Endocrinology 132, 1305-1312. [DOI] [PubMed] [Google Scholar]
- 10.Cornish V. W., Benson, D. R., Altenbach, C. A., Hideg, K., Hubbell, W. L. & Schultz, P. G. (1994) Proc. Natl. Acad. Sci. USA 91, 2910-2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanamori T., Nishikawa, S.-I., Shin, I., Schultz, P. G. & Endo, T. (1997) Proc. Natl. Acad. Sci. USA 94, 485-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L., Magliery, T. J., Liu, D. R. & Schultz, P. G. (2000) J. Am. Chem. Soc. 122, 5010-5011. [Google Scholar]
- 13.Wang L. & Schultz, P. G. (2001) Chem. Biol. 8, 883-890. [DOI] [PubMed] [Google Scholar]
- 14.Brick P., Bhat, T. N. & Blow, D. M. (1989) J. Mol. Biol. 208, 83-98. [DOI] [PubMed] [Google Scholar]
- 15.Maru Y., Afar, D. E., Witte, O. N. & Shibuya, M. (1996) J. Biol. Chem. 271, 15353-15357. [DOI] [PubMed] [Google Scholar]
- 16.McTigue M. A., Williams, D. R. & Tainer, J. A. (1995) J. Mol. Biol. 246, 21-27. [DOI] [PubMed] [Google Scholar]